
Clinical Pharmacology of Bacteriophage Therapy: A Focus on Multidrug-Resistant Pseudomonas aeruginosa Infections

Abstract
1. Introduction
2. Overview of Pseudomonas aeruginosa: Clinical Impact
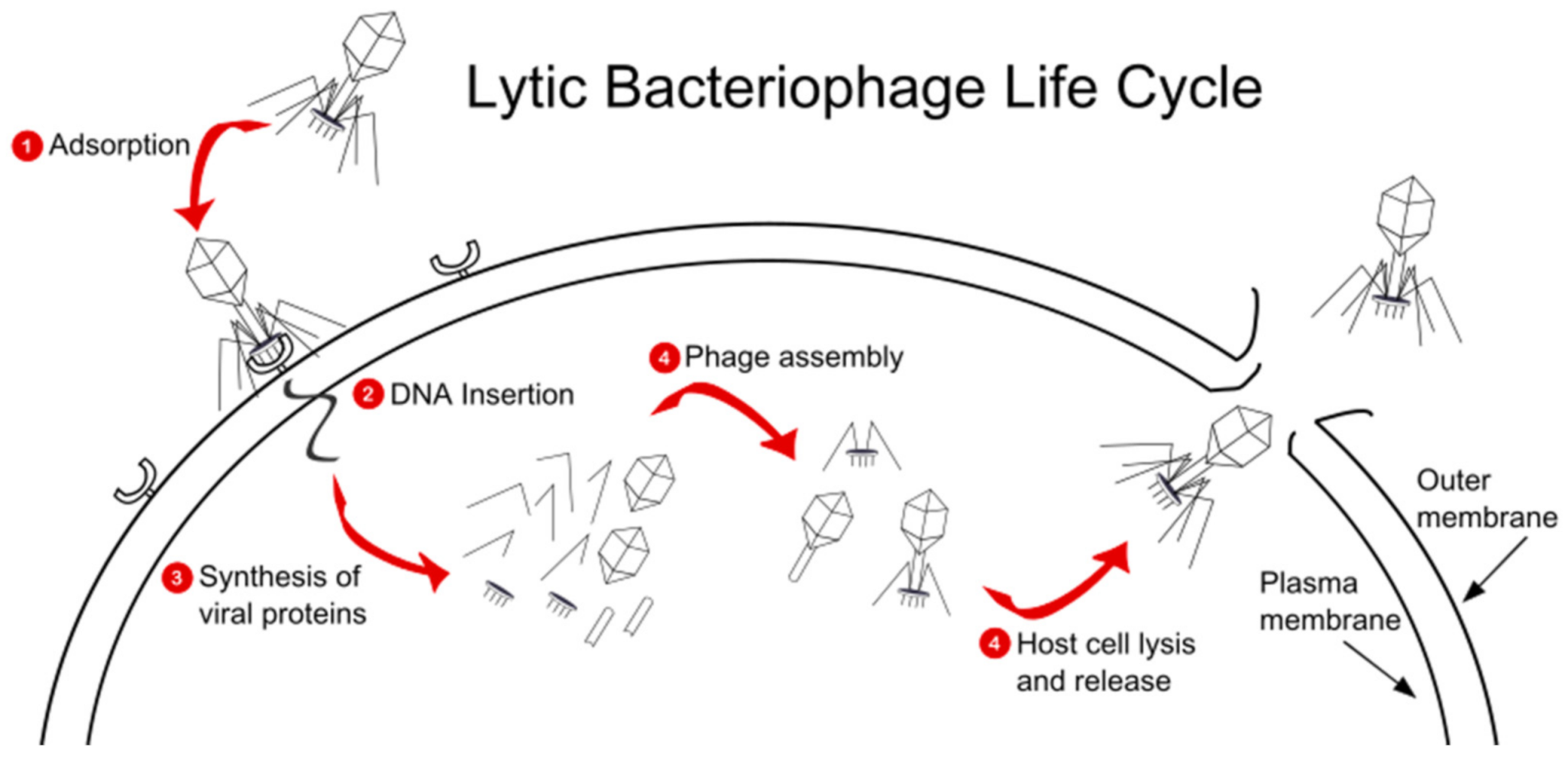
3. Pseudomonas Bacteriophages Background
4. Benefits of Pseudomonas Phage Therapy
5. Phage-Dosing Strategies for MDR Pseudomonas aeruginosa Infections
6. Phage Selection for MDR Pseudomonas aeruginosa Infections
7. Pharmacokinetics of Pseudomonas Phage Therapy
8. Formulation Considerations
9. Efficacy of Phage Therapy against MDR Pseudomonas aeruginosa
10. Remaining Gaps in Literature
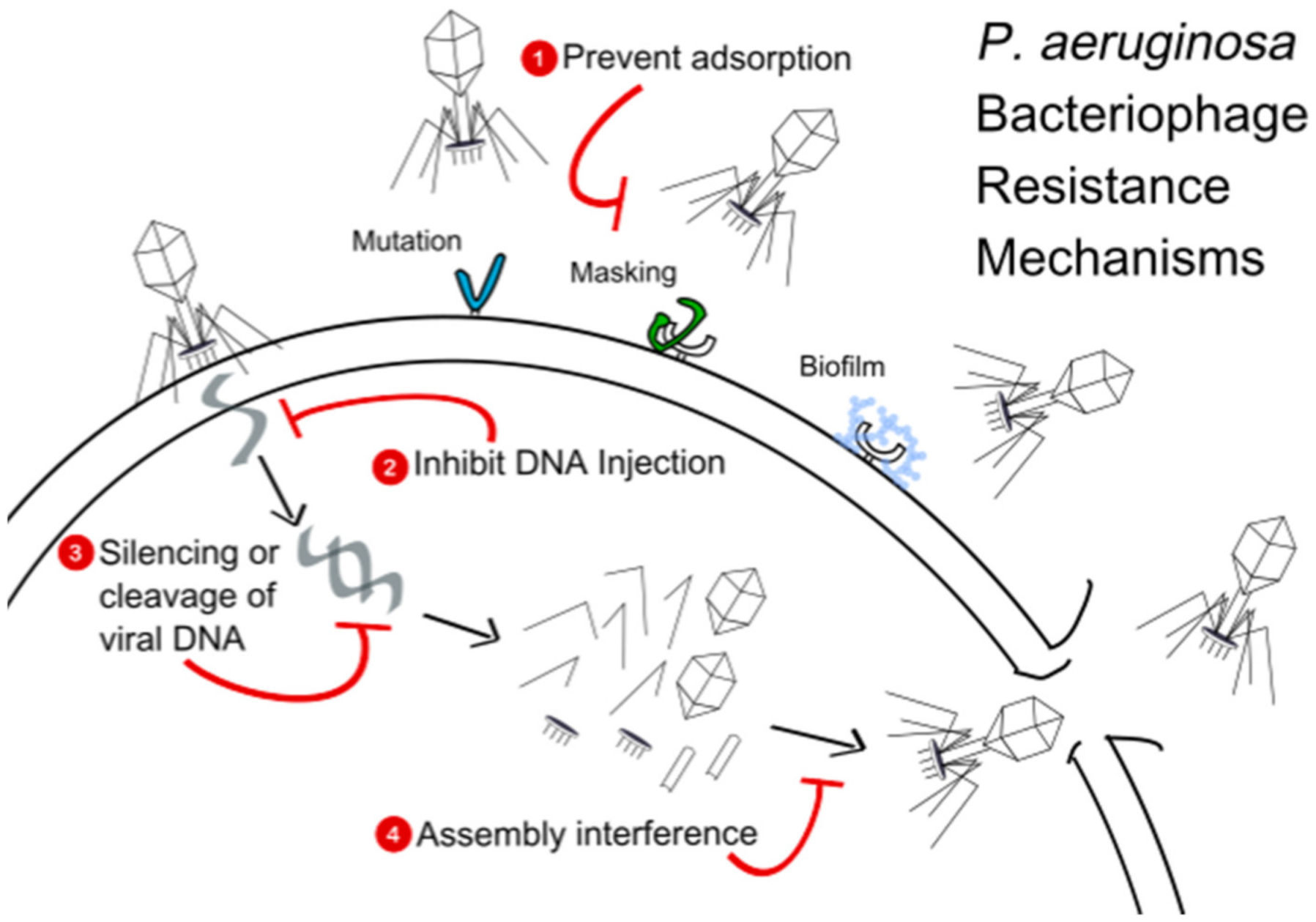
11. Challenges Ahead
11.1. Possibility of the Infected Organism Acquiring Virulence Traits from the Phage
11.2. Adsorption Inhibition
12. Potential of Pseudomonas Phage Therapy
13. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- CDC. Antibiotic Resistance Threats in the United States; U.S. Department of Health and Human Services, CDC: Atlanta, GA, USA, 2019.
- Bonomo, R.A.; Szabo, D. Mechanisms of Multidrug Resistance in Acinetobacter Species and Pseudomonas aeruginosa. Clin. Infect. Dis. 2006, 43 (Suppl. 2), S49–S56. [Google Scholar] [CrossRef] [PubMed]
- Tkhilaishvili, T.; Wang, L.; Perka, C.; Trampuz, A.; Gonzalez Moreno, M. Using Bacteriophages as a Trojan Horse to the Killing of Dual-Species Biofilm Formed by Pseudomonas aeruginosa and Methicillin Resistant Staphylococcus aureus. Front. Microbiol. 2020, 11, 695. [Google Scholar] [CrossRef] [PubMed]
- Sader, H.S.; Castanheira, M.; Duncan, L.R.; Flamm, R.K. Antimicrobial Susceptibility of Enterobacteriaceae and Pseudomonas aeruginosa Isolates from United States Medical Centers Stratified by Infection Type: Results from the International Network for Optimal Resistance Monitoring (INFORM) Surveillance Program, 2015–2016. Diagn. Microbiol. Infect. Dis. 2018, 92, 69–74. [Google Scholar] [CrossRef]
- World Health Organization. 2019 Antibacterial Agents in Clinical Development: An Analysis of the Antibacterial Clinical Development Pipeline; Licence: CC BY-NC-SA 3.0 IGO; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Hill, C.; Mills, S.; Ross, R.P. Phages & antibiotic resistance: Are the most abundant entities on earth ready for a comeback? Future Microbiol. 2018, 13, 711–726. [Google Scholar] [CrossRef]
- Wittebole, X.; Roock, S.D.; Opal, S.M. A historical overview of bacteriophage therapy as an alternative to antibiotics for the treatment of bacterial pathogens. Virulence 2014, 5, 226–235. [Google Scholar] [CrossRef]
- Taati Moghadam, M.; Khoshbayan, A.; Chegini, Z.; Farahani, I.; Shariati, A. Bacteriophages, a New Therapeutic Solution for Inhibiting Multidrug-Resistant Bacteria Causing Wound Infection: Lesson from Animal Models and Clinical Trials. Drug Des. Dev. Ther. 2020, 14, 1867–1883. [Google Scholar] [CrossRef]
- Myelnikov, D. An Alternative Cure: The Adoption and Survival of Bacteriophage Therapy in the USSR, 1922–1955. J. Hist. Med. Allied Sci. 2018, 73, 385–411. [Google Scholar] [CrossRef] [PubMed]
- Antoine, C.; Laforêt, F.; Blasdel, B.; Glonti, T.; Kutter, E.; Pirnay, J.P.; Mainil, J.; Delcenserie, V.; Thiry, D. Efficacy assessment of PEV2 phage on Galleria mellonella larvae infected with a Pseudomonas aeruginosa dog otitis isolate. Res. Vet. Sci. 2021, 15, 598–601. [Google Scholar] [CrossRef]
- Chen, F.; Cheng, X.; Li, J.; Yuan, X.; Huang, X.; Lian, M.; Li, W.; Huang, T.; Xie, Y.; Liu, J.; et al. Novel Lytic Phages Protect Cells and Mice against Pseudomonas aeruginosa Infection. J. Virol. 2021, 8, e01832-20. [Google Scholar]
- Aslam, S.; Lampley, E.; Wooten, D.; Karris, M.; Benson, C.; Strathdee, S.; Schooley, R.T. Lessons Learned from the First 10 Consecutive Cases of Intravenous Bacteriophage Therapy to Treat Multidrug-Resistant Bacterial Infections at a Single Center in the United States. Open Forum Infect. Dis. 2020, 7, ofaa389. [Google Scholar] [CrossRef]
- Wright, A.; Hawkins, C.H.; Anggård, E.E.; Harper, D.R. A controlled clinical trial of a therapeutic bacteriophage preparation in chronic otitis due to antibiotic-resistant Pseudomonas aeruginosa; a preliminary report of efficacy. Clin. Otolaryngol. 2009, 34, 349–357. [Google Scholar] [CrossRef]
- Markoishvili, K.; Tsitlanadze, G.; Katsarava, R.; Glenn, J.; Sulakvelidze, A. A novel sustained-release matrix based on biodegradable poly(ester amide)s and impregnated with bacteriophages and an antibiotic shows promise in management of infected venous stasis ulcers and other poorly healing wounds. Int. J. Dermatol. 2002, 41, 453–458. [Google Scholar] [CrossRef]
- Gupta, P.; Singh, H.S.; Shukla, V.K.; Nath, G.; Bhartiya, S.K. Bacteriophage Therapy of Chronic Nonhealing Wound: Clinical Study. Int. J. Low. Extrem. Wounds. 2019, 18, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Weber-Dąbrowska, B.; Mulczyk, M.; Górski, A. Bacteriophages as an efficient therapy for antibiotic-resistant septicemia in man. Transplant. Proc. 2003, 35, 1385–1386. [Google Scholar] [CrossRef]
- Weber-Dabrowska, B.; Mulczyk, M.; Górski, A. Bacteriophage therapy for infections in cancer patients. Clin. Appl. Immunol. Rev. 2001, 1, 131–134. [Google Scholar] [CrossRef]
- Jończyk-Matysiak, E.; Weber-Dąbrowska, B.; Owczarek, B.; Międzybrodzki, R.; Łusiak-Szelachowska, M.; Łodej, N.; Górski, A. Phage-Phagocyte Interactions and Their Implications for Phage Application as Therapeutics. Viruses 2017, 9, 150. [Google Scholar] [CrossRef]
- Kantoch, M. The role of phagocytes in virus infections. Arch. Immunol. Ther. Exp. 1961, 9, 261–340. [Google Scholar]
- WHO Publishes List of Bacteria for which New Antibiotics are Urgently Needed. Available online: https://www.who.int/news-room/detail/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-neneed (accessed on 11 September 2020).
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef]
- Daly, J.A.; Boshard, R.; Matsen, J.M. Differential primary plating medium for enhancement of pigment production by Pseudomonas aeruginosa. J. Clin. Microbiol. 1984, 19, 742–743. [Google Scholar] [CrossRef]
- Lister, P.D.; Wolter, D.J.; Hanson, N.D. Antibacterial-Resistant Pseudomonas aeruginosa: Clinical Impact and Complex Regulation of Chromosomally Encoded Resistance Mechanisms. Clin. Microbiol. Rev. 2009, 22, 582–610. [Google Scholar] [CrossRef]
- Diggle, S.P.; Whiteley, M. Microbe Profile: Pseudomonas aeruginosa: Opportunistic pathogen and lab rat. Microbiology 2020, 166, 30–33. [Google Scholar] [CrossRef]
- Lalancette, C.; Charron, D.; Laferrière, C.; Dolcé, P.; Déziel, E.; Prévost, M.; Bédard, E. Hospital Drains as Reservoirs of Pseudomonas aeruginosa: Multiple-Locus Variable-Number of Tandem Repeats Analysis Genotypes Recovered from Faucets, Sink Surfaces and Patients. Pathogens 2017, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.N.; Bidell, M.R.; Lodise, T.P. Approach to the Treatment of Patients with Serious Multidrug-Resistant Pseudomonas aeruginosa Infections. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2020, 40, 952–969. [Google Scholar] [CrossRef] [PubMed]
- Healthcare-Associated Infections—Community Interface (HAIC): Multi-site Gram-Negative Surveillance Initiative. Available online: https://www.cdc.gov/hai/eip/mugsi.html (accessed on 13 October 2020).
- Peña, C.; Cabot, G.; Gómez-Zorrilla, S.; Zamorano, L.; Ocampo-Sosa, A.; Murillas, J.; Almirante, B.; Pomar, V.; Aguilar, M.; Granados, A.; et al. Influence of Virulence Genotype and Resistance Profile in the Mortality of Pseudomonas aeruginosa Bloodstream Infections. Clin. Infect. Dis. 2015, 60, 539–548. [Google Scholar] [CrossRef]
- Sader, H.S.; Huband, M.D.; Castanheira, M.; Flamm, R.K. Pseudomonas aeruginosa Antimicrobial Susceptibility Results from Four Years (2012 to 2015) of the International Network for Optimal Resistance Monitoring Program in the United States. Antimicrob. Agents Chemother. 2017, 61, e02252-16. [Google Scholar] [CrossRef] [PubMed]
- Walkty, A.; Lagace-Wiens, P.; Adam, H.; Baxter, M.; Karlowsky, J.; Mulvey, M.R.; McCracken, M.; Zhanel, G.G. Antimicrobial susceptibility of 2906 Pseudomonasaeruginosa clinical isolates obtained from patients in Canadian hospitals over a period of 8 years: Results of the Canadian Ward surveillance study (CANWARD), 2008–2015. Diagn. Microbiol. Infect. Dis. 2017, 87, 60–63. [Google Scholar] [CrossRef]
- Donlan, R.M. Biofilms: Microbial Life on Surfaces. Emerg. Infect. Dis. 2002, 8, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Jakutytė, L.; Baptista, C.; São-José, C.; Daugelavičius, R.; Carballido-López, R.; Tavares, P. Bacteriophage Infection in Rod-Shaped Gram-Positive Bacteria: Evidence for a Preferential Polar Route for Phage SPP1 Entry in Bacillus subtilis. J. Bacteriol. 2011, 193, 4893–4903. [Google Scholar] [CrossRef]
- Criscuolo, E.; Spadini, S.; Lamanna, J.; Ferro, M.; Burioni, R. Bacteriophages and Their Immunological Applications against Infectious Threats. J. Immunol. Res. 2017, 2017, 3780697. [Google Scholar] [CrossRef] [PubMed]
- Jurczak-Kurek, A.; Gąsior, T.; Nejman-Faleńczyk, B.; Bloch, S.; Dydecka, A.; Topka, G.; Necel, A.; Jakubowska-Deredas, M.; Narajczyk, M.; Richert, M.; et al. Biodiversity of bacteriophages: Morphological and biological properties of a large group of phages isolated from urban sewage. Sci. Rep. 2016, 6, 34338. [Google Scholar] [CrossRef]
- Duckworth, D.H. Who discovered bacteriophage? Bacteriol. Rev. 1976, 40, 793–802. [Google Scholar] [CrossRef]
- Herelle, F.; Smith, G.H. The Bacteriophage, Its Rôle in Immunity; Williams & Wilkins: Philadelphia, PA, USA, 1922. [Google Scholar]
- Kellenberger, G.; Kellenberger, E. Electron microscopical studies of phage multiplication: III. Observation of single cell bursts. Virology 1957, 3, 275–285. [Google Scholar] [CrossRef]
- Holloway, B.W.; Egan, J.B.; Monk, M. Lysogeny in Pseudomonas Aeruginosa. Aust. J. Exp. Biol. Med. Sci. 1960, 38, 321–330. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04803708 (accessed on 13 April 2021).
- Pires, D.P.; Boas, D.V.; Sillankorva, S.; Azeredo, J. Phage Therapy: A Step Forward in the Treatment of Pseudomonas aeruginosa Infections. J. Virol. 2015, 89, 7449–7456. [Google Scholar] [CrossRef] [PubMed]
- Davies, D. Understanding biofilm resistance to antibacterial agents. Nat. Rev. Drug Discov. 2003, 2, 114–122. [Google Scholar] [CrossRef]
- Kirby, A.E.; Garner, K.; Levin, B.R. The Relative Contributions of Physical Structure and Cell Density to the Antibiotic Susceptibility of Bacteria in Biofilms. Antimicrob. Agents Chemother. 2012, 56, 2967–2975. [Google Scholar] [CrossRef]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial biofilms: A common cause of persistent infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.K.; Das, T.; Manos, J.; Kutter, E.; Morales, S.; Chan, H.K. Bacteriophage PEV20 and ciprofloxacin combination treatment enhances removal of P. aeruginosa biofilm isolated from cystic fibrosis and wound patients. AAPS J. 2019, 21, 49. [Google Scholar] [CrossRef]
- Alemayehu, D.; Casey, P.G.; McAuliffe, O.; Guinane, C.M.; Martin, J.G.; Shanahan, F.; Coffey, A.; Ross, R.P.; Hill, C. Bacteriophages ϕMR299-2 and ϕNH-4 Can Eliminate Pseudomonas aeruginosa in the Murine Lung and on Cystic Fibrosis Lung Airway Cells. mBio 2012, 3. [Google Scholar] [CrossRef]
- Law, N.; Logan, C.; Yung, G.; Furr, C.L.; Lehman, S.M.; Morales, S.; Rosas, F.; Gaidamaka, A.; Bilinsky, I.; Grint, P.; et al. Successful adjunctive use of bacteriophage therapy for treatment of multidrug-resistant Pseudomonas aeruginosa infection in a cystic fibrosis patient. Infection 2019, 47, 665–668. [Google Scholar] [CrossRef]
- Skurnik, M.; Pajunen, M.; Kiljunen, S. Biotechnological challenges of phage therapy. Biotechnol. Lett. 2007, 29, 995–1003. [Google Scholar] [CrossRef]
- Loc-Carrillo, C.; Abedon, S.T. Pros and cons of phage therapy. Bacteriophage 2011, 1, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Tamma, P.D.; Aitken, S.L.; Bonomo, R.A.; Mathers, A.J.; van Duin, D.; Clancy, C.J. Infectious Diseases Society of America Antimicrobial Resistant Treatment Guidance: Gram-Negative Bacterial Infections. Clin. Infect. Dis. 2020, ciaa1478. [Google Scholar] [CrossRef] [PubMed]
- Mentzelopoulos, S.D.; Pratikaki, M.; Platsouka, E.; Kraniotaki, H.; Zervakis, D.; Koutsoukou, A.; Nanas, S.; Paniara, O.; Roussos, C.; Giamarellos-Bourboulis, E.; et al. Prolonged use of carbapenems and colistin predisposes to ventilator-associated pneumonia by pandrug-resistant Pseudomonas aeruginosa. Intensive Care Med. 2007, 33, 1524–1532. [Google Scholar] [CrossRef]
- Falagas, M.E.; Bliziotis, I.A.; Kasiakou, S.K.; Samonis, G.; Athanassopoulou, P.; Michalopoulos, A. Outcome of infections due to pandrug-resistant (PDR) Gram-negative bacteria. BMC Infect. Dis. 2005, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Sistrom, M.; Wertz, J.E.; Kortright, K.E.; Narayan, D.; Turner, P.E. Phage selection restores antibiotic sensitivity in MDR Pseudomonas aeruginosa. Sci. Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, D.; Wolcott, R.; Kuskowski, M.; Wolcott, B.M.; Ward, L.S.; Sulakvelidze, A. Bacteriophage therapy of venous leg ulcers in humans: Results of a phase I safety trial. J. Wound Care. 2009, 18, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Jault, P.; Leclerc, T.; Jennes, S.; Pirnay, J.P.; Que, Y.A.; Resch, G.; Rousseau, A.F.; Ravat, F.; Carsin, H.; Le Floch, R.; et al. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): A randomised, controlled, double-blind phase 1/2 trial. Lancet Infect. Dis. 2019, 19, 35–45. [Google Scholar] [CrossRef]
- Torres-Barceló, C.; Hochberg, M.E. Evolutionary Rationale for Phages as Complements of Antibiotics. Trends Microbiol. 2016, 24, 249–256. [Google Scholar] [CrossRef]
- Comeau, A.M.; Tétart, F.; Trojet, S.N.; Prère, M.F.; Krisch, H.M. Phage-Antibiotic Synergy (PAS): β-Lactam and Quinolone Antibiotics Stimulate Virulent Phage Growth. PLoS ONE 2007, 2, e799. [Google Scholar] [CrossRef]
- Ryan, E.M.; Alkawareek, M.Y.; Donnelly, R.F.; Gilmore, B.F. Synergistic phage-antibiotic combinations for the control of Escherichia coli biofilms in vitro. FEMS Immunol. Med. Microbiol. 2012, 65, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Kebriaei, R.; Lev, K.; Morrisette, T.; Stamper, K.C.; Abdul-Mutakabbir, J.C.; Lehman, S.M.; Morales, S.; Rybak, M.J. Bacteriophage-Antibiotic Combination Strategy: An Alternative against Methicillin-Resistant Phenotypes of Staphylococcus aureus. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef] [PubMed]
- Morrisette, T.; Lev, K.; Kebriaei, R.; Abdul-Mutakabbir, J.C.; Stamper, K.C.; Morales, S.; Lehman, S.M.; Canfield, G.S.; Duerkop, B.A.; Arias, C.A.; et al. Bacteriophage-Antibiotic Combinations for Enterococcus faecium with Varying Bacteriophage and Daptomycin Susceptibilities. Antimicrob. Agents Chemother. 2020, 64, e00993-20. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.; Wahida, A.; Latz, S.; Krüttgen, A.; Häfner, H.; Buhl, E.M.; Ritter, K.; Horz, H.P. Enhanced antibacterial effect of the novel T4-like bacteriophage KARL-1 in combination with antibiotics against multi-drug resistant Acinetobacter baumannii. Sci. Rep. 2018, 8, 14140. [Google Scholar] [CrossRef] [PubMed]
- Cafora, M.; Deflorian, G.; Forti, F.; Ferrari, L.; Binelli, G.; Briani, F.; Ghisotti, D.; Pistocchi, A. Phage therapy against Pseudomonas aeruginosa infections in a cystic fibrosis zebrafish model. Sci. Rep. 2019, 9, 1527. [Google Scholar] [CrossRef]
- Quantitative Models of Phage-Antibiotic Combination Therapy. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7002117/ (accessed on 15 September 2020).
- Chan, B.K.; Turner, P.E.; Kim, S.; Mojibian, H.R.; Elefteriades, J.A.; Narayan, D. Phage treatment of an aortic graft infected with Pseudomonas aeruginosa. Evol. Med. Public Health 2018, 2018, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Viruses in the sea. Nature 2005, 437, 356–361. [Google Scholar] [CrossRef]
- Azizian, R.; Nasser, A.; Askari, H.; Taheri Kalani, M.; Sadeghifard, N.; Pakzad, I.; Amini, R.; Mozaffari Nejad, A.S.; Azizi Jalilian, F. Sewage as a rich source of phage study against Pseudomonas aeruginosa PAO. Biologicals 2015, 43, 238–241. [Google Scholar] [CrossRef]
- Chhibber, S.; Kaur, P.; Gondil, V.S. Simple drop cast method for enumeration of bacteriophages. J. Virol. Methods. 2018, 262, 1–5. [Google Scholar] [CrossRef]
- Fernández, L.; Gutiérrez, D.; García, P.; Rodríguez, A. The Perfect Bacteriophage for Therapeutic Applications—A Quick Guide. Antibiotics 2019, 8, 126. [Google Scholar] [CrossRef]
- Clokie, M.R.; Millard, A.D.; Letarov, A.V.; Heaphy, S. Phages in nature. Bacteriophage 2011, 1, 31–45. [Google Scholar] [CrossRef]
- Wright, R.T.; Friman, V.P.; Smith, M.M.; Brockhurst, M.A. Resistance Evolution against Phage Combinations Depends on the Timing and Order of Exposure. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef] [PubMed]
- León, M.; Bastías, R. Virulence reduction in bacteriophage resistant bacteria. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef]
- Burmeister, A.R.; Fortier, A.; Roush, C.; Lessing, A.J.; Bender, R.G.; Barahman, R.; Grant, R.; Chan, B.K.; Turner, P.E. Pleiotropy complicates a trade-off between phage resistance and antibiotic resistance. Proc. Natl. Acad. Sci. USA 2020, 117, 11207–11216. [Google Scholar] [CrossRef]
- Chibeu, A.; Ceyssens, P.J.; Hertveldt, K.; Volckaert, G.; Cornelis, P.; Matthijs, S.; Lavigne, R. The adsorption of Pseudomonas aeruginosa bacteriophage φKMV is dependent on expression regulation of type IV pili genes. FEMS Microbiol. Lett. 2009, 296, 210–218. [Google Scholar] [CrossRef]
- Le, S.; Yao, X.; Lu, S.; Tan, Y.; Rao, X.; Li, M.; Jin, X.; Wang, J.; Zhao, Y.; Wu, N.C.; et al. Chromosomal DNA deletion confers phage resistance to Pseudomonas aeruginosa. Sci. Rep. 2014, 4, 4738. [Google Scholar] [CrossRef]
- Park, S.C.; Shimamura, I.; Fukunaga, M.; Mori, K.I.; Nakai, T. Isolation of Bacteriophages Specific to a Fish Pathogen, Pseudomonas plecoglossicida, as a Candidate for Disease Control. Appl. Environ. Microbiol. 2000, 66, 1416–1422. [Google Scholar] [CrossRef] [PubMed]
- Forti, F.; Roach, D.R.; Cafora, M.; Pasini, M.E.; Horner, D.S.; Fiscarelli, E.V.; Rossitto, M.; Cariani, L.; Briani, F.; Debarbieux, L.; et al. Design of a Broad-Range Bacteriophage Cocktail That Reduces Pseudomonas aeruginosa Biofilms and Treats Acute Infections in Two Animal Models. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef]
- Yang, Y.; Shen, W.; Zhong, Q.; Chen, Q.; He, X.; Baker, J.L.; Xiong, K.; Jin, X.; Wang, J.; Hu, F.; et al. Development of a Bacteriophage Cocktail to Constrain the Emergence of Phage-Resistant Pseudomonas aeruginosa. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Latz, S.; Krüttgen, A.; Häfner, H.; Buhl, E.M.; Ritter, K.; Horz, H.P. Differential Effect of Newly Isolated Phages Belonging to PB1-Like, phiKZ-Like and LUZ24-Like Viruses against Multi-Drug Resistant Pseudomonas aeruginosa under Varying Growth Conditions. Viruses 2017, 9, 315. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Abedon, S.T.; Loc-Carrillo, C. Phage cocktails and the future of phage therapy. Future Microbiol. 2013, 8, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Lehman, S.M.; Mearns, G.; Rankin, D.; Cole, R.A.; Smrekar, F.; Branston, S.D.; Morales, S. Design and Preclinical Development of a Phage Product for the Treatment of Antibiotic-Resistant Staphylococcus aureus Infections. Viruses 2019, 11, 88. [Google Scholar] [CrossRef]
- Gordillo Altamirano, F.L.; Barr, J.J. Unlocking the next generation of phage therapy: The key is in the receptors. Curr. Opin. Biotechnol. 2021, 68, 115–123. [Google Scholar] [CrossRef]
- Baker, P.; Hill, P.J.; Snarr, B.D.; Alnabelseya, N.; Pestrak, M.J.; Lee, M.J.; Jennings, L.K.; Tam, J.; Melnyk, R.A.; Parsek, M.R.; et al. Exopolysaccharide biosynthetic glycoside hydrolases can be utilized to disrupt and prevent Pseudomonas aeruginosa biofilms. Sci. Adv. 2016, 2, e1501632. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.W.; Chang, R.Y.; Rao, G.G.; Jermain, B.; Han, M.-L.; Zhao, J.X.; Chen, K.; Wang, J.P.; Barr, J.J.; Schooley, R.; et al. Pharmacokinetics/pharmacodynamics of antipseudomonal bacteriophage therapy in rats: A proof-of-concept study. Clin. Microbiol. Infect. 2020, 26, 1229–1235. [Google Scholar] [CrossRef]
- Barbu, E.M.; Cady, K.C.; Hubby, B. Phage Therapy in the Era of Synthetic Biology. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Smith, H.W.; Huggins, M.B. Successful treatment of experimental Escherichia coli infections in mice using phage: Its general superiority over antibiotics. J. Gen. Microbiol. 1982, 128, 307–318. [Google Scholar] [CrossRef]
- Reynaud, A.; Cloastre, L.; Bernard, J.; Laveran, H.; Ackermann, H.W.; Licois, D.; Joly, B. Characteristics and diffusion in the rabbit of a phage for Escherichia coli 0103. Attempts to use this phage for therapy. Vet. Microbiol. 1992, 30, 203–212. [Google Scholar] [CrossRef]
- Muir, W.R.; Blakemore, W.S. Staphylococcal bacteriophage in experimental infection in mice. Surg. Forum 1960, 10, 339–342. [Google Scholar]
- Weber-Dabrowska, B.; Dabrowski, M.; Slopek, S. Studies on bacteriophage penetration in patients subjected to phage therapy. Arch. Immunol. Ther. Exp. 1987, 35, 563–568. [Google Scholar]
- Schultz, I.; Neva, F.A. Relationship between Blood Clearance and Viruria after Intravenous Injection of Mice And Rats with Bacteriophage and Polioviruses. J. Immunol. 1965, 94, 833–841. [Google Scholar] [PubMed]
- Pouillot, F.; Chomton, M.; Blois, H.; Courroux, C.; Noelig, J.; Bidet, P.; Bingen, E.; Bonacorsi, S. Efficacy of bacteriophage therapy in experimental sepsis and meningitis caused by a clone O25b:H4-ST131 Escherichia coli strain producing CTX-M-15. Antimicrob. Agents Chemother. 2012, 56, 3568–3575. [Google Scholar] [CrossRef]
- Danis-Wlodarczyk, K.; Dąbrowska, K.; Abedon, S.T. Phage Therapy: The Pharmacology of Antibacterial Viruses. Curr. Issues Mol. Biol. 2021, 40, 81–164. [Google Scholar] [CrossRef] [PubMed]
- McVay, C.S.; Velásquez, M.; Fralick, J.A. Phage Therapy of Pseudomonas aeruginosa Infection in a Mouse Burn Wound Model. Antimicrob. Agents Chemother. 2007, 51, 1934–1938. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, K.; Rørbo, N.; Rybtke, M.L.; Martinet, M.G.; Tolker-Nielsen, T.; Høiby, N.; Middelboe, M.; Ciofu, O. P. aeruginosa flow-cell biofilms are enhanced by repeated phage treatments but can be eradicated by phage–ciprofloxacin combination: —monitoring the phage–P. aeruginosa biofilms interactions. Pathog. Dis. 2019, 77. [Google Scholar] [CrossRef]
- Bartell, P.F.; Geffen, A.; Orr, T.; Blakemore, W.S. Staphylococcus Phage-Bacterium In Vivo Interaction. Nature 1965, 205, 474–475. [Google Scholar] [CrossRef]
- Dąbrowska, K. Phage therapy: What factors shape phage pharmacokinetics and bioavailability? Systematic and critical review. Med. Res. Rev. 2019, 39, 2000–2025. [Google Scholar] [CrossRef]
- Kantoch, M. Studies on the Phagocytosis of Bacterial Viruses. I. Arch. Immunol. Ter. Dosw. 1958, 6, 63–84. [Google Scholar]
- Schultz, I.; Frohlich, E. Viruria and Viraliquoria in the Dog after Intravenous Injection of T5 Bacteriophage. Proc. Soc. Exp. Biol. Med. Soc. 1965, 118, 136–138. [Google Scholar] [CrossRef]
- Taylor, R.P.; Sutherland, W.M.; Martin, E.N.; Ferguson, P.J.; Reinagel, M.L.; Gilbert, E.; Lopez, K.; Incardona, N.L.; Ochs, H.D. Bispecific monoclonal antibody complexes bound to primate erythrocyte complement receptor 1 facilitate virus clearance in a monkey model. J. Immunol. 1997, 158, 842–850. [Google Scholar] [PubMed]
- Inchley, C.J. The actvity of mouse Kupffer cells following intravenous injection of T4 bacteriophage. Clin. Exp. Immunol. 1969, 5, 173–187. [Google Scholar] [PubMed]
- Gill, J.J.; Pacan, J.C.; Carson, M.E.; Leslie, K.E.; Griffiths, M.W.; Sabour, P.M. Efficacy and Pharmacokinetics of Bacteriophage Therapy in Treatment of Subclinical Staphylococcus aureus Mastitis in Lactating Dairy Cattle. Antimicrob. Agents Chemother. 2006, 50, 2912–2918. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, B.R.; Kim, S.; Rahman, M.; Kim, J. Antibacterial efficacy of lytic Pseudomonas bacteriophage in normal and neutropenic mice models. J. Microbiol. 2011, 49, 994–999. [Google Scholar] [CrossRef]
- Molenaar, T.M.; Michon, I.; de Haas, S.A.; van Berkel, T.C.; Kuiper, J.; Biessen, E.L. Uptake and processing of modified bacteriophage M13 in mice: Implications for phage display. Virology 2002, 293, 182–191. [Google Scholar] [CrossRef]
- Hájek, P. The elimination of bacteriophages phiX 174 and T2 from the circulating blood of newborn precolostral pigs. Folia Microbiol. (Praha) 1970, 15, 125–128. [Google Scholar] [CrossRef]
- Mukerjee, S.; Ghosh, S.N. Localization of cholera bacterio-phage after intravenous injection. Ann. Biochem. Exp. Med. 1962, 22, 73–76. [Google Scholar]
- Capparelli, R.; Nocerino, N.; Iannaccone, M.; Ercolini, D.; Parlato, M.; Chiara, M.; Iannelli, D. Bacteriophage therapy of Salmonella enterica: A fresh appraisal of bacteriophage therapy. J. Infect. Dis. 2010, 201, 52–61. [Google Scholar] [CrossRef]
- Watanabe, R.; Matsumoto, T.; Sano, G.; Ishii, Y.; Tateda, K.; Sumiyama, Y.; Uchiyama, J.; Sakurai, S.; Matsuzaki, S.; Imai, S. Efficacy of Bacteriophage Therapy against Gut-Derived Sepsis Caused by Pseudomonas aeruginosa in Mice. Antimicrob. Agents Chemother. 2007, 51, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Gabig, M.; Herman-Antosiewicz, A.; Kwiatkowska, M.; Los, M.; Thomas, M.S.; Wegrzyn, G. The cell surface protein Ag43 facilitates phage infection of Escherichia coli in the presence of bile salts and carbohydrates. Microbiol. Read. Engl. 2002, 148, 1533–1542. [Google Scholar] [CrossRef][Green Version]
- Rus, H.; Cudrici, C.; Niculescu, F. The role of the complement system in innate immunity. Immunol. Res. 2005, 33, 103–112. [Google Scholar] [CrossRef]
- Dąbrowska, K.; Miernikiewicz, P.; Piotrowicz, A.; Hodyra, K.; Owczarek, B.; Lecion, D.; Kaźmierczak, Z.; Letarov, A.; Górski, A. Immunogenicity studies of proteins forming the T4 phage head surface. J. Virol. 2014, 88, 12551–12557. [Google Scholar] [CrossRef]
- Wang, J.; Hu, B.; Xu, M.; Yan, Q.; Liu, S.; Zhu, X.; Sun, Z.; Reed, E.; Ding, L.; Gong, J.; et al. Use of bacteriophage in the treatment of experimental animal bacteremia from imipenem-resistant Pseudomonas aeruginosa. Int. J. Mol. Med. 2006, 17, 309–317. [Google Scholar] [CrossRef]
- Altamirano, F.G.; Barr, J.J. Phage Therapy in the Postantibiotic Era. Clin. Microbiol. Rev. 2019, 32. [Google Scholar] [CrossRef]
- Colom, J.; Cano-Sarabia, M.; Otero, J.; Cortés, P.; Maspoch, D.; Llagostera, M. Liposome-Encapsulated Bacteriophages for Enhanced Oral Phage Therapy against Salmonella spp. Appl. Environ. Microbiol. 2015, 81, 4841–4849. [Google Scholar] [CrossRef]
- Malik, D.J.; Sokolov, I.J.; Vinner, G.K.; Mancuso, F.; Cinquerrui, S.; Vladisavljevic, G.T.; Clokie, M.J.; Garton, N.J.; Stapley, A.F.; Kirpichnikova, A. Formulation, stabilisation and encapsulation of bacteriophage for phage therapy. Adv. Colloid Interface Sci. 2017, 249, 100–133. [Google Scholar] [CrossRef]
- Stanford, K.; McAllister, T.A.; Niu, Y.D.; Stephens, T.P.; Mazzocco, A.; Waddell, T.E.; Johnson, R.P. Oral delivery systems for encapsulated bacteriophages targeted at Escherichia coli O157:H7 in feedlot cattle. J. Food Prot. 2010, 73, 1304–1312. [Google Scholar] [CrossRef]
- Doogue, M.P.; Polasek, T.M. The ABCD of clinical pharmacokinetics. Ther. Adv. Drug Saf. 2013, 4, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Astudillo, A.; Leung, S.Y.; Kutter, E.; Morales, S.; Chan, H.K. Nebulization effects on structural stability of bacteriophage PEV 44. Eur. J. Pharm. Biopharm. 2018, 125, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Afrin, S.; Gupta, V. Pharmaceutical Formulation. Available online: https://www.ncbi.nlm.nih.gov/books/NBK562239/ (accessed on 10 September 2020).
- Morrisette, T.; Kebriaei, R.; Lev, K.L.; Morales, S.; Rybak, M.J. Bacteriophage Therapeutics: A Primer for Clinicians on Phage-Antibiotic Combinations. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2020, 40, 153–168. [Google Scholar] [CrossRef]
- Vandenheuvel, D.; Lavigne, R.; Brüssow, H. Bacteriophage Therapy: Advances in Formulation Strategies and Human Clinical Trials. Annu. Rev. Virol. 2015, 2, 599–618. [Google Scholar] [CrossRef] [PubMed]
- Kutateladze, M.; Adamia, R. Phage therapy experience at the Eliava Institute. Méd. Mal. Infect. 2008, 38, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Rose, T.; Verbeken, G.; Vos, D.D.; Merabishvili, M.; Vaneechoutte, M.; Lavigne, R.; Jennes, S.; Zizi, M.; Pirnay, J.P. Experimental phage therapy of burn wound infection: Difficult first steps. Int. J. Burns Trauma 2014, 4, 66–73. [Google Scholar] [PubMed]
- Jennes, S.; Merabishvili, M.; Soentjens, P.; Pang, K.W.; Rose, T.; Keersebilck, E.; Soete, O.; François, P.M.; Teodorescu, S.; Verween, G. Use of bacteriophages in the treatment of colistin-only-sensitive Pseudomonas aeruginosa septicaemia in a patient with acute kidney injury—A case report. Crit. Care 2017, 21. [Google Scholar] [CrossRef]
- Marza, J.S.; Soothill, J.S.; Boydell, P.; Collyns, T.A. Multiplication of therapeutically administered bacteriophages in Pseudomonas aeruginosa infected patients. Burns 2006, 32, 644–646. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Semenkovich, N.P.; Whiteson, K.; Rohwer, F.; Gordon, J.I. Going viral: Next-generation sequencing applied to phage populations in the human gut. Nat. Rev. Microbiol. 2012, 10, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Wu, M.; McNulty, N.P.; Rohwer, F.L.; Gordon, J.I. Gnotobiotic mouse model of phage-bacterial host dynamics in the human gut. Proc. Natl. Acad. Sci. USA 2013, 110, 20236–20241. [Google Scholar] [CrossRef]
- Uhr, J.W.; Finkelstein, M.S.; Baumann, J.B. Antibody formation. III. The primary and secondary antibody response to bacteriophage phi X 174 in guinea pigs. J. Exp. Med. 1962, 115, 655–670. [Google Scholar] [CrossRef]
- Uhr, J.W.; Finkelstein, M.S. Antibody formation. IV. Formation of rapidly and slowly sedimenting antibodies and immunological memory to bacteriophage phi-X 174. J. Exp. Med. 1963, 117, 457–477. [Google Scholar] [CrossRef]
- Hájek, P.; Mandel, L. Antibody response of young animals to bacteriophages of different immunological behaviour: Phi X 174 and T2. Folia Microbiol. 1966, 11, 282–289. [Google Scholar] [CrossRef]
- Hájek, P. Neutralization of bacterial viruses by antibodies of young animals. 3. The development of the avidity of 19S and 7S neutralizing antibodies in the course of primary and secondary response in young rabbits immunized with PhiX 174 bacteriophage. Folia Microbiol. 1970, 15, 9–16. [Google Scholar] [CrossRef]
- Górski, A.; Międzybrodzki, R.; Jończyk-Matysiak, E.; Żaczek, M.; Borysowski, J. Phage-specific diverse effects of bacterial viruses on the immune system. Future Microbiol. 2019, 14, 1171–1174. [Google Scholar] [CrossRef]
- Van Belleghem, J.D.; Clement, F.; Merabishvili, M.; Lavigne, R.; Vaneechoutte, M. Pro- and anti-inflammatory responses of peripheral blood mononuclear cells induced by Staphylococcus aureus and Pseudomonas aeruginosa phages. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Sweere, J.M.; Van Belleghem, J.D.; Ishak, H.; Bach, M.S.; Popescu, M.; Sunkari, V.; Kaber, G.; Manasherob, R.; Suh, G.A.; Cao, X.; et al. Bacteriophage trigger antiviral immunity and prevent clearance of bacterial infection. Science 2019, 363. [Google Scholar] [CrossRef] [PubMed]
- Żaczek, M.; Łusiak-Szelachowska, M.; Jończyk-Matysiak, E.; Weber-Dąbrowska, B.; Międzybrodzki, R.; Owczarek, B.; Kopciuch, A.; Fortuna, W.; Rogóż, P.; Górski, A. Antibody Production in Response to Staphylococcal MS-1 Phage Cocktail in Patients Undergoing Phage Therapy. Front. Microbiol. 2016, 7, 1681. [Google Scholar] [CrossRef]
- Łusiak-Szelachowska, M.; Zaczek, M.; Weber-Dąbrowska, B.; Międzybrodzki, R.; Kłak, M.; Fortuna, W.; Letkiewicz, S.; Rogóż, P.; Szufnarowski, K.; Jończyk-Matysiak, E.; et al. Phage neutralization by sera of patients receiving phage therapy. Viral Immunol. 2014, 27, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Roach, D.R.; Leung, C.Y.; Henry, M.; Morello, E.; Singh, D.; Di Santo, J.P.; Weitz, J.S.; Debarbieux, L. Synergy between the Host Immune System and Bacteriophage Is Essential for Successful Phage Therapy against an Acute Respiratory Pathogen. Cell Host Microbe 2017, 22, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Międzybrodzki, R.; Fortuna, W.; Weber-Dąbrowska, B.; Górski, A. A retrospective analysis of changes in inflammatory markers in patients treated with bacterial viruses. Clin. Exp. Med. 2009, 9, 303. [Google Scholar] [CrossRef] [PubMed]
- Górski, A.; Jończyk-Matysiak, E.; Łusiak-Szelachowska, M.; Weber-Dąbrowska, B.; Międzybrodzki, R.; Borysowski, J. Therapeutic potential of phages in autoimmune liver diseases. Clin. Exp. Immunol. 2018, 192, 1–6. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04191148 (accessed on 10 September 2020).
- Reyes-Robles, T.; Dillard, R.S.; Cairns, L.S.; Silva-Valenzuela, C.A.; Housman, M.; Ali, A.; Wright, E.R.; Camilli, A. Vibrio cholerae Outer Membrane Vesicles Inhibit Bacteriophage Infection. J. Bacteriol. 2018, 200. [Google Scholar] [CrossRef]
- Manning, A.J.; Kuehn, M.J. Contribution of bacterial outer membrane vesicles to innate bacterial defense. BMC Microbiol. 2011, 11, 258. [Google Scholar] [CrossRef]
- Issa, R.; Chanishvili, N.; Caplin, J.; Kakabadze, E.; Bakuradze, N.; Makalatia, K.; Cooper, I. Antibiofilm potential of purified environmental bacteriophage preparations against early stage Pseudomonas aeruginosa biofilms. J. Appl. Microbiol. 2019, 126, 1657–1667. [Google Scholar] [CrossRef]
- Gallet, R.; Kannoly, S.; Wang, I.N. Effects of bacteriophage traits on plaque formation. BMC Microbiol. 2011, 11, 181. [Google Scholar] [CrossRef]
- Parracho, H.M.; Burrowes, B.H.; Enright, M.C.; McConville, M.L.; Harper, D.R. The role of regulated clinical trials in the development of bacteriophage therapeutics. J. Mol. Genet. Med. Int. J. Biomed. Res. 2012, 6, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, A.S. Pharmacological limitations of phage therapy. Ups. J. Med. Sci. 2019, 124, 218–227. [Google Scholar] [CrossRef]
- Reardon, S. Phage therapy gets revitalized. Nat. News 2014, 510, 15. [Google Scholar] [CrossRef]
- Danis-Wlodarczyk, K.; Vandenheuvel, D.; Jang, H.B.; Briers, Y.; Olszak, T.; Arabski, M.; Wasik, S.; Drabik, M.; Higgins, G.; Tyrrell, J.; et al. A proposed integrated approach for the preclinical evaluation of phage therapy in Pseudomonas infections. Sci. Rep. 2016, 6, 28115. [Google Scholar] [CrossRef] [PubMed]
- Mumford, R.; Friman, V. Bacterial competition and quorum-sensing signalling shape the eco-evolutionary outcomes of model in vitro phage therapy. Evol. Appl. 2016, 10, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Casas, V.; Maloy, S. Role of bacteriophage-encoded exotoxins in the evolution of bacterial pathogens. Future Microbiol. 2011, 6, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Canchaya, C.; Fournous, G.; Brüssow, H. The impact of prophages on bacterial chromosomes. Mol. Microbiol. 2004, 53, 9–18. [Google Scholar] [CrossRef]
- Casjens, S. Prophages and bacterial genomics: What have we learned so far? Mol. Microbiol. 2003, 49, 277–300. [Google Scholar] [CrossRef]
- Canchaya, C.; Desiere, F.; McShan, W.M.; Ferretti, J.J.; Parkhill, J.; Brüssow, H. Genome analysis of an inducible prophage and prophage remnants integrated in the Streptococcus pyogenes strain SF370. Virology 2002, 302, 245–258. [Google Scholar] [CrossRef]
- Abedon, S.T.; Lejeune, J.T. Why bacteriophage encode exotoxins and other virulence factors. Evol. Bioinform. Online 2007, 1, 97–110. [Google Scholar] [CrossRef] [PubMed]
- D’Herelle, F. The Bacteriophage and its Clinical Applications; Charles, C., Ed.; Thoams: Baltimore, MD, USA, 1930. [Google Scholar]
- Boyd, E.F.; Davis, B.M.; Hochhut, B. Bacteriophage–bacteriophage interactions in the evolution of pathogenic bacteria. Trends Microbiol. 2001, 9, 137–144. [Google Scholar] [CrossRef]
- Wagner, P.L.; Waldor, M.K. Bacteriophage Control of Bacterial Virulence. Infect. Immun. 2002, 70, 3985–3993. [Google Scholar] [CrossRef]
- Frobisher, M.; Brown, J. Transmissible toxicogenicity of streptococci. Bull. Johns Hopkins Hosp. 1927, 41, 167–173. [Google Scholar]
- Freeman, V.J. Studies on the Virulence of Bacteriophage-Infected Strains of Corynebacterium Diphtheriae. J. Bacteriol. 1951, 61, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Matsumoto, H.; Ohnishi, M.; Terawaki, Y. Molecular analysis of a cytotoxin-converting phage, φCTX, of Pseudomonas aeruginosa: Structure of the attP–cos–ctx region and integration into the serine tRNA gene. Mol. Microbiol. 1993, 7, 657–667. [Google Scholar] [CrossRef]
- Kakabadze, E.; Makalatia, K.; Grdzelishvili, N.; Bakuradze, N.; Goderdzishvili, M.; Kusradze, I.; Phoba, M.F.; Lunguya, O.; Lood, C.; Lavigne, R.; et al. Selection of Potential Therapeutic Bacteriophages that Lyse a CTX-M-15 Extended Spectrum β-Lactamase Producing Salmonella enterica Serovar Typhi Strain from the Democratic Republic of the Congo. Viruses 2018, 10, 172. [Google Scholar] [CrossRef]
- Djebara, S.; Maussen, C.; De Vos, D.; Merabishvili, M.; Damanet, B.; Pang, K.W.; De Leenheer, P.; Strachinaru, I.; Soentjens, P.; Pirnay, J.P. Processing Phage Therapy Requests in a Brussels Military Hospital: Lessons Identified. Viruses 2019, 11, 265. [Google Scholar] [CrossRef]
- Vaca, S.; Arce, J.; Oliver, G.; Arenas, D.; Argüello, F. FIZ15 bacteriophage increases the adhesion of pseudomonas aeruginosa to human buccal epithelial cells. Rev. Latinoam. Microbiol. 1989, 31, 1–5. [Google Scholar]
- Holloway, B.W.; Cooper, G.N. Lysogenic Conversion in Pseudomonas Aeruginosa. J. Bacteriol. 1962, 84, 1321–1324. [Google Scholar] [CrossRef]
- Matinkhoo, S.; Lynch, K.H.; Dennis, J.J.; Finlay, W.H.; Vehring, R. Spray-dried respirable powders containing bacteriophages for the treatment of pulmonary infections. J. Pharm. Sci. 2011, 100, 5197–5205. [Google Scholar] [CrossRef] [PubMed]
- Sahota, J.S.; Smith, C.M.; Radhakrishnan, P.; Winstanley, C.; Goderdzishvili, M.; Chanishvili, N.; Kadioglu, A.; O’Callaghan, C.; Clokie, M.R. Bacteriophage Delivery by Nebulization and Efficacy Against Phenotypically Diverse Pseudomonas aeruginosa from Cystic Fibrosis Patients. J. Aerosol Med. Pulm. Drug Deliv. 2015, 28, 353–360. [Google Scholar] [CrossRef]
- Briggiler Marcó, M.; Reinheimer, J.; Quiberoni, A. Phage adsorption and lytic propagation in Lactobacillus plantarum: Could host cell starvation affect them? BMC Microbiol. 2015, 15, 273. [Google Scholar] [CrossRef] [PubMed]
- Tokman, J.I.; Kent, D.J.; Wiedmann, M.; Denes, T. Temperature Significantly Affects the Plaquing and Adsorption Efficiencies of Listeria Phages. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Nordström, K.; Forsgren, A.; Cox, P. Prevention of Bacteriophage Adsorption to Staphylococcus aureus by Immunoglobulin G. J. Virol. 1974, 14, 203–206. [Google Scholar] [CrossRef]
- Luckey, M.; Neilands, J.B. Iron Transport in Salmonella typhimurium LT-2: Prevention, by Ferrichrome, of Adsorption of Bacteriophages ES18 and ES18.hl to a Common Cell Envelope Receptor. J. Bacteriol. 1976, 127, 1036–1037. [Google Scholar] [CrossRef]
- Li, G.; Shen, M.; Yang, Y.; Le, S.; Li, M.; Wang, J.; Zhao, Y.; Tan, Y.; Hu, F.; Lu, S. Adaptation of Pseudomonas aeruginosa to Phage PaP1 Predation via O-Antigen Polymerase Mutation. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Hall-Stoodley, L.; Costerton, J.W.; Stoodley, P. Bacterial biofilms: From the natural environment to infectious diseases. Nat. Rev. Microbiol. 2004, 2, 95–108. [Google Scholar] [CrossRef]
- Chaudhry, W.N.; Concepción-Acevedo, J.; Park, T.; Andleeb, S.; Bull, J.J.; Levin, B.R. Synergy and Order Effects of Antibiotics and Phages in Killing Pseudomonas aeruginosa Biofilms. PLoS ONE 2017, 12, e0168615. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Romero, M.A.; Casadesús, J. Contribution of phenotypic heterogeneity to adaptive antibiotic resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 355–360. [Google Scholar] [CrossRef]
- Akanda, Z.Z.; Taha, M.; Abdelbary, H. Current review—The rise of bacteriophage as a unique therapeutic platform in treating peri-prosthetic joint infections. J. Orthop. Res. 2018, 36, 1051–1060. [Google Scholar] [CrossRef]
- Clinical Microbiology Reviews: Prosthetic Joint Infection. Available online: https://cmr.asm.org/content/27/2/302 (accessed on 10 January 2021).
- Ferry, T.; Boucher, F.; Fevre, C.; Perpoint, T.; Chateau, J.; Petitjean, C.; Josse, J.; Chidiac, C.; L’hostis, G.; Leboucher, G.; et al. Innovations for the treatment of a complex bone and joint infection due to XDR Pseudomonas aeruginosa including local application of a selected cocktail of bacteriophages. J. Antimicrob. Chemother. 2018, 73, 2901–2903. [Google Scholar] [CrossRef]
- Burgener, E.B.; Sweere, J.M.; Bach, M.S.; Secor, P.R.; Haddock, N.; Jennings, L.K.; Marvig, R.L.; Johansen, H.K.; Rossi, E.; Cao, X.; et al. Filamentous bacteriophages are associated with chronic Pseudomonas lung infections and antibiotic resistance in cystic fibrosis. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.; Gibson, R.L.; McNamara, S.; Emerson, J.; Burns, J.L.; Castile, R.; Hiatt, P.; McCoy, K.; Wilson, C.B.; Inglis, A.; et al. Early pulmonary infection, inflammation, and clinical outcomes in infants with cystic fibrosis. Pediatr. Pulmonol. 2001, 32, 356–366. [Google Scholar] [CrossRef]
- Emerson, J.; Rosenfeld, M.; McNamara, S.; Ramsey, B.; Gibson, R.L. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr. Pulmonol. 2002, 34, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Mitri, C.; Xu, Z.; Bardin, P.; Corvol, H.; Touqui, L.; Tabary, O. Novel Anti-Inflammatory Approaches for Cystic Fibrosis Lung Disease: Identification of Molecular Targets and Design of Innovative Therapies. Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04596319 (accessed on 7 January 2021).
- Lin, D.M.; Koskella, B.; Lin, H.C. Phage therapy: An alternative to antibiotics in the age of multi-drug resistance. World J. Gastrointest. Pharmacol. Ther. 2017, 8, 162–173. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Indication | Phage Dose | Phage | n | Main Outcome | Ref. |
|---|---|---|---|---|---|
| Randomized Controlled Trials | |||||
| Leg ulcers | 4 mL (1 × 109 PFU/mL) topically applied once weekly for 12 weeks | Phage cocktail (WPP-201) | 42 | Phase I safety trial: no significant difference was reported between the phage-treated group and the control group for the frequency of adverse events or the frequency of healing. | [53] |
| Burn wounds | 1 mL (1 × 102 PFU/mL), topically applied once daily for 7 days | Phage cocktail (PP1131) | 27 | Patients in the phage group experienced a longer time to sustained reduction in bacterial burden compared to the SOC group (144 vs. 47 h). | [54] |
| Chronic otitis | 0.2 mL (6 × 104 PFU) of antibiotic application once | Phage cocktail (Biophage-PA) | 24 | Phase I/II controlled clinical trial: statistically significant clinical improvements from baseline in the phage-treated group compared with the control group. | [13] |
| Prospective Trials | |||||
| Burn wounds | 1 mL (109 PFU/mL) per 50 cm2 topically applied once | Phage cocktail (BFC-1) | 9 | For all patients, the bacterial load remained unchanged after phage application, as well as after standard treatment. | [121] |
| Case Series | |||||
| Skin ulcers and wounds | 1 × 106 PFU/cm2 topically applied until ulcer healed (6 days to 15 months) | Phage cocktail (Pyophage) | 96 | Wounds/ulcers completely healed in 70% of patients. | [14] |
| Septicemia | 10 mL (PFU/mL: NR) orally three times daily (median duration: 29 days) | NR | 94 | Complete recovery in 80 patients. Phage therapy was ineffective in 14 patients. No significant difference reported for phage therapy alone (n = 23) or PAC (n = 71). | [16] |
| Systemic infections | 2 × 105–4 × 1010 PFU/mL IV +/− nebulization for 4–12 weeks | Phage cocktail | 5 | Intravenous BT was safe, with a successful outcome in 3/5 patients with antibiotic-recalcitrant P. aeruginosa infections. | [12] |
| Chronic wounds | 0.1 mL/cm2 (1 × 109 PFU/mL) topically applied on alternate days x 3–5 doses | Phage cocktail | 20 | All wounds became sterile within 13 days, and 7 cases achieved complete wound healing by day 21. | [15] |
| Systemic infections | 1 × 108 PFU/mL orally TID for 2–9 weeks (median duration: 32 days) | Single phage or phage cocktail | 20 | The cure of infection was achieved in all cases. | [17] |
| Case Reports | |||||
| CF pneumonia | 5 mL (4 × 109 PFU/5 mL) IV every 6 h for 8 weeks | Phage cocktail (AB-PA01) | 1 | Clinical resolution of infection without the recurrence of pneumonia due to Pseudomonas or CF exacerbation within 100 days following the end of BT. | [46] |
| Septicemia and wounds | 50 mL (109 PFU/mL) of IV infusion once daily and 50 mL of irrigation every 8 h for 10 days | Phage cocktail (BFC-1) | 1 | Pathogen eradicated from blood, CRP levels dropped, fever disappeared, and kidney function returned after a few days. | [122] |
| Burn wound | 0.2 mL (1 × 103 PFU/mL) topically applied once | NR | 1 | Three days after phage application, P. aeruginosa was not isolated from tissue swabs. Extensive grafting following phage therapy was successful. | [123] |
| Aortic graft infection | 10 mL (1 × 107 PFU/mL) one injection into fistula | Phage OMKO1 | 1 | Following the application of phage and ceftazidime, the infection appeared to resolve with no signs of recurrence. | [63] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holger, D.; Kebriaei, R.; Morrisette, T.; Lev, K.; Alexander, J.; Rybak, M. Clinical Pharmacology of Bacteriophage Therapy: A Focus on Multidrug-Resistant Pseudomonas aeruginosa Infections. Antibiotics 2021, 10, 556. https://doi.org/10.3390/antibiotics10050556
Holger D, Kebriaei R, Morrisette T, Lev K, Alexander J, Rybak M. Clinical Pharmacology of Bacteriophage Therapy: A Focus on Multidrug-Resistant Pseudomonas aeruginosa Infections. Antibiotics. 2021; 10(5):556. https://doi.org/10.3390/antibiotics10050556
Chicago/Turabian StyleHolger, Dana, Razieh Kebriaei, Taylor Morrisette, Katherine Lev, Jose Alexander, and Michael Rybak. 2021. "Clinical Pharmacology of Bacteriophage Therapy: A Focus on Multidrug-Resistant Pseudomonas aeruginosa Infections" Antibiotics 10, no. 5: 556. https://doi.org/10.3390/antibiotics10050556
APA StyleHolger, D., Kebriaei, R., Morrisette, T., Lev, K., Alexander, J., & Rybak, M. (2021). Clinical Pharmacology of Bacteriophage Therapy: A Focus on Multidrug-Resistant Pseudomonas aeruginosa Infections. Antibiotics, 10(5), 556. https://doi.org/10.3390/antibiotics10050556