
New Chloramphenicol Derivatives with a Modified Dichloroacetyl Tail as Potential Antimicrobial Agents

 , and
, and 
Abstract
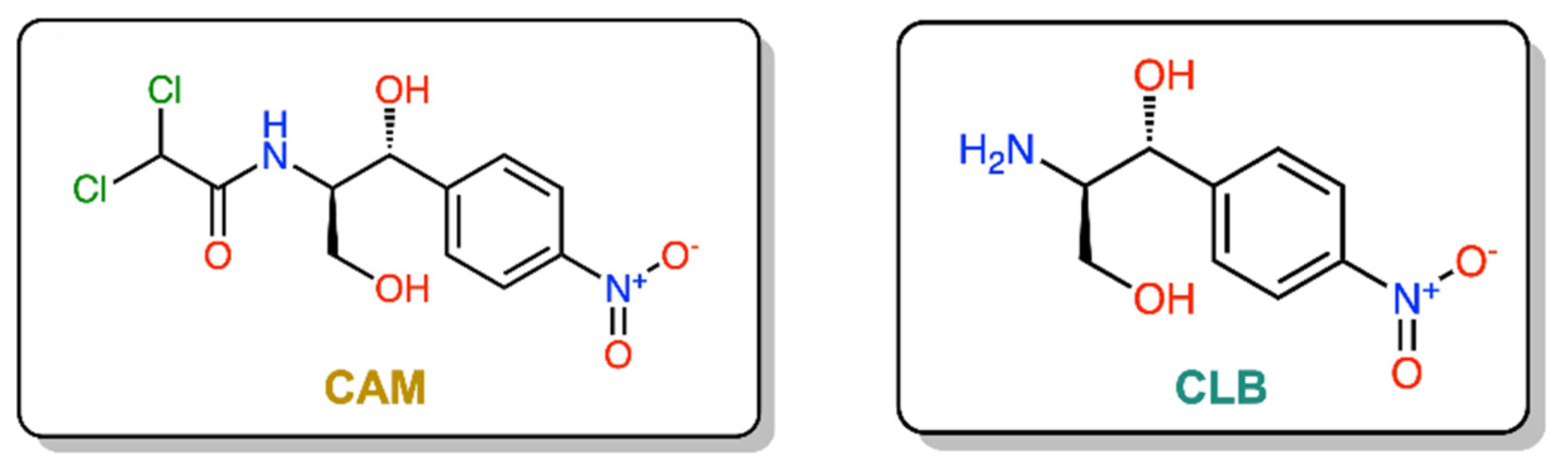
1. Introduction
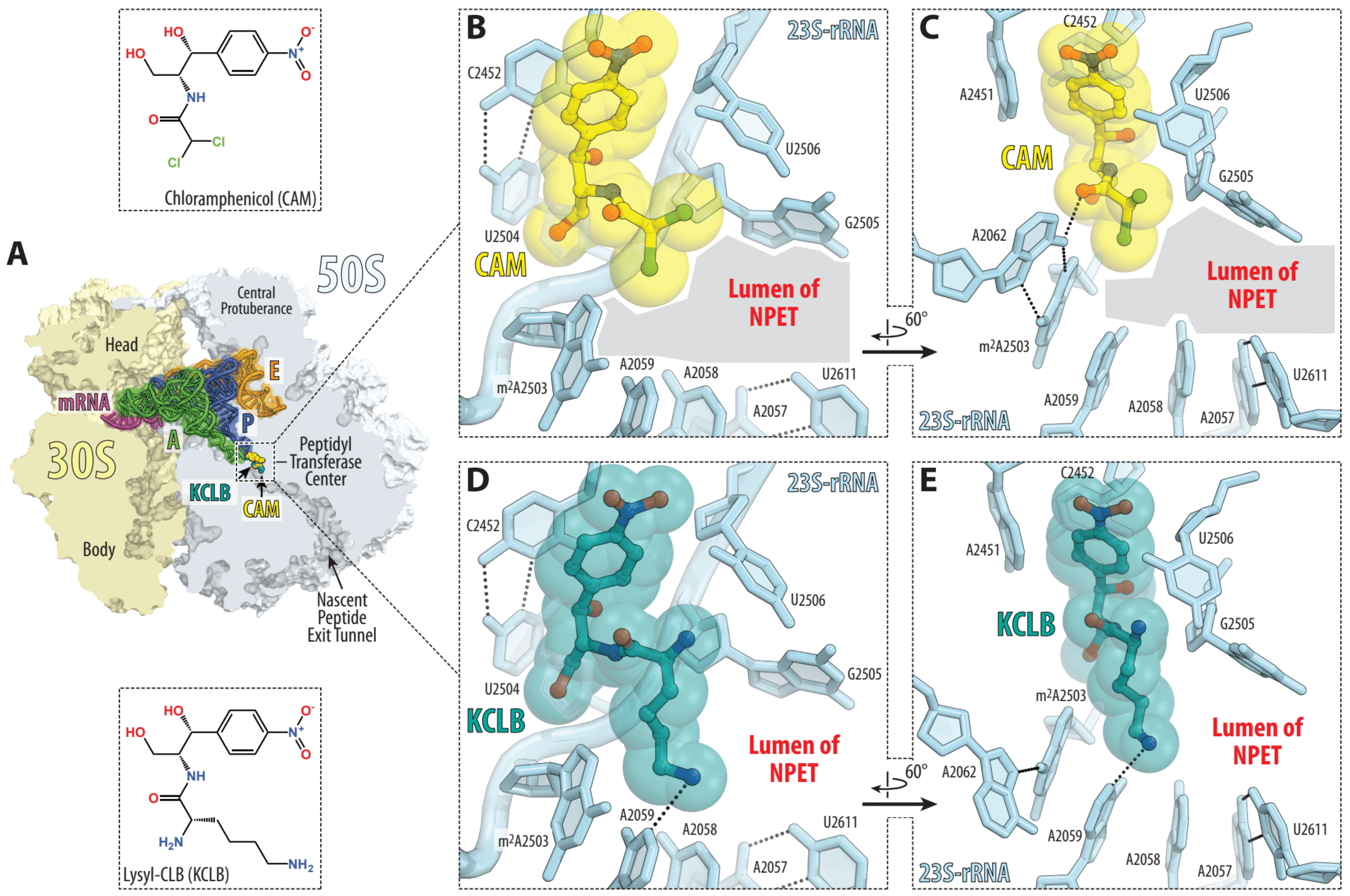
2. Results and Discussion
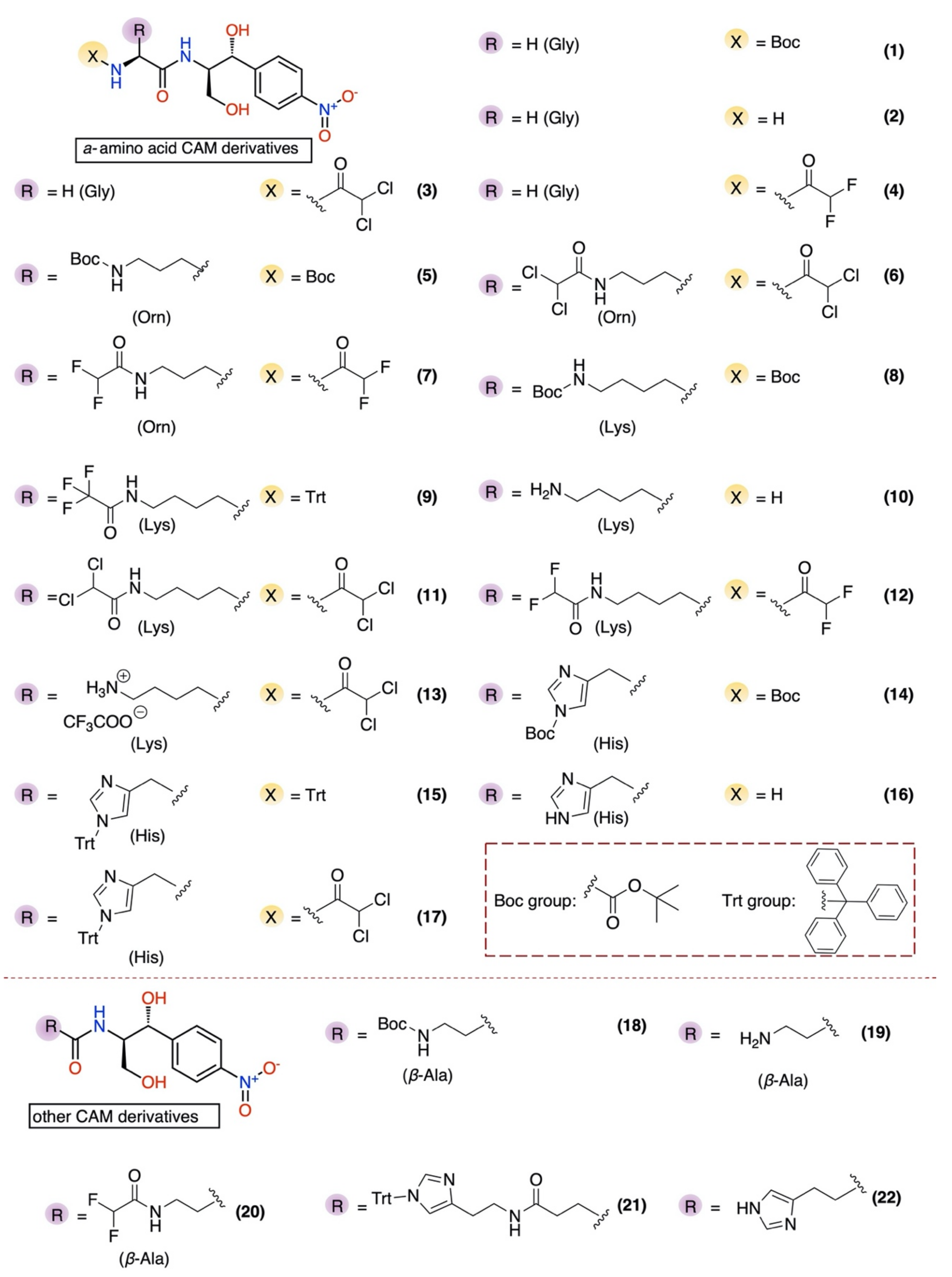
2.1. Chemical Synthesis
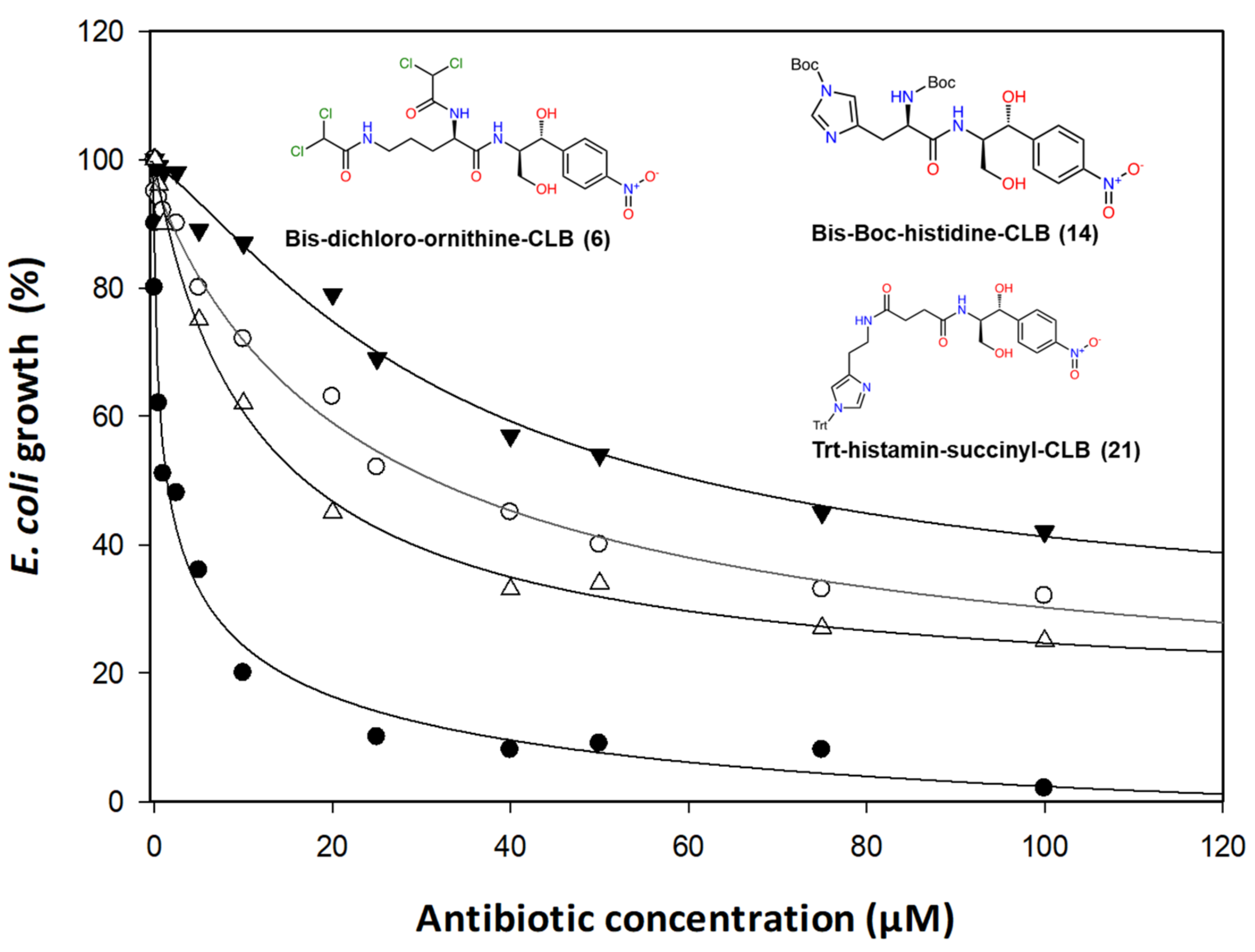
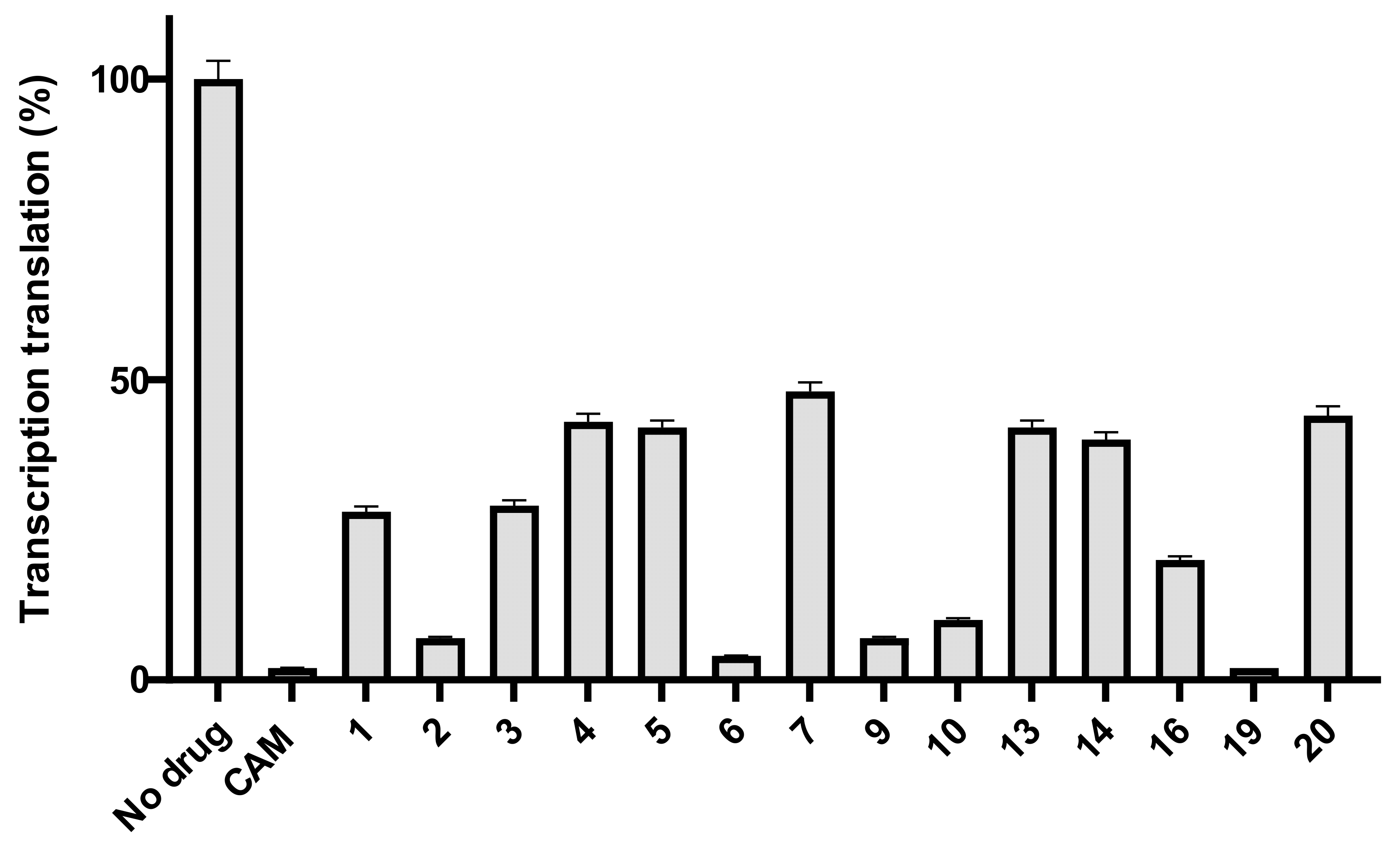
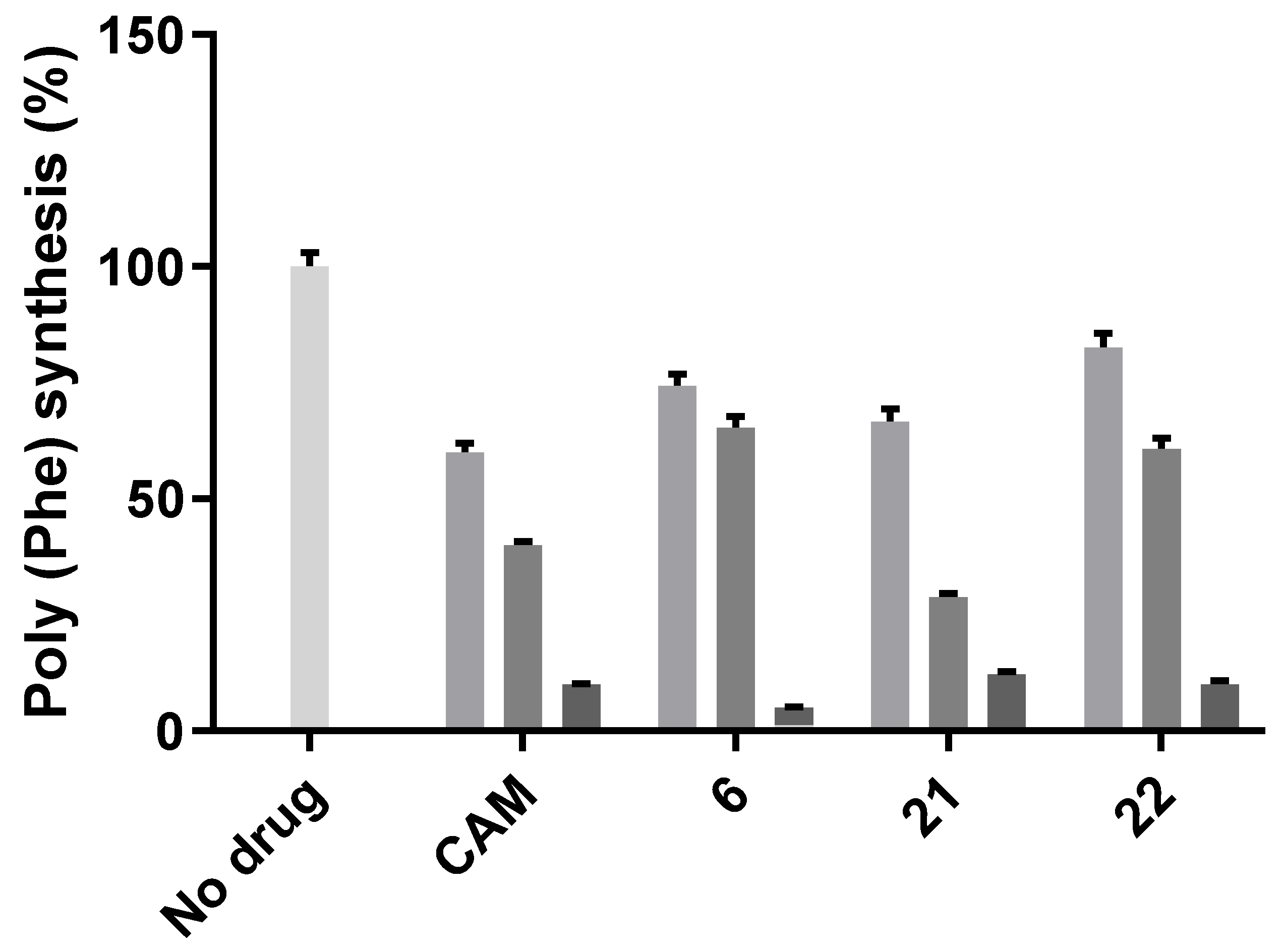
2.2. Antibacterial Activity
- Basic amino acids ornithine, histidine and lysine as well as glycine but not β-alanine recover the inhibitory activity lost after removal of the dichloroacetyl tail of the chloramphenicol molecule.
- The imidazole ring is also important as we can see from histidine replacement with dihydro-urocanic acid or in the case of Trt-histamine–succinyl–CLB, compounds 22 and 21, respectively.
- Dichloroacetylation of free amino groups really helps in the case of ornithine, but not in the case of lysine or histidine.
- The length of the side chain carbon skeleton of an amino acid is very important for binding and accommodation as we can see comparing dichloroacetyl-ornithine- or lysine–CLB.
- Protected versus free amine groups assist in recovering the compound’s antimicrobial activity, but not for all cases. For each individual case, it is important to monitor the surrounding interactions and their correlation with the effect to binding forces and inhibitory activity.
3. Materials and Methods
3.1. Materials
3.2. Bacterial Strains
3.3. Biochemical Preperations
3.4. Inhibition of Translation Using an E. coli-Based In Vitro Cell-Free Expression System
3.5. EC50 Determination
3.6. Poly(U)-Dependent Poly(Phe) Synthesis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gould, K. Antibiotics: From prehistory to the present day. J. Antimicrob. Chemother. 2016, 71, 572–575. [Google Scholar] [CrossRef]
- Rončević, T.; Puizina, J.; Tossi, A. Antimicrobial peptides as anti-infective agents in pre-post-antibiotic era? Int. J. Mol. Sci. 2019, 20, 5713. [Google Scholar] [CrossRef]
- Lewis, K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 2013, 12, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.M.; Seiple, I.B.; Myers, A.G. The evolving role of chemical synthesis in antibacterial drug discovery. Angew. Chem. Int. Ed. 2014, 53, 8840–8869. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, J.; Bartz, Q.R.; Smith, R.M.; Joslyn, D.A.; Burkholder, P.R. Chloromycetin, a New Antibiotic from a Soil Actinomycete. Science 1947, 106, 417. [Google Scholar] [CrossRef]
- Schroeter, W. Hematologic side effects of chloramphenicol. Neuropadiatrie 1974, 5, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Eliakim-Raz, N.; Lador, A.; Leibovici-Weissman, Y.; Elbaz, M.; Paul, M.; Leibovici, L. Efficacy and safety of chloramphenicol: Joining the revival of old antibiotics? Systematic review and meta-analysis of randomized controlled trials. J. Antimicrob. Chemother. 2014, 70, 979–996. [Google Scholar] [CrossRef]
- Hansen, J.L.; Moore, P.B.; Steitz, T.A. Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J. Mol. Biol. 2003, 330, 1061–1075. [Google Scholar] [CrossRef]
- Bulkley, D.; Innis, C.A.; Blaha, G.; Steitz, T.A. Revisiting the structures of several antibiotics bound to the bacterial ribosome. Proc. Natl. Acad. Sci. USA 2010, 107, 17158–17163. [Google Scholar] [CrossRef]
- Dunkle, J.A.; Xiong, L.; Mankin, A.S.; Cate, J.H. Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc. Natl. Acad. Sci. USA 2010, 107, 17152–17157. [Google Scholar] [CrossRef]
- Svetlov, M.S.; Plessa, E.; Chen, C.W.; Bougas, A.; Krokidis, M.G.; Dinos, G.P.; Polikanov, Y.S. High-resolution crystal structures of ribosome-bound chloramphenicol and erythromycin provide the ultimate basis for their competition. RNA 2019, 25, 600–606. [Google Scholar] [CrossRef]
- Marks, J.; Kannan, K.; Roncase, E.J.; Klepacki, D.; Kefi, A.; Orelle, C.; Vázquez-Laslop, N.; Mankin, A.S. Context-specific inhibition of translation by ribosomal antibiotics targeting the peptidyl transferase center. Proc. Natl. Acad. Sci. USA 2016, 113, 12150–12155. [Google Scholar] [CrossRef]
- Choi, J.; Marks, J.; Zhang, J.; Chen, D.H.; Wang, J.; Vázquez-Laslop, N.; Mankin, A.S.; Puglisi, J.D. Dynamics of the context-specific translation arrest by chloramphenicol and linezolid. Nat. Chem. Biol. 2020, 16, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, S.; Shen, J.; Kadlec, K.; Wang, Y.; Brenner Michael, G.; Feßler, A.T.; Vester, B. Lincosamides, Streptogramins, Phenicols, and Pleuromutilins: Mode of Action and Mechanisms of Resistance. Cold Spring Harb. Perspect. Med. 2016, 6, a027037. [Google Scholar] [CrossRef] [PubMed]
- Dinos, G.P.; Athanassopoulos, C.M.; Missiri, D.A.; Giannopoulou, P.C.; Vlachogiannis, I.A.; Papadopoulos, G.E.; Papaioannou, D.; Kalpaxis, D.L. Chloramphenicol derivatives as antibacterial and anticancer agents: Historic problems and current solutions. Antibiotics 2016, 5, 20. [Google Scholar] [CrossRef]
- Chen, H.; Liu, C.; Chen, D.; Madrid, K.; Peng, S.; Dong, X.; Zhang, M.; Gu, Y. Bacteria-Targeting Conjugates Based on Antimicrobial Peptide for Bacteria Diagnosis and Therapy. Mol. Pharm. 2015, 12, 2505–2516. [Google Scholar] [CrossRef]
- Dong, F.; Li, L.; Lin, L.; He, D.; Chen, J.; Wei, W.; Wei, D. Transesterification synthesis of chloramphenicol esters with the lipase from bacillus amyloliquefaciens. Molecules 2017, 22, 1523. [Google Scholar] [CrossRef]
- Louzoun Zada, S.; Green, K.D.; Shrestha, S.K.; Herzog, I.M.; Garneau-Tsodikova, S.; Fridman, M. Derivatives of Ribosome-Inhibiting Antibiotic Chloramphenicol Inhibit the Biosynthesis of Bacterial Cell Wall. ACS Infect. Dis. 2018, 4, 1121–1149. [Google Scholar] [CrossRef] [PubMed]
- Tereshchenkov, A.G.; Dobosz-Bartoszek, M.; Osterman, I.A.; Marks, J.; Sergeeva, V.A.; Kasatsky, P.; Komarova, E.S.; Stavrianidi, A.N.; Rodin, I.A.; Konevega, A.L.; et al. Binding and Action of Amino Acid Analogs of Chloramphenicol upon the Bacterial Ribosome. J. Mol. Biol. 2018, 16, 842–852. [Google Scholar] [CrossRef]
- Khairullina, Z.Z.; Tereshchenkov, A.G.; Zavyalova, S.A.; Komarova, D.A.; Lukianov, V.N.; Tashlitsky, I.A.; Osterman, N.V. Interaction of Chloramphenicol Cationic Peptide Analogues with the Ribosome. Biochemistry 2020, 85, 1443–1457. [Google Scholar] [CrossRef]
- Mardirossian, M.; Barrière, Q.; Timchenko, T.; Müller, C.; Pacor, S.; Mergaert, P.; Scocchi, M.; Wilson, D.N. Fragments of the nonlytic proline-rich antimicrobial peptide Bac5 kill Escherichia coli cells by inhibiting protein synthesis. Antimicrob. Agents Chemother. 2018, 62, e00534-18. [Google Scholar] [CrossRef] [PubMed]
- Drainas, D.; Mamos, P.; Coutsogeorgopoulos, C. Aminoacyl Analogs of Chloramphenicol: Examination of the Kinetics of Inhibition of Peptide Bond Formation. J. Med. Chem. 1993, 36, 3542–3545. [Google Scholar] [CrossRef]
- Picking, W.D.; Odom, O.W.; Tsalkova, T.; Serdyuk, I.; Hardesty, B. The conformation of nascent polylysine and polyphenylalanine peptides on ribosomes. J. Biol. Chem. 1991, 266, 1534–1542. [Google Scholar] [CrossRef]
- Starosta, A.L.; Karpenko, V.V.; Shishkina, A.V.; Mikolajka, A.; Sumbatyan, N.V.; Schluenzen, F.; Korshunova, G.A.; Bogdanov, A.A.; Wilson, D.N. Interplay between the ribosomal tunnel, nascent chain, and macrolides influences drug inhibition. Chem. Biol. 2010, 17, 504–514. [Google Scholar] [CrossRef]
- Blaha, G.; Stelzl, U.; Spahn, C.M.; Agrawal, R.K.; Frank, J.; Nierhaus, K.H. Preparation of functional ribosomal complexes and effect of buffer conditions on tRNA positions observed by cryoelectron microscopy. Methods Enzymol. 2000, 317, 292–309. [Google Scholar] [CrossRef] [PubMed]
- Rheinberger, H.J.; Geigenmüller, U.; Wedde, M.; Nierhaus, K.H. Parameters for the Preparation of Escherichia Coli Ribosomes and Ribosomal Subunits Active in tRNA Binding. Methods Enzymol. 1988, 164, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Bommer, U.; Burkhardt, N.; Junemann, R.; Spahn, C.M.; Triana-Alonso, F.J.; Nierhaus, K.H. Ribosomes and polysomes. In Subcellular Fractionation. A Practical Approach; Graham, J., Rickwoods, D., Eds.; IRL Press at Oxford University Press: Oxford, UK, 1996; Volume 9, pp. 787–793. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibiotic | μM | μg/mL |
|---|---|---|
| Chloramphenicol | 2.36 | 0.76 |
| Bis-dichloroacetyl-ornithine | 12.25 | 6.70 |
| Histamine–succinyl–CLB | 25.02 | 13.71 |
| Bis-Boc-histidine–CLB | 34.00 | 18.66 |
| Compound | Structure |
|---|---|
| 2 |  |
| 6 |  |
| 9 |  |
| 10 |  |
| 19 |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsirogianni, A.; Kournoutou, G.G.; Bougas, A.; Poulou-Sidiropoulou, E.; Dinos, G.; Athanassopoulos, C.M. New Chloramphenicol Derivatives with a Modified Dichloroacetyl Tail as Potential Antimicrobial Agents. Antibiotics 2021, 10, 394. https://doi.org/10.3390/antibiotics10040394
Tsirogianni A, Kournoutou GG, Bougas A, Poulou-Sidiropoulou E, Dinos G, Athanassopoulos CM. New Chloramphenicol Derivatives with a Modified Dichloroacetyl Tail as Potential Antimicrobial Agents. Antibiotics. 2021; 10(4):394. https://doi.org/10.3390/antibiotics10040394
Chicago/Turabian StyleTsirogianni, Artemis, Georgia G. Kournoutou, Anthony Bougas, Eleni Poulou-Sidiropoulou, George Dinos, and Constantinos M. Athanassopoulos. 2021. "New Chloramphenicol Derivatives with a Modified Dichloroacetyl Tail as Potential Antimicrobial Agents" Antibiotics 10, no. 4: 394. https://doi.org/10.3390/antibiotics10040394
APA StyleTsirogianni, A., Kournoutou, G. G., Bougas, A., Poulou-Sidiropoulou, E., Dinos, G., & Athanassopoulos, C. M. (2021). New Chloramphenicol Derivatives with a Modified Dichloroacetyl Tail as Potential Antimicrobial Agents. Antibiotics, 10(4), 394. https://doi.org/10.3390/antibiotics10040394