
Tracking Reservoirs of Antimicrobial Resistance Genes in a Complex Microbial Community Using Metagenomic Hi-C: The Case of Bovine Digital Dermatitis


Abstract
1. Introduction
2. Results
2.1. Microbiome Profiles
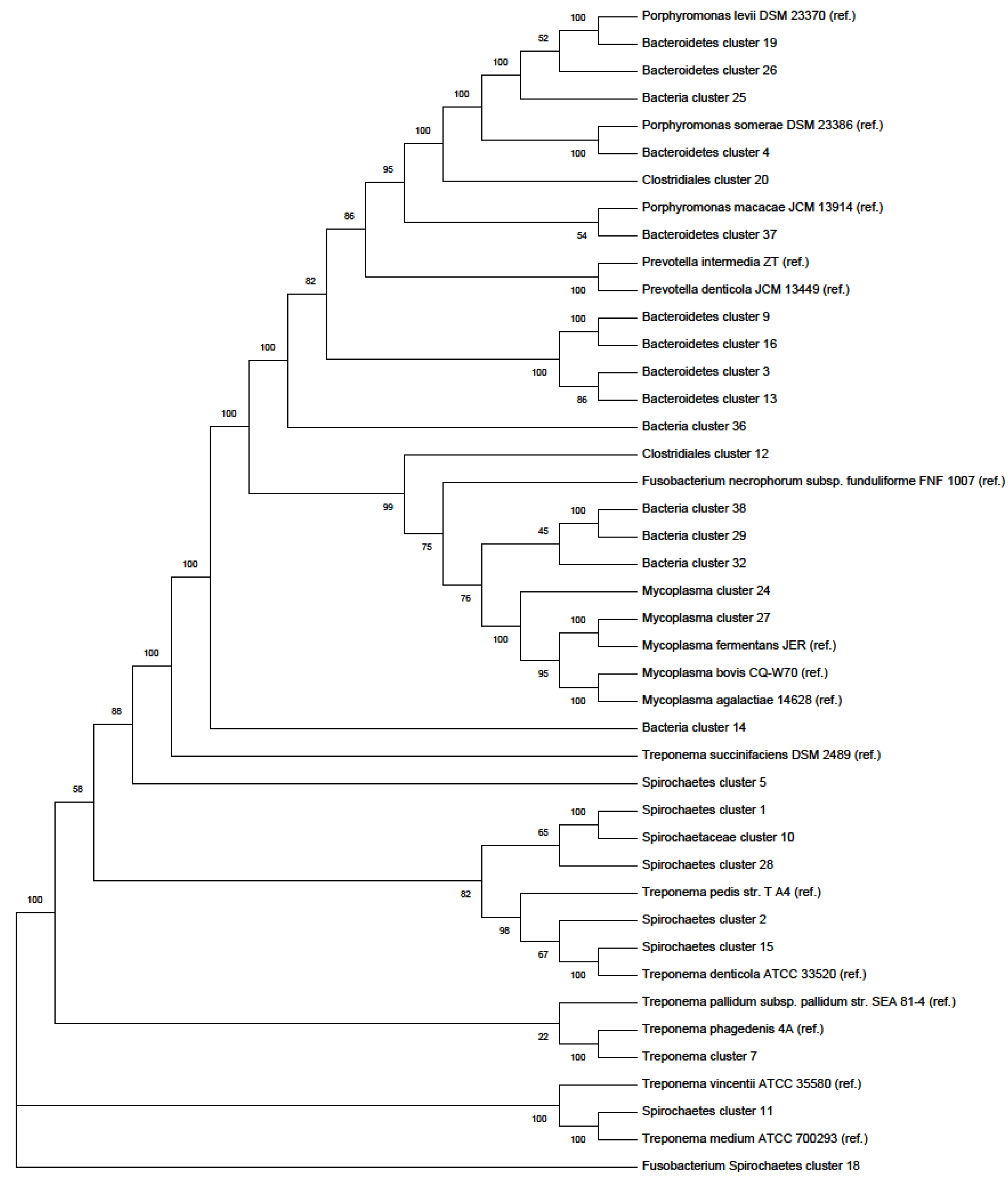
Phylogenetic Tree
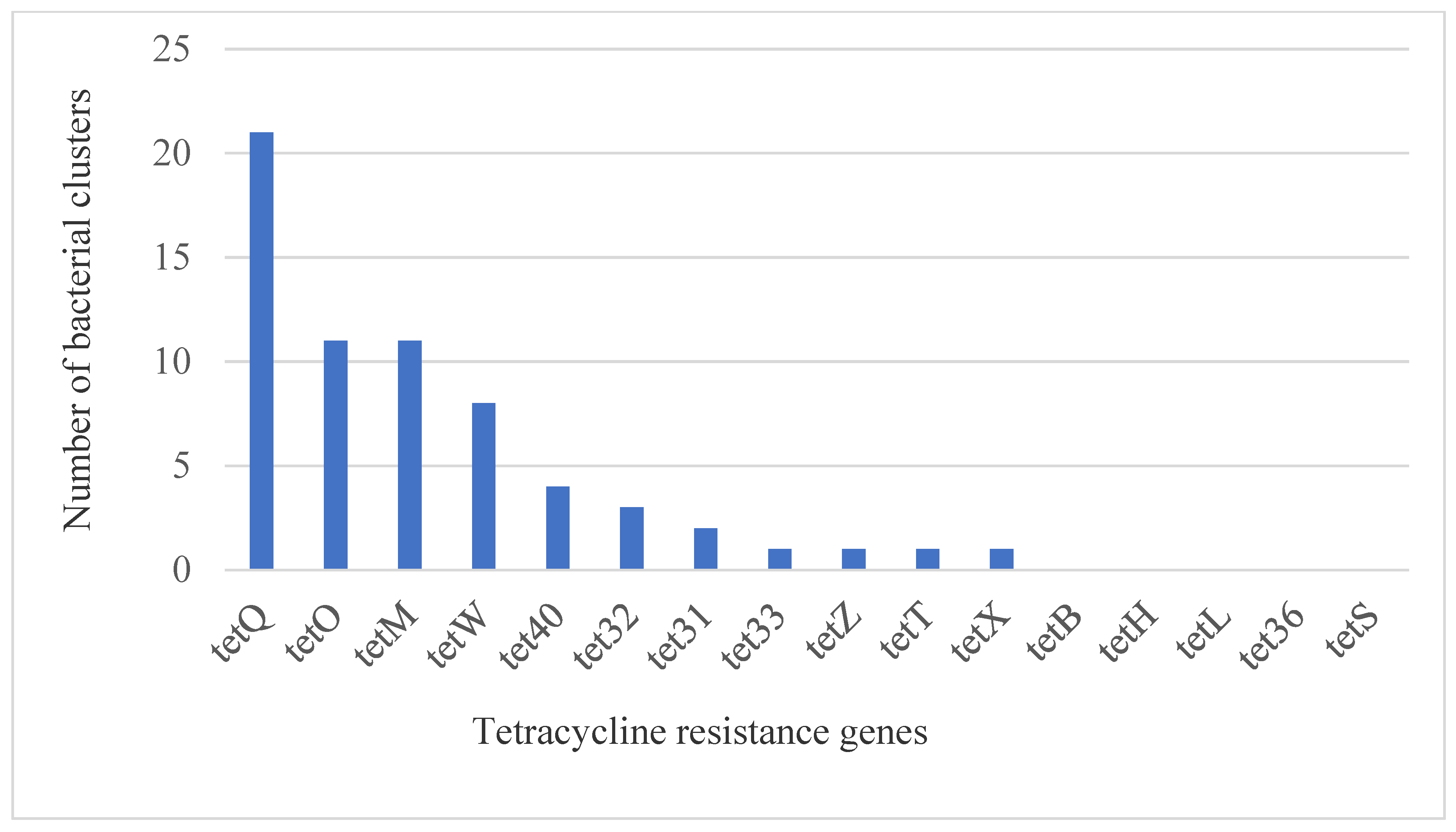
2.2. Resistome Profiles
3. Discussion
4. Materials and Methods
4.1. Description of the Sample
4.2. DNA Extraction and Library Preparation
4.2.1. DNA Extraction for Shotgun Metagenomics
4.2.2. DNA Extraction and Library Preparation for ProxiMeta Hi-C Metagenomics
4.2.3. Metagenomic Data Analysis
4.3. 16S rRNA BLASTing and Phylogenetic Tree Plotting
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van de Gucht, T.; Saeys, W.; van Meensel, J.; van Nuffel, A.; Vangeyte, J.; Lauwers, L. Farm-specific economic value of automatic lameness detection systems in dairy cattle: From concepts to operational simulations. J. Dairy Sci. 2018, 101, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Relun, A.; Lehebel, A.; Chesnin, A.; Guatteo, R.; Bareille, N. Association between digital dermatitis lesions and test-day milk yield of Holstein cows from 41 French dairy farms. J. Dairy Sci. 2013, 96, 2190–2200. [Google Scholar] [CrossRef] [PubMed]
- Terrell, S.P.; Reinhardt, C.D.; Larson, C.K.; Vahl, C.I.; Thomson, D.U. Incidence of lameness and association of cause and severity of lameness on the outcome for cattle on six commercial beef feedlots. J. Am. Vet. Med. Assoc. 2017, 250, 437–445. [Google Scholar] [CrossRef]
- Sullivan, L.E.; Carter, S.D.; Blowey, R.; Duncan, J.S.; Grove-White, D.; Evans, N.J. Digital dermatitis in beef cattle. Vet. Rec. 2013, 173, 582. [Google Scholar] [CrossRef] [PubMed]
- Wilson-Welder, J.H.; Alt, D.P.; Nally, J.E. The etiology of digital dermatitis in ruminants: Recent perspectives. Vet. Med.-Res. Rep. 2015, 6, 155–164. [Google Scholar]
- Plummer, P.J.; Krull, A. Clinical Perspectives of Digital Dermatitis in Dairy and Beef Cattle. Vet. Clin. N. Am.-Food A 2017, 33, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, S.L.; Malchiodi, F.; Thompson-Crispi, K.; Miglior, F.; Mallard, B.A. Short communication: Prevalence of digital dermatitis in Canadian dairy cattle classified as high, average, or low antibody and cell-mediated immune responders. J. Dairy Sci. 2017, 100, 8409–8413. [Google Scholar] [CrossRef] [PubMed]
- Holzhauer, M.; Hardenberg, C.; Bartels, C.J.M.; Frankena, K. Herd- and cow-level prevalence of digital dermatitis in the Netherlands and associated factors. J. Dairy Sci. 2006, 89, 580–588. [Google Scholar] [CrossRef]
- Hesseling, J.; Legione, A.R.; Stevenson, M.A.; McCowan, C.I.; Pyman, M.F.; Finochio, C.; Nguyen, D.; Roic, C.L.; Thiris, O.L.; Zhang, A.J.; et al. Bovine digital dermatitis in Victoria, Australia. Aust. Vet. J. 2019, 97, 404–413. [Google Scholar] [CrossRef]
- Read, D.H.; Walker, R.L. Papillomatous digital dermatitis (footwarts) in California dairy cattle: Clinical and gross pathologic findings. J. Vet. Diagn. Invest. 1998, 10, 67–76. [Google Scholar] [CrossRef]
- van Amstel, S.R.; van Vuuren, S.; Tutt, C.L.C. Digital dermatitis - report of an outbreak. J. S. Afr. Vet. Assoc. 1995, 66, 177–181. [Google Scholar]
- Cha, E.; Hertl, J.A.; Bar, D.; Grohn, Y.T. The cost of different types of lameness in dairy cows calculated by dynamic programming. Prev. Vet. Med. 2010, 97, 1–8. [Google Scholar] [CrossRef]
- Refaai, W.; Van Aert, M.; Abd El-Aal, A.M.; Behery, A.E.; Opsomer, G. Infectious diseases causing lameness in cattle with a main emphasis on digital dermatitis (Mortellaro disease). Livest. Sci. 2013, 156, 53–63. [Google Scholar] [CrossRef]
- Krull, A.C.; Shearer, J.K.; Gorden, P.J.; Cooper, V.L.; Phillips, G.J.; Plummer, P.J. Deep Sequencing Analysis Reveals Temporal Microbiota Changes Associated with Development of Bovine Digital Dermatitis. Infect. Immun. 2014, 82, 3359–3373. [Google Scholar] [CrossRef]
- Trott, D.J.; Moeller, M.R.; Zuerner, R.L.; Goff, J.P.; Waters, W.R.; Alt, D.P.; Walker, R.L.; Wannemuehler, M.J. Characterization of Treponema phagedenis-like spirochetes isolated from papillomatous digital dermatitis lesions in dairy cattle. J. Clin. Microbiol. 2003, 41, 2522–2529. [Google Scholar] [CrossRef]
- Rebhun, W.C.; Payne, R.M.; King, J.M.; Wolfe, M.; Begg, S.N. Interdigital papillomatosis in dairy-cattle. J. Am. Vet. Med. Assoc. 1980, 177, 437–440. [Google Scholar] [PubMed]
- Rebhun, W.C. Interdigital papillomatosis in dairy cattle. Proc. XIIth World Congress Diseases Cattle Neth. 1982, II, 833–837. [Google Scholar]
- Nielsen, M.W.; Strube, M.L.; Isbrand, A.; Al-Medrasi, W.D.H.M.; Boye, M.; Jensen, T.K.; Klitgaard, K. Potential bacterial core species associated with digital dermatitis in cattle herds identified by molecular profiling of interdigital skin samples. Vet. Microbiol. 2016, 186, 139–149. [Google Scholar] [CrossRef]
- Zinicola, M.; Higgins, H.; Lima, S.; Machado, V.; Guard, C.; Bicalho, R. Shotgun Metagenomic Sequencing Reveals Functional Genes and Microbiome Associated with Bovine Digital Dermatitis. PLoS ONE 2015, 10, e0133674. [Google Scholar] [CrossRef] [PubMed]
- Klitgaard, K.; Boye, M.; Capion, N.; Jensen, T.K. Evidence of multiple Treponema phylotypes involved in bovine digital dermatitis as shown by 16S rRNA gene analysis and fluorescence in situ hybridization. J. Clin. Microbiol. 2008, 46, 3012–3020. [Google Scholar] [CrossRef]
- Yano, T.; Moe, K.K.; Yamazaki, K.; Ooka, T.; Hayashi, T.; Misawa, N. Identification of candidate pathogens of papillomatous digital dermatitis in dairy cattle from quantitative 16S rRNA clonal analysis. Vet. Microbiol. 2010, 143, 352–362. [Google Scholar] [CrossRef]
- Evans, N.J.; Brown, J.M.; Demirkan, I.; Singh, P.; Getty, B.; Timofte, D.; Vink, W.D.; Murray, R.D.; Blowey, R.W.; Birtles, R.J.; et al. Association of unique, isolated treponemes with bovine digital dermatitis lesions. J. Clin. Microbiol. 2009, 47, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Zinicola, M.; Lima, F.; Lima, S.; Machado, V.; Gomez, M.; Doepfer, D.; Guard, C.; Bicalho, R. Altered microbiomes in bovine digital dermatitis lesions, and the gut as a pathogen reservoir. PLoS ONE 2015, 10, e0120504. [Google Scholar] [CrossRef]
- Mamuad, L.L.; Seo, B.J.; Al Faruk, M.S.; Espiritu, H.M.; Jin, S.J.; Kim, W.-I.; Lee, S.-S.; Cho, Y.-I. Treponema spp., the dominant pathogen in the lesion of bovine digital dermatitis and its characterization in dairy cattle. Vet. Microbiol. 2020, 245, 108696. [Google Scholar] [CrossRef]
- Espiritu, H.M.; Mamuad, L.L.; Jin, S.-j.; Kim, S.-h.; Kwon, S.-w.; Lee, S.-s.; Lee, S.-m.; Cho, Y.-i. Genotypic and phenotypic characterization of Treponema phagedenis from bovine digital dermatitis. Microorganisms 2020, 8, 1520. [Google Scholar] [CrossRef] [PubMed]
- Santos, T.M.A.; Pereira, R.V.; Caixeta, L.S.; Guard, C.L.; Bicalho, R.C. Microbial diversity in bovine papillomatous digital dermatitis in Holstein dairy cows from upstate New York. Fems Microbiol. Ecol. 2012, 79, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Beninger, C.; Naqvi, S.A.; Naushad, S.; Orsel, K.; Luby, C.; Derakhshani, H.; Khafipour, E.; De Buck, J. Associations between digital dermatitis lesion grades in dairy cattle and the quantities of four Treponema species. Vet. Res. 2018, 49, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Manske, T.; Hultgren, J.; Bergsten, C. Topical treatment of digital dermatitis associated with severe heel-horn erosion in a Swedish dairy herd. Prev. Vet. Med. 2002, 53, 215–231. [Google Scholar] [CrossRef]
- Watts, K.M.; Lahiri, P.; Arrazuria, R.; De Buck, J.; Knight, C.G.; Orsel, K.; Barkema, H.W.; Cobo, E.R. Oxytetracycline reduces inflammation and Treponeme burden whereas vitamin D-3 promotes beta-defensin expression in bovine infectious digital dermatitis. Cell Tissue Res. 2020, 379, 337–348. [Google Scholar] [CrossRef]
- Shearer, J.K.; Hernandez, J. Efficacy of two modified nonantibiotic formulations (Victory) for treatment of papillomatous digital dermatitis in dairy cows. J. Dairy Sci. 2000, 83, 741–745. [Google Scholar] [CrossRef]
- Yano, T.; Moe, K.K.; Chuma, T.; Misawa, N. Antimicrobial susceptibility of Treponema phagedenis-like spirochetes isolated from dairy cattle with papillomatous digital dermatitis lesions in Japan. J. Vet. Med. Sci. 2010, 72, 379–382. [Google Scholar] [CrossRef]
- Roberts, M.C. Tetracycline resistance determinants: Mechanisms of action, regulation of expression, genetic mobility, and distribution. Fems Microbiol. Rev. 1996, 19, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.L.; Caffrey, N.P.; Nobrega, D.B.; Cork, S.C.; Ronksley, P.E.; Barkema, H.W.; Polachek, A.J.; Ganshorn, H.; Sharma, N.; Kellner, J.D.; et al. Restricting the use of antibiotics in food-producing animals and its associations with antibiotic resistance in food-producing animals and human beings: A systematic review and meta-analysis. Lancet Planet Hlth. 2017, 1, e316–e327. [Google Scholar] [CrossRef]
- Evans, N.J.; Brown, J.M.; Demirkan, I.; Birtles, R.; Hart, C.A.; Carter, S.D. In vitro susceptibility of bovine digital dermatitis associated spirochaetes to antimicrobial agents. Vet. Microbiol. 2009, 136, 115–120. [Google Scholar] [CrossRef][Green Version]
- Schmieder, R.; Edwards, R. Insights into antibiotic resistance through metagenomic approaches. Future Microbiol. 2012, 7, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Bickhart, D.; Watson, M.; Koren, S.; Panke-Buisse, K.; Cersosimo, L.M.; Press, M.O.; Van Tassell, C.P.; Van Kessel, J.A.S.; Haley, B.J.; Kim, S.W.; et al. Assignment of virus and antimicrobial resistance genes to microbial hosts in a complex microbial community by combined long-read assembly and proximity ligation. Genome Biol. 2019, 20, 153. [Google Scholar] [CrossRef] [PubMed]
- Press, M.O.; Wiser, A.H.; Kronenberg, Z.N.; Langford, K.W.; Shakya, M.; Lo, C.C.; Liachko, I. Hi-C deconvolution of a human gut microbiome yields high-quality draft genomes and reveals plasmid-genome interactions. BioRxiv 2017. [Google Scholar] [CrossRef]
- Stewart, R.D.; Auffret, M.D.; Warr, A.; Wiser, A.H.; Press, M.O.; Langford, K.W.; Liachko, I.; Snelling, T.J.; Dewhurst, R.J.; Walker, A.W.; et al. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Burton, J.N.; Liachko, I.; Dunham, M.J.; Shendure, J. Species-Level Deconvolution of Metagenome Assemblies with Hi-C-Based Contact Probability Maps. G3 (Bethesda) 2014, 4, 1339–1346. [Google Scholar] [CrossRef]
- Stalder, T.; Press, M.O.; Sullivan, S.; Liachko, I.; Top, E.M. Linking the resistome and plasmidome to the microbiome. ISME J. 2019, 13, 2437–2446. [Google Scholar] [CrossRef]
- Staton, G.J.; Sullivan, L.E.; Blowey, R.W.; Carter, S.D.; Evans, N.J. Surveying bovine digital dermatitis and non-healing bovine foot lesions for the presence of Fusobacterium necrophorum, Porphyromonas endodontalis and Treponema pallidum. Vet. Rec. 2020, 186, 450. [Google Scholar] [CrossRef]
- Koniarova, I.; Orsag, A.; Ledecky, V. The role of anaerobe in the occurrence of dermatitis digitalis-et-interdigitalis in cattle. Vet. Med.-Czech. 1993, 38, 589–596. [Google Scholar]
- Jacobs, C.; Orsel, K.; Mason, S.; Barkema, H.W. Comparison of effects of routine topical treatments in the milking parlor on digital dermatitis lesions. J. Dairy Sci. 2018, 101, 5255–5266. [Google Scholar] [CrossRef]
- Cutler, J.H.H.; Cramer, G.; Walter, J.J.; Millman, S.T.; Kelton, D.F. Randomized clinical trial of tetracycline hydrochloride bandage and paste treatments for resolution of lesions and pain associated with digital dermatitis in dairy cattle. J. Dairy Sci. 2013, 96, 7550–7557. [Google Scholar] [CrossRef]
- Kyselkova, M.; Jirout, J.; Vrchotova, N.; Schmitt, H.; Elhottova, D. Spread of tetracycline resistance genes at a conventional dairy farm. Front. Microbiol. 2015, 6, 536. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.C. Update on acquired tetracycline resistance genes. Fems Microbiol. Lett. 2005, 245, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Kobashi, Y.; Hasebe, A.; Nishio, M.; Uchiyama, H. Diversity of tetracycline resistance genes in bacteria isolated from various agricultural environments. Microbes Environ. 2007, 22, 44–51. [Google Scholar] [CrossRef]
- Cheng, W.X.; Chen, H.; Su, C.; Yan, S.H. Abundance and persistence of antibiotic resistance genes in livestock farms: A comprehensive investigation in eastern China. Environ. Int. 2013, 61, 1–7. [Google Scholar] [CrossRef]
- Fabian, N.; Starosta, A.L.; Arenz, S.; Sohmen, D.; Doenhoefer, A.; Wilson, D.N. Tetracycline antibiotics and resistance mechanisms. Biol. Chem. 2014, 395, 559–575. [Google Scholar]
- Shoemaker, N.B.; Vlamakis, H.; Hayes, K.; Salyers, A.A. Evidence for extensive resistance gene transfer among Bacteroides spp. and among Bacteroides and other genera in the human colon. Appl. Environ. Microbiol. 2001, 67, 561–568. [Google Scholar] [CrossRef]
- Nikolich, M.P.; Shoemaker, N.B.; Salyers, A.A. A Bacteroides tetracycline resistance gene represents a new class of ribosome protection tetracycline resistance. Antimicrob. Agents Chemother. 1992, 36, 1005–1012. [Google Scholar] [CrossRef]
- Speer, B.S.; Bedzyk, L.; Salyers, A.A. Evidence that a novel tetracycline resistance gene found on 2 Bacteroides transposons encodes an NADP-requiring oxidoreductase. J. Bacteriol. 1991, 173, 176–183. [Google Scholar] [CrossRef]
- Nikolich, M.P.; Hong, G.; Shoemaker, N.B.; Salyers, A.A. Evidence for natural horizontal transfer of tetQ between bacteria that normally colonize humans and bacteria that normally colonize livestock. Appl. Environ. Microb. 1994, 60, 3255–3260. [Google Scholar] [CrossRef] [PubMed]
- Whittle, G.; Shoemaker, N.B.; Salyers, A.A. The role of Bacteroides conjugative transposons in the dissemination of antibiotic resistance genes. Cell. Mol. Life Sci. 2002, 59, 2044–2054. [Google Scholar] [CrossRef] [PubMed]
- Quesada-Gomez, C. Bacteroides mobilizable and conjugative genetic elements: Antibiotic resistance among clinical isolates. Rev. Esp. Quim. 2011, 24, 184–190. [Google Scholar]
- Volkers, G.; Palm, G.J.; Weiss, M.S.; Wright, G.D.; Hinrichs, W. Structural basis for a new tetracycline resistance mechanism relying on the TetX monooxygenase. Febs Lett. 2011, 585, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.C.; Chung, W.O.; Roe, D.E. Characterization of tetracycline and erythromycin resistance determinants in Treponema denticola. Antimicrob. Agents Chemother. 1996, 40, 2236. [Google Scholar] [CrossRef] [PubMed]
- Stamm, L.V. Global challenge of antibiotic-resistant Treponema pallidum. Antimicrob. Agents Chemother. 2010, 54, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Robinson, D.; Bebear, C. Antibiotic susceptibilities of mycoplasmas and treatment of mycoplasmal infections. J. Antimicrob. Chemoth. 1997, 40, 622–630. [Google Scholar] [CrossRef]
- Gautier-Bouchardon, A.V. Antimicrobial resistance in Mycoplasma spp. Microbiol. Spectrum 2018, 6, ARBA–0030–2018. [Google Scholar]
- Siugzdaite, J.; Gabinaitiene, A.; Kerziene, S. Susceptibility of Mycoplasma bovis field isolates to antimicrobial agents. Vet. Med-Czech 2012, 57, 575–582. [Google Scholar] [CrossRef]
- Laven, R.A.; Logue, D.N. Treatment strategies for digital dermatitis for the UK. Vet. J. 2006, 171, 79–88. [Google Scholar] [CrossRef]
- FDA 2018 Summary Report on Antimicrobials Sold or Distributed for Use in Food-Producing Animals. FDA; 2019. Available online: https://www.fda.gov/media/133411/download (accessed on 10 December 2019).
- Schrag, N.F.D.; Godden, S.M.; Apley, M.D.; Singer, R.S.; Lubbers, B.V. Antimicrobial use quantification in adult dairy cows - Part 3-Use measured by standardized regimens and grams on 29 dairies in the United States. Zoonoses Public Hlth. 2020, 67, 82–93. [Google Scholar] [CrossRef]
- Huber, H.; Giezendanner, N.; Stephan, R.; Zweifel, C. Genotypes, antibiotic resistance profiles and microarray-based characterization of methicillin-resistant Staphylococcus aureus strains isolated from livestock and veterinarians in Switzerland. Zoonoses Public Hlth 2011, 58, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.A.; Hao, L.J.; Guo, X.Y.; Wang, N.; Ye, B.P. Prevalence of antibiotic resistance genes of wastewater and surface water in livestock farms of Jiangsu province, China. Environ. Sci. Pollut. R. 2015, 22, 13950–13959. [Google Scholar] [CrossRef] [PubMed]
- Mu, Q.H.; Li, J.; Sun, Y.X.; Mao, D.Q.; Wang, Q.; Luo, Y. Occurrence of sulfonamide-, tetracycline-, plasmid-mediated quinolone- and macrolide-resistance genes in livestock feedlots in northern China. Environ. Sci. Pollut. R. 2015, 22, 6932–6940. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Yang, X.H.; Jiao, S.J.; Zhang, J.; Ye, B.P.; Gao, S.X. Sulfonamide-resistant bacteria and their resistance genes in soils fertilized with manures from Jiangsu province, southeastern China. PLoS ONE 2014, 9, e112626. [Google Scholar] [CrossRef]
- Pruden, A.; Pei, R.T.; Storteboom, H.; Carlson, K.H. Antibiotic resistance genes as emerging contaminants: Studies in northern Colorado. Env. Sci. Tech. 2006, 40, 7445–7450. [Google Scholar] [CrossRef]
- Krull, A.C.; Cooper, V.L.; Coatney, J.W.; Shearer, J.K.; Gorden, P.J.; Plummer, P.J. A highly effective protocol for the rapid and consistent induction of digital dermatitis in Holstein calves. PLoS ONE 2016, 11, e0154481. [Google Scholar] [CrossRef]
- Bushnell, B.; Rood, J.; Singer, E. BBMerge – Accurate paired shotgun read merging via overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Li, D.; Luo, R.; Liu, C.-M.; Leung, C.-M.; Ting, H.-F.; Sadakane, K.; Yamashita, H.; Lam, T.-W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Faust, G.G.; Hall, I.M. SAMBLASTER: Fast duplicate marking and structural variant read extraction. Bioinformatics 2014, 30, 2503–2505. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 genome project data processing subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment fo r nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Phyla | Lowest Level Identification | Classification | Relative Abundance (%) |
|---|---|---|---|
| Bacteroidetes | Phylum | 5.27 | |
| Porphyromonas somerae | Species | 0.36 | |
| Porphyromonas levii | Species | 1.04 | |
| Porphyromonadaceae (unclassified) | Family | 0.88 | |
| Bacteroidetes (unclassified) | Phylum | 2.99 | |
| Spirochaetes | Phylum | 4.87 | |
| Treponema phagedenis | Species | 1.00 | |
| Treponema medium | Species | 0.51 | |
| Spirochaetaceae (unclassified) | Family | 3.36 | |
| Firmicutes | Phylum | 3.83 | |
| Streptococcus henryi | Species | 0.25 | |
| Lactobacillales (unclassified) | Order | 1.15 | |
| Clostridiales (unclassified) | Order | 2.43 | |
| Tenericutes | Phylum | 0.84 | |
| Mycoplasma fermentans | Species | 0.63 | |
| Mycoplasma (unclassified) | Genus | 0.21 | |
| Proteobacteria | Phylum | 0.45 | |
| Betaproteobacteria (unclassified) | Class | 0.26 | |
| Gammaproteobacteria (unclassified) | Class | 0.19 |
| Cluster-ID | Top References | Best Match | Identity (%) | 16S rRNA Length (Bases) |
|---|---|---|---|---|
| cluster_3 | p_Bacteroidetes | Uncultured bacterium clone gls269 | 84.42 | 1511 |
| cluster_4 | Porphyromonas_somerae_DSM_23386 | Porphyromonas somerae DSM 23386 strain JCM 13867 | 98.35 | 567 |
| cluster_5 | f_Spirochaetaceae | Treponemarefringens | 97.23 | 1365 |
| cluster_6 | p_Bacteroidetes | Bacteroidia bacterium feline oral taxon 312 clone UI046 | 93.38 | 707 |
| cluster_8 | c_Betaproteobacteria | Oligella ureolytica DSM 18253 | 96.51 | 1540 |
| cluster_12 | o_Clostridiales | Ezakiella sp. Marseille-P2951 strain Marseille-P2951T | 99.73 | 747 |
| cluster_13 | p_Bacteroidetes | Porphyromonas somerae DSM 23386 strain JCM 13867 | 89.96 | 1113 |
| cluster_14 | k_Bacteria | Spirochaeta sp. canine oral taxon 314 clone 1A090 | 99.92 | 1234 |
| cluster_16 | p_Bacteroidetes | Bacteroidia bacterium feline oral taxon 312 clone UI046 | 85.81 | 721 |
| cluster_17 | o_Clostridiales | Catonella sp. oral clone BR063 | 94.1 | 874 |
| cluster_18 | f_Spirochaetaceae | Treponema sp. canine oral taxon 233 clone QB043 | 99.01 | 414 |
| cluster_19 | Porphyromonas_levii_DSM_23370 | Porphyromonas levii strain Israel | 99.4 | 368 |
| cluster_21 | o_Clostridiales | Uncultured rumen bacterium | 98.4 | 566 |
| cluster_23 | Streptococcus_henryi_DSM_19005 | Streptococcus henryi strain OZK31 | 98.45 | 916 |
| cluster_24 | g_Mycoplasma | Mycoplasma agalactiae 5632 | 93.52 | 802 |
| cluster_25 | k_Bacteria | Porphyromonas levii DSM 23370 strain JCM 13866 | 94.59 | 398 |
| cluster_26 | f_Porphyromonadaceae | Porphyromonas levii DSM 23370 strain JCM 13866 | 94.48 | 372 |
| cluster_27 | Mycoplasma_fermentans_JER | Mycoplasma fermentans M64 | 99.87 | 1522 |
| cluster_29 | k_Bacteria | Acholeplasmatales bacterium canine oral taxon 172 clone QC046 | 98.6 | 1555 |
| cluster_30 | c_Gammaproteobacteria | Pseudomonas sp. M-08 gene | 97.87 | 1324 |
| cluster_31 | o_Clostridiales | Uncultured bacterium clone 1101352040638 | 93.16 | 1536 |
| cluster_32 | k_Bacteria | Erysipelothrix rhusiopathiae | 92.17 | 611 |
| cluster_33 | o_Clostridiales | Peptoniphilaceae bacterium SIT14 | 96.86 | 1536 |
| cluster_35 | o_Clostridiales | Uncultured Firmicutes bacterium clone P-07 | 99.68 | 1510 |
| cluster_36 | k_Bacteria | Uncultured Bacteroidetes bacterium clone BL2_5 | 98.27 | 1536 |
| cluster_38 | k_Bacteria | Uncultured Tenericutes bacterium clone P-06 | 100 | 1542 |
| cluster_39 | o_Clostridiales | Clostridium sticklandii str. DSM 519 | 85.56 | 471 |
| Phyla | Bacterial Taxa | Efflux Pump | Ribosomal Protection Proteins | Inactivation Enzyme | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| tet31 | tet33 | tetB | tetH | tetL | tetZ | tet32 | tet36 | tet40 | tetM | tetO | tetO | tetS | tetT | tetW | tetX | ||
| Bacteroidetes | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 3 | 3 | 435 | 0 | 0 | 2 | 0 | |
| Porphyromonas somerae | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 19 | 0 | 0 | 0 | 0 | |
| Porphyromonas levii | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 86 | 0 | 0 | 1 | 0 | |
| Porphyromonadaceae (unclass. *) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 68 | 0 | 0 | 0 | 0 | |
| Bacteroidetes (unclass.) | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 2 | 262 | 0 | 0 | 1 | 0 | |
| Spirochaetes | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 23 | 64 | 5 | 0 | 1 | 0 | 0 | |
| Treponema phagedenis | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 63 | 1 | 0 | 0 | 0 | 0 | |
| Treponema medium | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 14 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Spirochaetaceae (unclass.) | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 7 | 1 | 4 | 0 | 1 | 0 | 0 | |
| Firmicutes | 0 | 0 | 0 | 0 | 0 | 0 | 32 | 0 | 7 | 4 | 71 | 12 | 0 | 0 | 20 | 1 | |
| Streptococcus henryi | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | |
| Lactobacillales (unclass.) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Clostridiales (unclass.) | 0 | 0 | 0 | 0 | 0 | 0 | 32 | 0 | 7 | 2 | 71 | 12 | 0 | 0 | 19 | 1 | |
| Tenericutes | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Mycoplasma fermentans | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Mycoplasma (unclass.) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Proteobacteria | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 1 | 0 | |
| Betaproteobacteria (unclass.) | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Gammaproteobacteria (unclass.) | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 1 | 0 | |
| Total Resistance Genes in the Sample (n = 308 clusters) ** | |||||||||||||||||
| 13 | 4 | 1 | 22 | 4 | 6 | 39 | 14 | 41 | 112 | 224 | 1265 | 15 | 32 | 166 | 3 | ||
| Phyla | Bacterial Taxa | Aminoglycoside (aadA1, ant3, ant4, ant6, aph3, aph6) | Beta-Lactam (blaOXA, blaCARB) | Sulfonamide (sul1, sul2) | Phenicol (cmxAB, floR) | Lincosamide (lnuB, lsa) | Erythromycin (ermA, ermB, ermF, ermX, myrA) |
|---|---|---|---|---|---|---|---|
| Bacteroidetes | 8 | 0 | 3 | 0 | 1 | 4 | |
| Porphyromonas somerae | 0 | 0 | 3 | 0 | 0 | 0 | |
| Porphyromonas levii | 3 | 0 | 0 | 0 | 0 | 1 | |
| Porphyromonadaceae | 0 | 0 | 0 | 0 | 1 | 1 | |
| Bacteroidetes | 5 | 0 | 0 | 0 | 0 | 2 | |
| Spirochaetes | 85 | 0 | 0 | 0 | 0 | 0 | |
| Treponema phagedenis | 1 | 0 | 0 | 0 | 0 | 0 | |
| Treponema medium | 9 | 0 | 0 | 0 | 0 | 0 | |
| Spirochaetaceae | 75 | 0 | 0 | 0 | 0 | 0 | |
| Firmicutes | 9 | 0 | 0 | 0 | 28 | 2 | |
| Streptococcus henryi | 3 | 0 | 0 | 0 | 10 | 0 | |
| Lactobacillales | 1 | 0 | 0 | 0 | 5 | 0 | |
| Clostridiales | 5 | 0 | 0 | 0 | 13 | 2 | |
| Tenericutes | 0 | 0 | 0 | 0 | 0 | 0 | |
| Mycoplasma fermentans | 0 | 0 | 0 | 0 | 0 | 0 | |
| Mycoplasma | 0 | 0 | 0 | 0 | 0 | 0 | |
| Proteobacteria | 11 | 1 | 1 | 1 | 0 | 0 | |
| Betaproteobacteria | 6 | 1 | 1 | 0 | 0 | 0 | |
| Gammaproteobacteria | 5 | 0 | 0 | 1 | 0 | 0 | |
| Total Resistance Genes in the Sample (n = 308 clusters) | 463 | 36 | 39 | 20 | 153 | 63 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beyi, A.F.; Hassall, A.; Phillips, G.J.; Plummer, P.J. Tracking Reservoirs of Antimicrobial Resistance Genes in a Complex Microbial Community Using Metagenomic Hi-C: The Case of Bovine Digital Dermatitis. Antibiotics 2021, 10, 221. https://doi.org/10.3390/antibiotics10020221
Beyi AF, Hassall A, Phillips GJ, Plummer PJ. Tracking Reservoirs of Antimicrobial Resistance Genes in a Complex Microbial Community Using Metagenomic Hi-C: The Case of Bovine Digital Dermatitis. Antibiotics. 2021; 10(2):221. https://doi.org/10.3390/antibiotics10020221
Chicago/Turabian StyleBeyi, Ashenafi F., Alan Hassall, Gregory J. Phillips, and Paul J. Plummer. 2021. "Tracking Reservoirs of Antimicrobial Resistance Genes in a Complex Microbial Community Using Metagenomic Hi-C: The Case of Bovine Digital Dermatitis" Antibiotics 10, no. 2: 221. https://doi.org/10.3390/antibiotics10020221
APA StyleBeyi, A. F., Hassall, A., Phillips, G. J., & Plummer, P. J. (2021). Tracking Reservoirs of Antimicrobial Resistance Genes in a Complex Microbial Community Using Metagenomic Hi-C: The Case of Bovine Digital Dermatitis. Antibiotics, 10(2), 221. https://doi.org/10.3390/antibiotics10020221