
Optimization of 2-Acylaminocycloalkylthiophene Derivatives for Activity against Staphylococcus aureus RnpA


Abstract
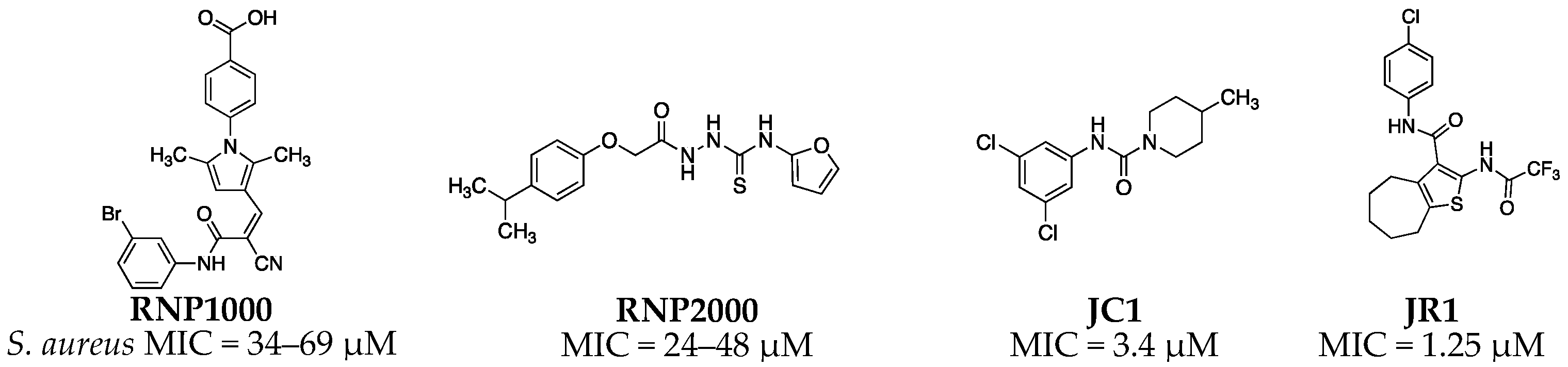
1. Introduction
2. Results
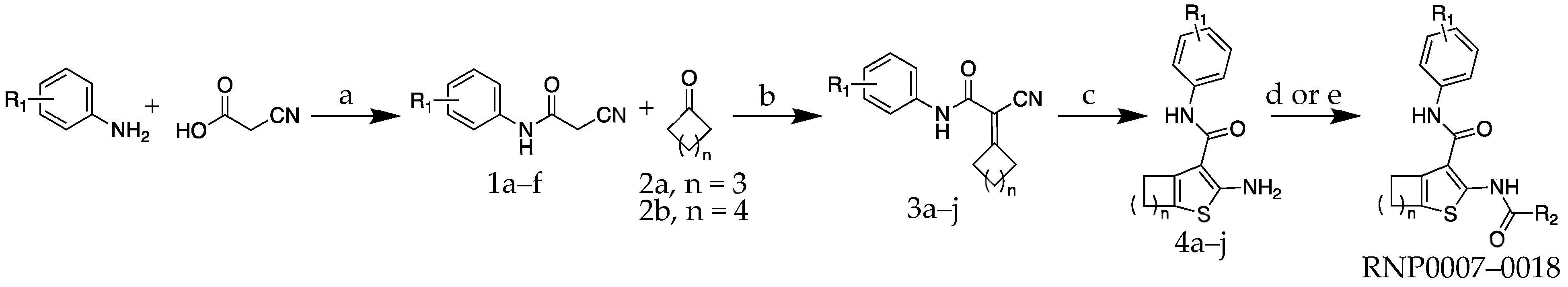
2.1. Chemistry
2.2. Antimicrobial Structure-Activity Relationship of 2-acylcycloalkylthiophene Analogs
2.3. In Vitro RNA Metabolism Activity for Analogs
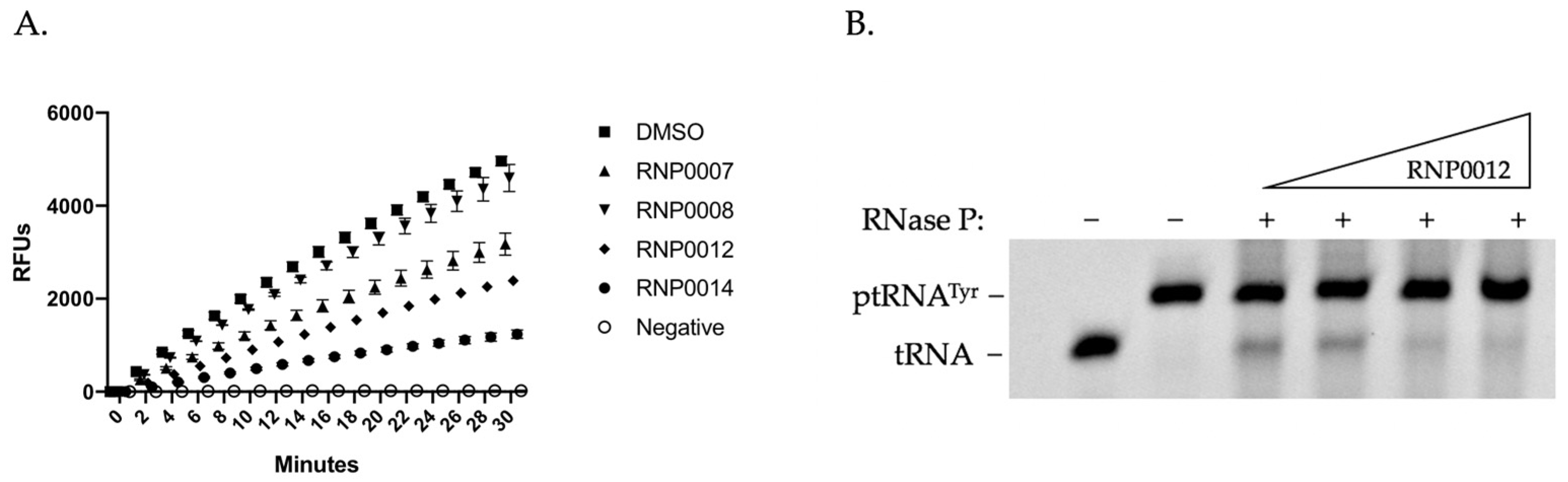
2.3.1. In Vitro mRNA Degradation Activity
2.3.2. In Vitro RnpA ptRNA Processing Activity of Analogs
2.4. Activity of RnpA Inhibitors on RNA Metabolism in Cellular Environment
2.4.1. Mupirocin Synergy Activity of Each Analog
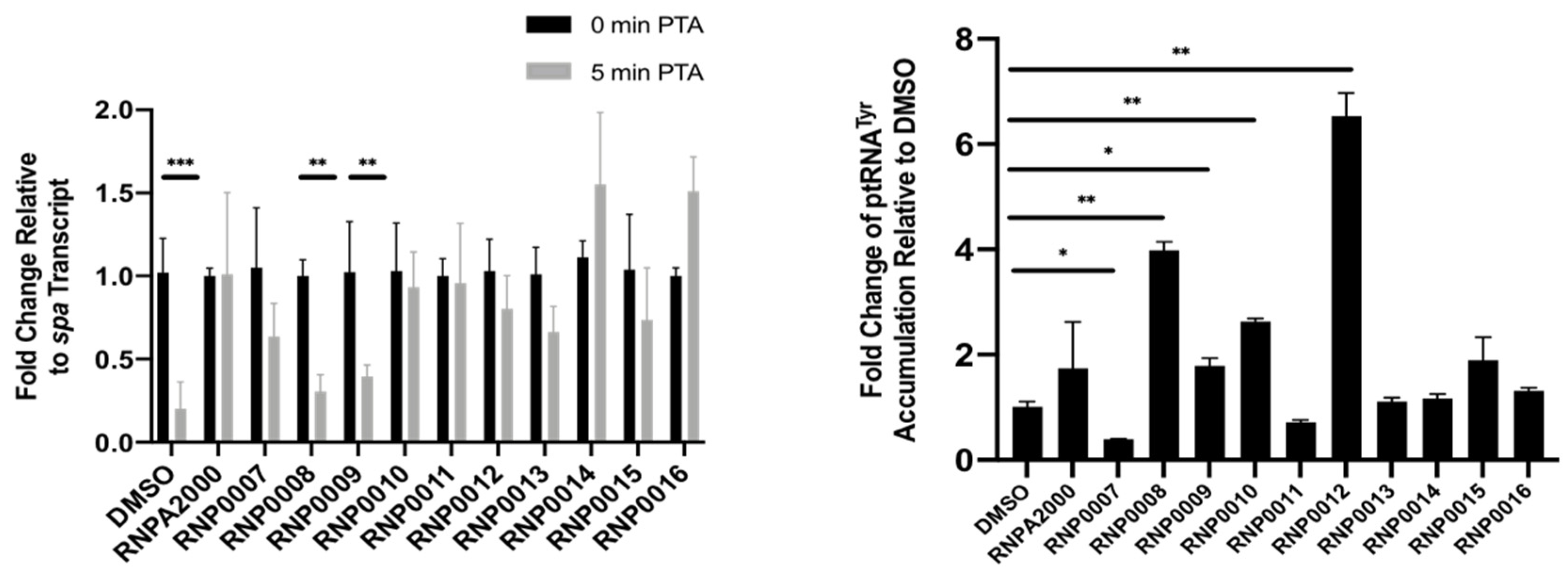
2.4.2. Inhibition of Intracellular mRNA Turnover
2.4.3. Inhibition of Intracellular prRNA Processing
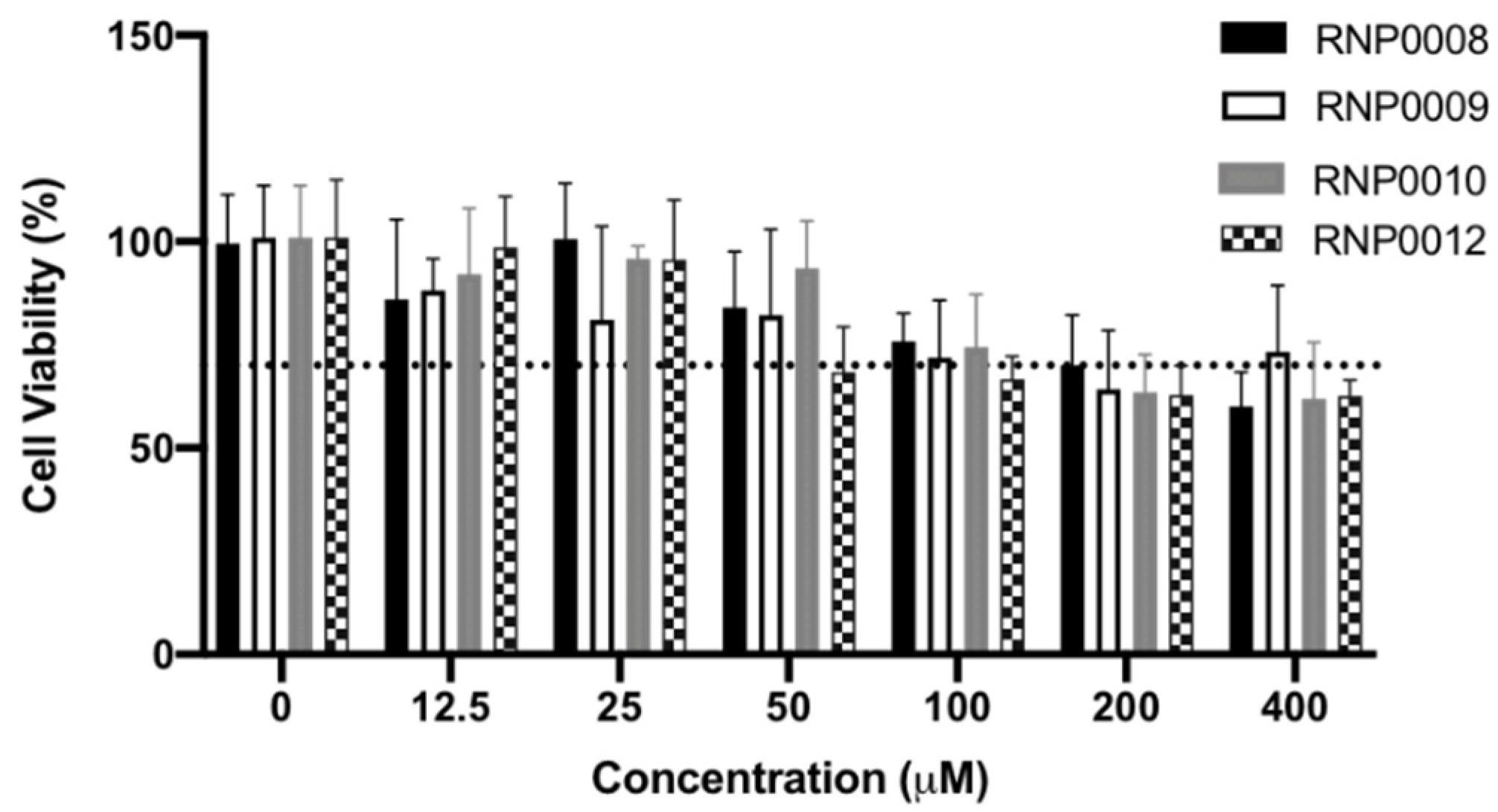
2.5. Evaluation of Analogs for Cytotoxicity on Human Cells
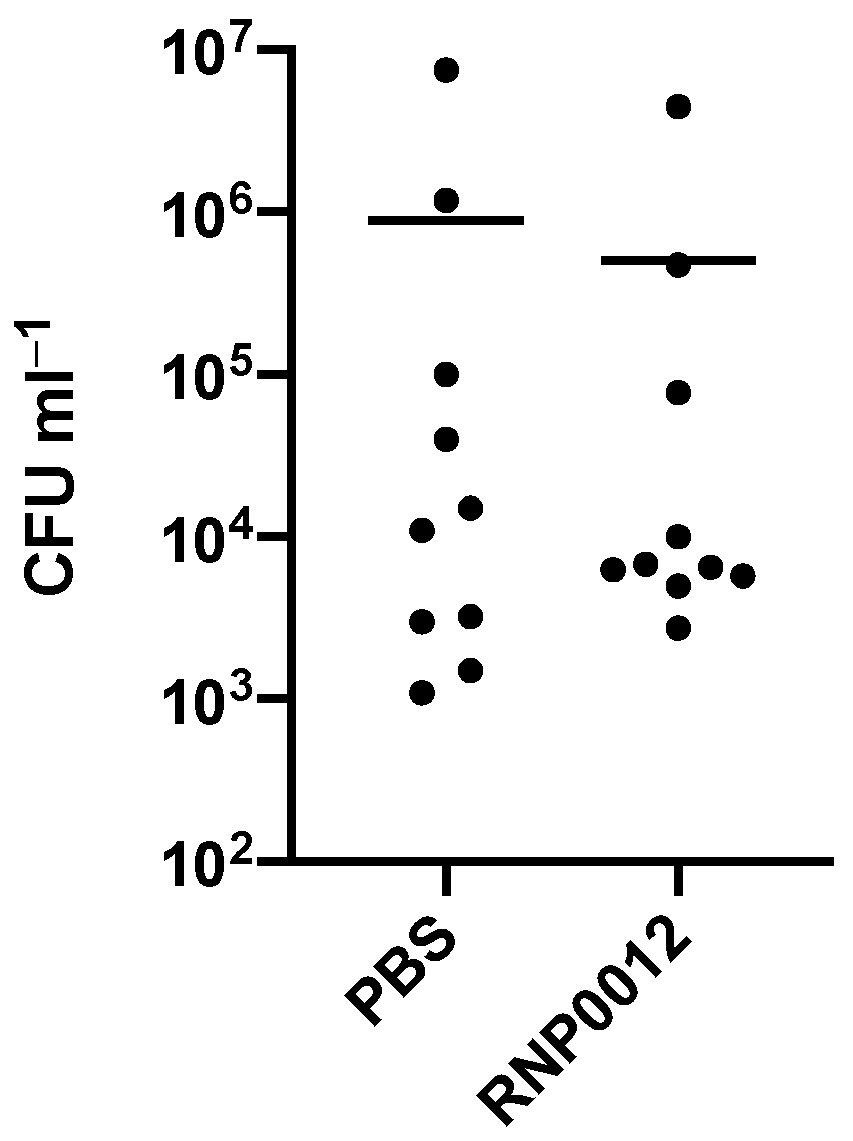
2.6. Evaluation of RNP0012 Efficacy in a Murine Model of Bacterial Keratitis
3. Discussion
4. Materials and Methods
4.1. Chemistry
General Experimental
4.2. Biological Evaluation
4.2.1. Bacterial Growth Conditions
4.2.2. Antimicrobial Susceptibility Testing
4.2.3. RnpA Protein Purification
4.2.4. In Vitro Transcription of RNA
4.2.5. In Vitro mRNA Degradation Assays
4.2.6. In Vitro ptRNA Processing Assays
4.2.7. Cellular mRNA Turnover Assays
4.2.8. Cellular tRNATyr Population Measures
4.2.9. Bacterial RNA isolation and Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
4.2.10. Cytotoxicity Testing
4.2.11. Murine Corneal Infection Model and Treatment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wertheim, H.F.L.; Melles, D.C.; Vos, M.C.; van Leeuwen, W.; van Belkum, A.; Verbrugh, H.A.; Nouwen, J.L. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis. 2005, 5, 751–762. [Google Scholar] [CrossRef]
- Becker, K.; Schaumburg, F.; Fegeler, C.; Friedrich, A.W.; Köck, R. Staphylococcus aureus from the German general population is highly diverse. Int. J. Med. Microbiol. 2017, 307, 21–27. [Google Scholar] [CrossRef]
- Roberts, S.; Chambers, S. Diagnosis and management of Staphylococcus aureus infections of the skin and soft tissue. Intern. Med. J. 2005, 35, S97–S105. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.H.; Howden, B.P. Diagnosis and management of Staphylococcus aureus bacteraemia. Intern. Med. J. 2005, 35, S17–S24. [Google Scholar] [CrossRef]
- Murdoch, D.R.; Corey, G.R.; Hoen, B.; Miró, J.M.; Fowler, V.G.; Bayer, A.S.; Karchmer, A.W.; Olaison, L.; Pappas, P.A.; Moreillon, P. Clinical presentation, etiology, and outcome of infective endocarditis in the 21st century: The International Collaboration on Endocarditis–Prospective Cohort Study. Arch. Intern. Med. 2009, 169, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Murray, R.J. Staphylococcus aureus infective endocarditis: Diagnosis and management guidelines. Intern. Med. J. 2005, 35, S25–S44. [Google Scholar] [CrossRef]
- Turner, N.A.; Sharma-Kuinkel, B.K.; Maskarinec, S.A.; Eichenberger, E.M.; Shah, P.P.; Carugati, M.; Holland, T.L.; Fowler, V.G. Methicillin-resistant Staphylococcus aureus: An overview of basic and clinical research. Nat. Rev. Microbiol. 2019, 17, 203–218. [Google Scholar] [CrossRef]
- Diekema, D.J.; Hsueh, P.-R.; Mendes, R.E.; Pfaller, M.A.; Rolston, K.V.; Sader, H.S.; Jones, R.N. The microbiology of bloodstream infection: 20-year trends from the SENTRY antimicrobial surveillance program. Antimicrob. Agents Chemother. 2019, 63, e00355-19. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.Y.C.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef]
- Fowler, V.G.; Miro, J.M.; Hoen, B.; Cabell, C.H.; Abrutyn, E.; Rubinstein, E.; Corey, G.R.; Spelman, D.; Bradley, S.F.; Barsic, B. Staphylococcus aureus endocarditis: A consequence of medical progress. JAMA 2005, 293, 3012–3021. [Google Scholar] [CrossRef]
- Huang, Y.; Jiao, Y.; Zhang, J.; Xu, J.; Cheng, Q.; Li, Y.; Liang, S.; Li, H.; Gong, J.; Zhu, Y. Microbial etiology and prognostic factors of ventilator-associated pneumonia: A multicenter retrospective study in Shanghai. Clin. Infect. Dis. 2018, 67, S146–S152. [Google Scholar] [CrossRef]
- Bonell, A.; Azarrafiy, R.; Huong, V.T.L.; Viet, T.L.; Phu, V.D.; Dat, V.Q.; Wertheim, H.; van Doorn, H.R.; Lewycka, S.; Nadjm, B. A systematic review and meta-analysis of ventilator-associated pneumonia in adults in Asia: An analysis of national income level on incidence and etiology. Clin. Infect. Dis. 2019, 68, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, V.D.; Maki, D.G.; Mehta, A.; Álvarez-Moreno, C.; Leblebicioglu, H.; Higuera, F.; Cuellar, L.E.; Madani, N.; Mitrev, Z.; Dueñas, L. International nosocomial infection control consortium report, data summary for 2002–2007, issued January 2008. Am. J. Infect. Control. 2008, 36, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, V.D.; Al-Abdely, H.M.; El-Kholy, A.A.; AlKhawaja, S.A.A.; Leblebicioglu, H.; Mehta, Y.; Rai, V.; Hung, N.V.; Kanj, S.S.; Salama, M.F. International Nosocomial Infection Control Consortium report, data summary of 50 countries for 2010–2015: Device-associated module. Am. J. Infect. Control. 2016, 44, 1495–1504. [Google Scholar] [CrossRef]
- Wałaszek, M.; Różańska, A.; Wałaszek, M.Z.; Wójkowska-Mach, J. Epidemiology of Ventilator-Associated Pneumonia, microbiological diagnostics and the length of antimicrobial treatment in the Polish Intensive Care Units in the years 2013–2015. BMC Infect. Dis. 2018, 18, 308. [Google Scholar] [CrossRef]
- Peng, M.Y.; Cevallos, V.; McLeod, S.D.; Lietman, T.M.; Rose-Nussbaumer, J. Bacterial keratitis: Isolated organisms and antibiotic resistance patterns in San Francisco. Cornea 2018, 37, 84. [Google Scholar] [CrossRef]
- Whitcher, J.P.; Srinivasan, M.; Upadhyay, M.P. Corneal blindness: A global perspective. Bull. World Health Organ. 2001, 79, 214–221. [Google Scholar]
- Planet, P.J. Life after USA300: The rise and fall of a superbug. J. Infect. Dis. 2017, 215, S71–S77. [Google Scholar] [CrossRef]
- Hiramatsu, K.; Hanaki, H.; Ino, T.; Yabuta, K.; Oguri, T.; Tenover, F.C. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J. Antimicrob. Chemother. 1997, 40, 135–136. [Google Scholar] [CrossRef]
- Liu, C.; Chambers, H.F. Staphylococcus aureus with heterogeneous resistance to vancomycin: Epidemiology, clinical significance, and critical assessment of diagnostic methods. Antimicrob. Agents Chemother. 2003, 47, 3040–3045. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Wang, M.-C.; Huang, I.-W.; Chen, F.-J.; Lauderdale, T.-L. Development of daptomycin nonsusceptibility with heterogeneous vancomycin-intermediate resistance and oxacillin susceptibility in methicillin-resistant Staphylococcus aureus during high-dose daptomycin treatment. Antimicrob. Agents Chemother. 2010, 54, 4038–4040. [Google Scholar] [CrossRef]
- Long, S.W.; Olsen, R.J.; Mehta, S.C.; Palzkill, T.; Cernoch, P.L.; Perez, K.K.; Musick, W.L.; Rosato, A.E.; Musser, J.M. PBP2a mutations causing high-level ceftaroline resistance in clinical methicillin-resistant Staphylococcus aureus isolates. Antimicrob. Agents Chemother. 2014, 58, 6668–6674. [Google Scholar] [CrossRef] [PubMed]
- Schaumburg, F.; Peters, G.; Alabi, A.; Becker, K.; Idelevich, E.A. Missense mutations of PBP2a are associated with reduced susceptibility to ceftaroline and ceftobiprole in African MRSA. J. Antimicrob. Chemother. 2016, 71, 41–44. [Google Scholar] [CrossRef]
- Mammina, C.; Bonura, C.; di Carlo, P.; Calà, C.; Aleo, A.; Monastero, R.; Palma, D.M. Daptomycin non-susceptible, vancomycin intermediate methicillin-resistant Staphylococcus aureus ST398 from a chronic leg ulcer, Italy. Scand. J. Infect. Dis. 2010, 42, 955–957. [Google Scholar] [CrossRef]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Spitzfaden, C.; Nicholson, N.; Jones, J.J.; Guth, S.; Lehr, R.; Prescott, C.D.; Hegg, L.A.; Eggleston, D.S. The structure of ribonuclease P protein from Staphylococcus aureus reveals a unique binding site for single-stranded RNA. J. Mol. Biol. 2000, 295, 105–115. [Google Scholar] [CrossRef]
- Kazantsev, A.V.; Pace, N.R. Bacterial RNase P: A new view of an ancient enzyme. Nat. Rev. Microbiol. 2006, 4, 729–740. [Google Scholar] [CrossRef]
- Olson, P.D.; Kuechenmeister, L.J.; Anderson, K.L.; Daily, S.; Beenken, K.E.; Roux, C.M.; Reniere, M.L.; Lewis, T.L.; Weiss, W.J.; Pulse, M.; et al. Small molecule inhibitors of staphylococcus aureus RnpA alter cellular mRNA turnover, exhibit antimicrobial activity, and attenuate pathogenesis. PLoS Pathog. 2011, 7. [Google Scholar] [CrossRef]
- Roux, C.M.; DeMuth, J.P.; Dunman, P.M. Characterization of components of the Staphylococcus aureus mRNA degradosome holoenzyme-like complex. J. Bacteriol. 2011, 193, 5520–5526. [Google Scholar] [CrossRef]
- Wang, X.; Wang, C.; Wu, M.; Tian, T.; Cheng, T.; Zhang, X.; Zang, J. Enolase binds to RnpA in competition with PNP ase in Staphylococcus aureus. FEBS Lett. 2017, 591, 3523–3535. [Google Scholar] [CrossRef]
- Ha, L.; Colquhoun, J.; Noinaj, N.; Das, C.; Dunman, P.M.; Flaherty, D.P. Crystal structure of the ribonuclease-P-protein subunit from Staphylococcus aureus research communications. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2018, 74 Pt 10, 632–637. [Google Scholar] [CrossRef]
- Blanchard, C.; Brooks, L.; Beckley, A.; Colquhoun, J.; Dewhurst, S.; Dunman, P.M. Neomycin sulfate improves the antimicrobial activity of mupirocin-based antibacterial ointments. Antimicrob. Agents Chemother. 2016, 60, 862–872. [Google Scholar] [CrossRef]
- Eidem, T.M.; Lounsbury, N.; Emery, J.F.; Bulger, J.; Smith, A.; Abou-Gharbia, M.; Childers, W.; Dunman, P.M. Small-Molecule Inhibitors of Staphylococcus aureus RnpA-Mediated RNA Turnover and tRNA Processing. Antimicrob. Agents Chemother. 2015, 59, 2016–2028. [Google Scholar] [CrossRef] [PubMed]
- Colquhoun, J.M.; Ha, L.; Beckley, A.; Meyers, B.; Flaherty, D.P.; Dunman, P.M. Identification of Small Molecule Inhibitors of Staphylococcus aureus RnpA. Antibiotics 2019, 8, 48. [Google Scholar] [CrossRef] [PubMed]
- Truong, E.C.; Phuan, P.W.; Reggi, A.L.; Ferrera, L.; Galietta, L.J.V.; Levy, S.E.; Moises, A.C.; Cil, O.; Diez-Cecilia, E.; Lee, S.; et al. Substituted 2-Acylaminocycloalkylthiophene-3-carboxylic Acid Arylamides as Inhibitors of the Calcium-Activated Chloride Channel Transmembrane Protein 16A (TMEM16A). J. Med. Chem. 2017, 60, 4626–4635. [Google Scholar] [CrossRef] [PubMed]
- Lounsbury, N.; Eidem, T.; Colquhoun, J.; Mateo, G.; Abou-gharbia, M.; Dunman, P.M.; Childers, W.E. Novel inhibitors of Staphylococcus aureus RnpA that synergize with mupirocin. Bioorg. Med. Chem. Lett. 2018, 28, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Potter, V.R. Sequential blocking of metabolic pathways in vivo. Proc. Soc. Exp. Biol. Med. 1951, 76, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Bushby, S.R. Synergy of trimethoprim-sulfamethoxazole. Can. Med. Assoc. J. 1975, 112, 63. [Google Scholar]
- Chojnacki, M.; Philbrick, A.; Wucher, B.; Reed, J.N.; Tomaras, A.; Dunman, P.M.; Wozniak, R.A.F. Development of a broad-spectrum antimicrobial combination for the treatment of Staphylococcus aureus and Pseudomonas aeruginosa corneal infections. Antimicrob. Agents Chemother. 2019, 63, e01929-18. [Google Scholar] [CrossRef]
- Kaur, J.; Cao, X.; Abutaleb, N.S.; Elkashif, A.; Graboski, A.L.; Krabill, A.D.; AbdelKhalek, A.H.; An, W.; Bhardwaj, A.; Seleem, M.N.; et al. Optimization of Acetazolamide-Based Scaffold as Potent Inhibitors of Vancomycin-Resistant Enterococcus. J. Med. Chem. 2020, 63, 9540–9562. [Google Scholar] [CrossRef] [PubMed]
- Odds, F.C. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 2003, 52, 1. [Google Scholar] [CrossRef] [PubMed]
- Blevins, J.S.; Beenken, K.E.; Elasri, M.O.; Hurlburt, B.K.; Smeltzer, M.S. Strain-dependent differences in the regulatory roles of sarA and agr in Staphylococcus aureus. Infect. Immun. 2002, 70, 470–480. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | R1 | R2 | n | MIC (µM) | MIC (µg/mL) |
|---|---|---|---|---|---|
| RNPA2000 | - | - | - | 44 | 15 |
| JR1 a | 4-Cl | CF3 | 4 | 1.25 | 0.52 |
| RNP0007 | 3-Cl | CF3 | 4 | 0.391 | 0.16 |
| RNP0008 | 3-F | CF3 | 4 | 0.391 | 0.16 |
| RNP0009 | 4-F | CF3 | 4 | 0.78 | 0.32 |
| RNP0010 | 3-CH3 | CF3 | 4 | 3.125 | 1.24 |
| RNP0011 | 3-CF3 | CF3 | 4 | 0.391 | 0.18 |
| RNP0012 | 4-CF3 | CF3 | 4 | 0.19 | 0.086 |
| RNP0013 | 3-F | CF3 | 3 | 6.25 | 2.4 |
| RNP0014 | 4-F | CF3 | 3 | 6.25 | 2.4 |
| RNP0015 | 4-CF3 | CF3 | 3 | 0.78 | 0.34 |
| RNP0016 | 3-CF3 | CF3 | 3 | 3.125 | 1.36 |
| RNP0017 | 3-Cl | CHF2 | 4 | >500 | >200 |
| RNP0018 | 3-Cl | CH2F | 4 | >500 | >200 |
| Compound | MIC (µM) | mRNA IC50 (µM) | ptRNA IC50 (µM) |
|---|---|---|---|
| RNPA2000 | 44 | 130 | 130 |
| JR1 a | 1.25 | 50 | 7 |
| RNP0007 | 0.391 | 175 | 98 |
| RNP0008 | 0.391 | 245 | 78 |
| RNP0009 | 0.78 | >250 | 79 |
| RNP0010 | 3.125 | 224 | 70 |
| RNP0011 | 0.391 | 59 | 57 |
| RNP0012 | 0.19 | 68 | 66 |
| RNP0013 | 6.25 | 50 | 95 |
| RNP0014 | 6.25 | 6.4 | 138 |
| RNP0015 | 0.78 | 43 | 46 |
| RNP0016 | 3.125 | 45 | 52 |
| RNP0017 | >500 | 43 | >250 |
| RNP0018 | >500 | 33 | 58 |
| Dosed-Individually | Dosed-Combination | FIC Index b | |||
|---|---|---|---|---|---|
| Cpd MIC a | Mup MIC a | Cpd MIC a | Mup MIC a | ||
| RNP0007 | 0.391 | 0.5 | 0.098 | 0.25 | 0.75 |
| RNP0008 | 0.391 | 0.5 | 0.196 | 0.25 | 1 |
| RNP0009 | 0.78 | 0.5 | 0.196 | 0.25 | 0.75 |
| RNP0010 | 3.125 | 0.5 | 0.78 | 0.25 | 0.75 |
| RNP0011 | 0.391 | 0.5 | 0.098 | 0.25 | 0.75 |
| RNP0012 | 0.19 | 0.5 | 0.048 | 0.25 | 0.75 |
| RNP0013 | 6.25 | 0.5 | 1.56 | 0.25 | 0.75 |
| RNP0014 | 6.25 | 0.5 | 1.56 | 0.125 | 0.5 |
| RNP0015 | 0.78 | 0.5 | 0.78 | 0.25 | 1.5 |
| RNP0016 | 3.125 | 0.5 | 1.56 | 0.25 | 1 |
| RNP0017 | >500 | 0.5 | - | - | - |
| RNP0018 | >500 | 0.5 | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chojnacki, M.; Cao, X.; Flaherty, D.P.; Dunman, P.M. Optimization of 2-Acylaminocycloalkylthiophene Derivatives for Activity against Staphylococcus aureus RnpA. Antibiotics 2021, 10, 369. https://doi.org/10.3390/antibiotics10040369
Chojnacki M, Cao X, Flaherty DP, Dunman PM. Optimization of 2-Acylaminocycloalkylthiophene Derivatives for Activity against Staphylococcus aureus RnpA. Antibiotics. 2021; 10(4):369. https://doi.org/10.3390/antibiotics10040369
Chicago/Turabian StyleChojnacki, Michaelle, Xufeng Cao, Daniel P. Flaherty, and Paul M. Dunman. 2021. "Optimization of 2-Acylaminocycloalkylthiophene Derivatives for Activity against Staphylococcus aureus RnpA" Antibiotics 10, no. 4: 369. https://doi.org/10.3390/antibiotics10040369
APA StyleChojnacki, M., Cao, X., Flaherty, D. P., & Dunman, P. M. (2021). Optimization of 2-Acylaminocycloalkylthiophene Derivatives for Activity against Staphylococcus aureus RnpA. Antibiotics, 10(4), 369. https://doi.org/10.3390/antibiotics10040369