
Targeting Mycobacterial F-ATP Synthase C-Terminal α Subunit Interaction Motif on Rotary Subunit γ

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
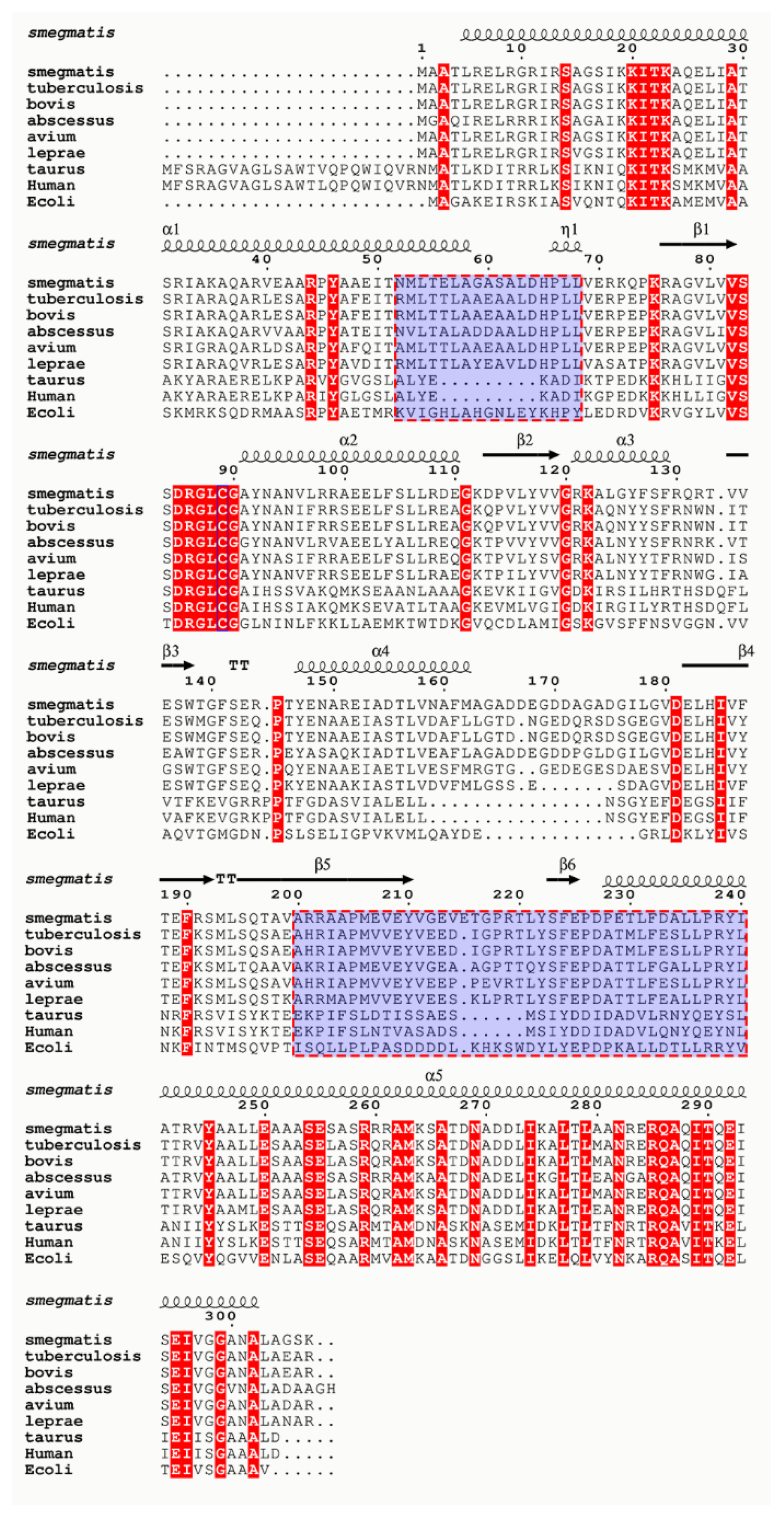
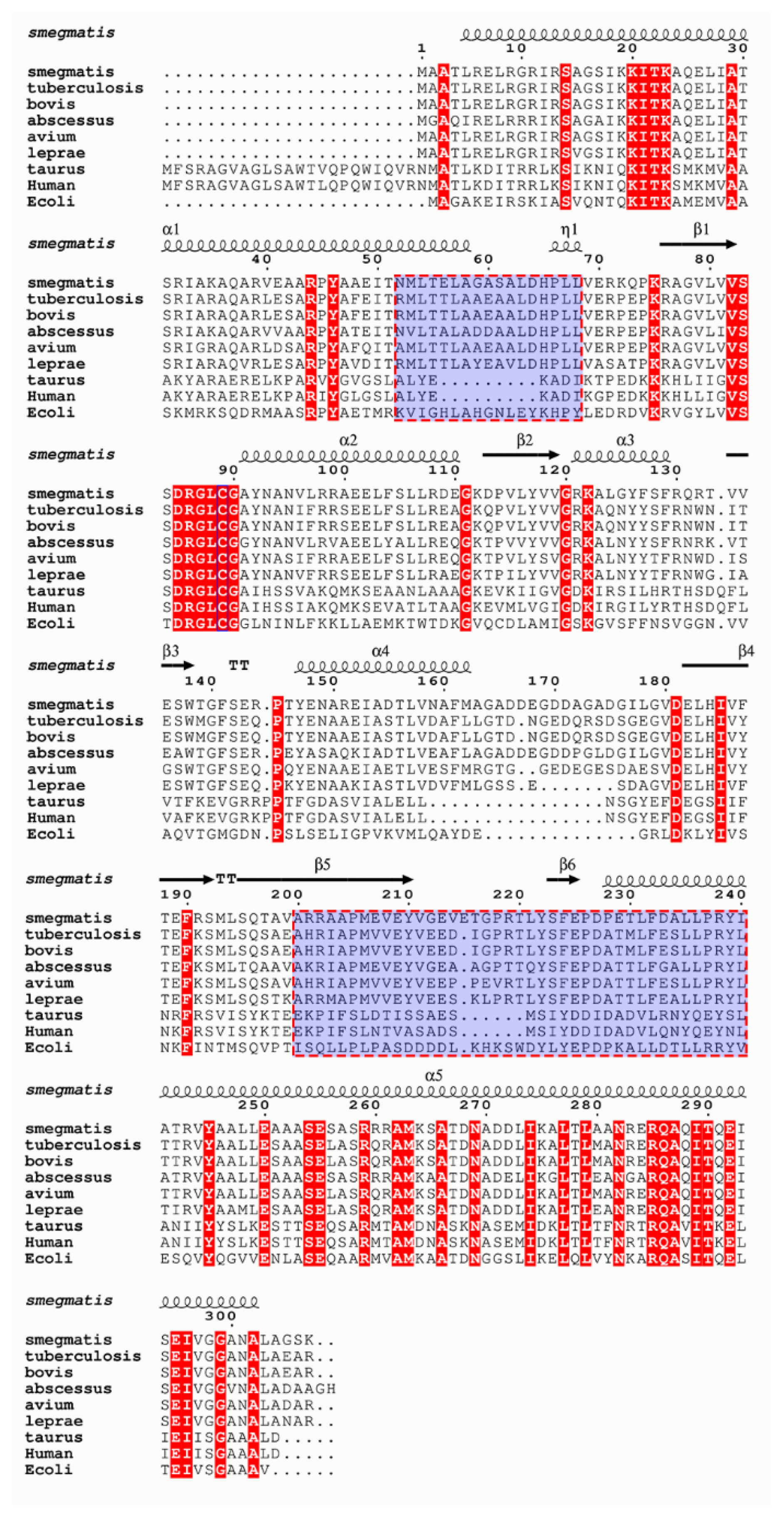
2.1. Homology Modeling
2.2. Receptor-Ligand-Based Pharmacophore Model
Validation of Pharmacophore and 3D Ligand Database Screening
2.3. Virtual Selection of Eleven Ligands
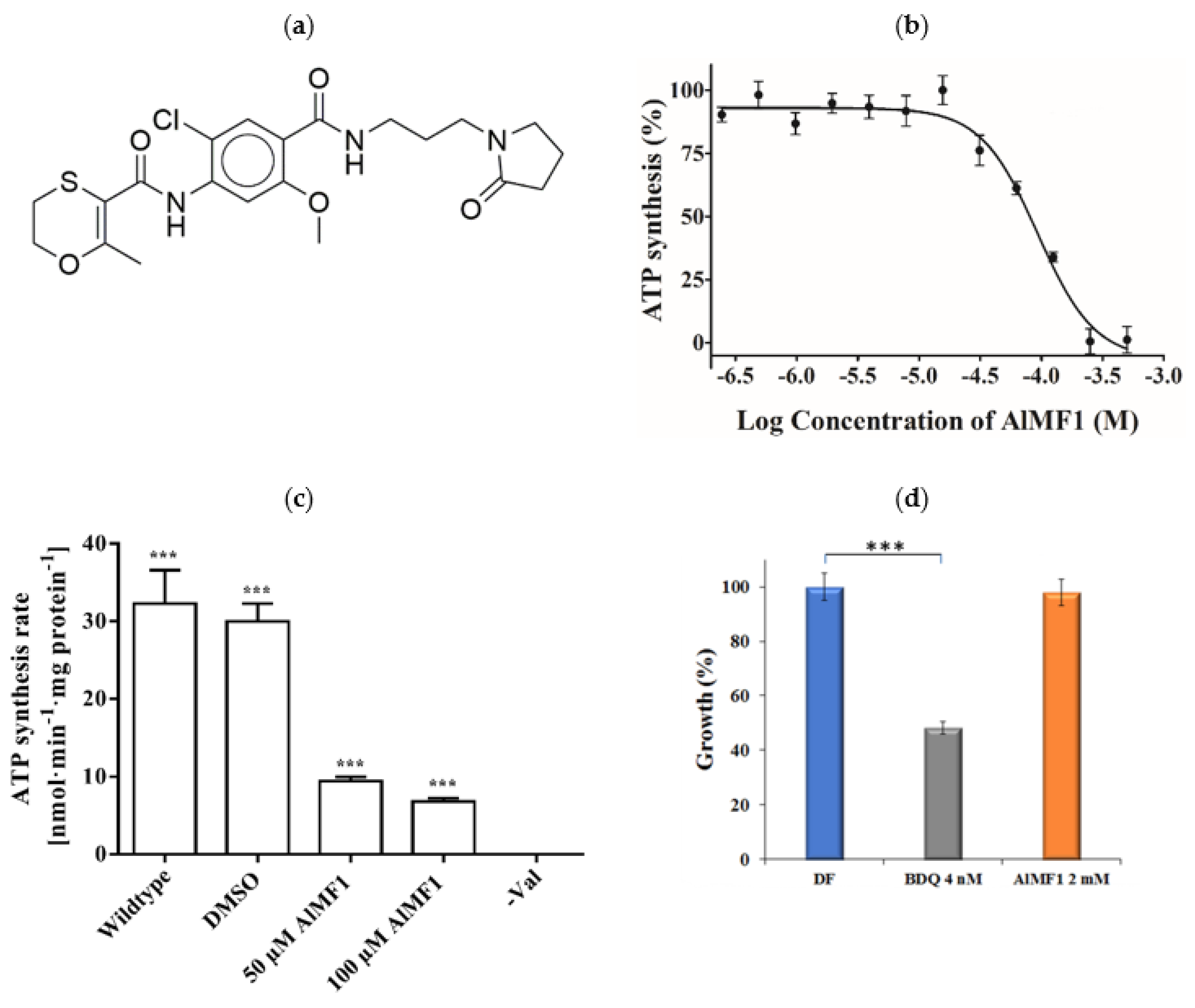
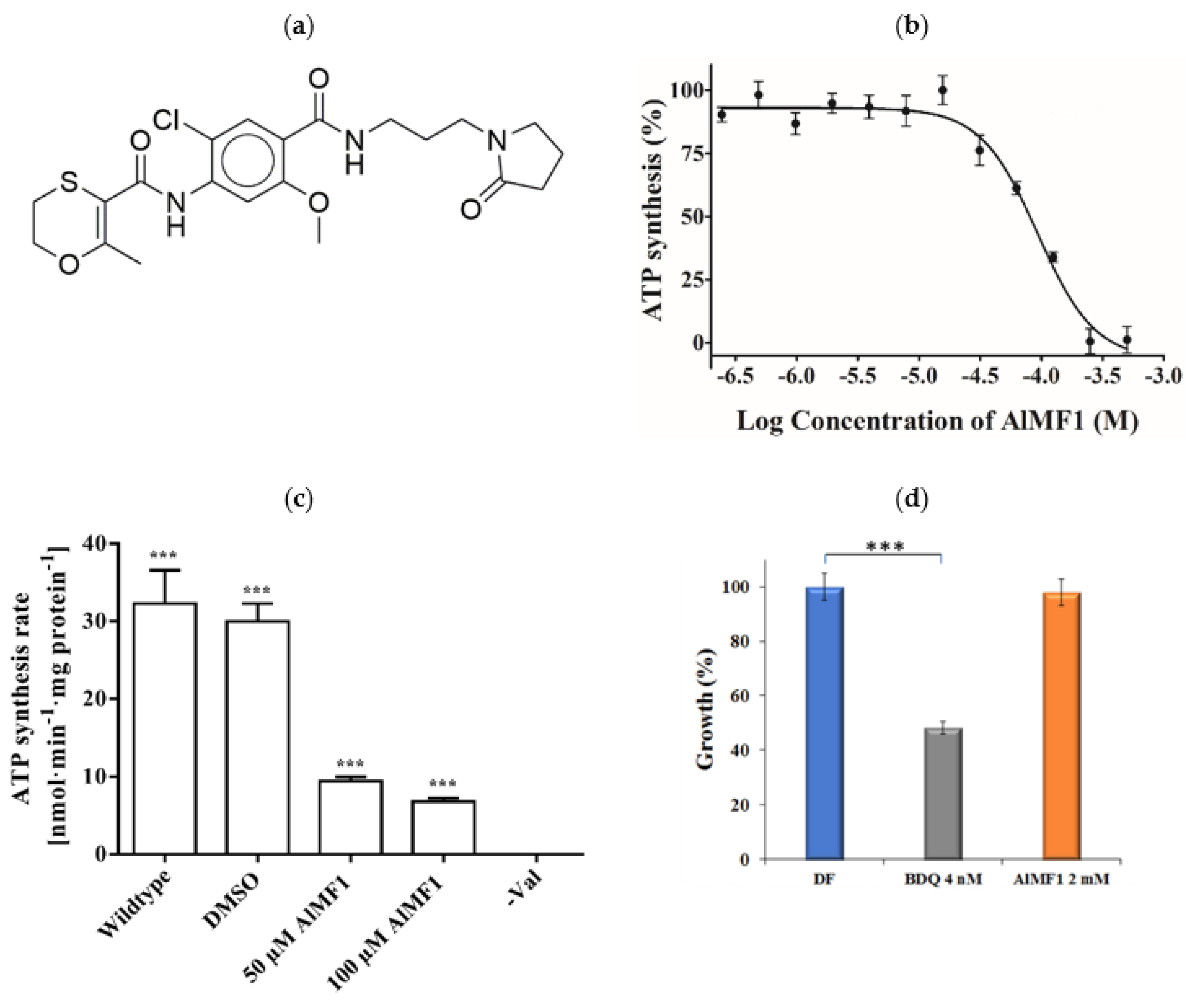
2.4. Potency and Target Specificity of Ligand AlMF1
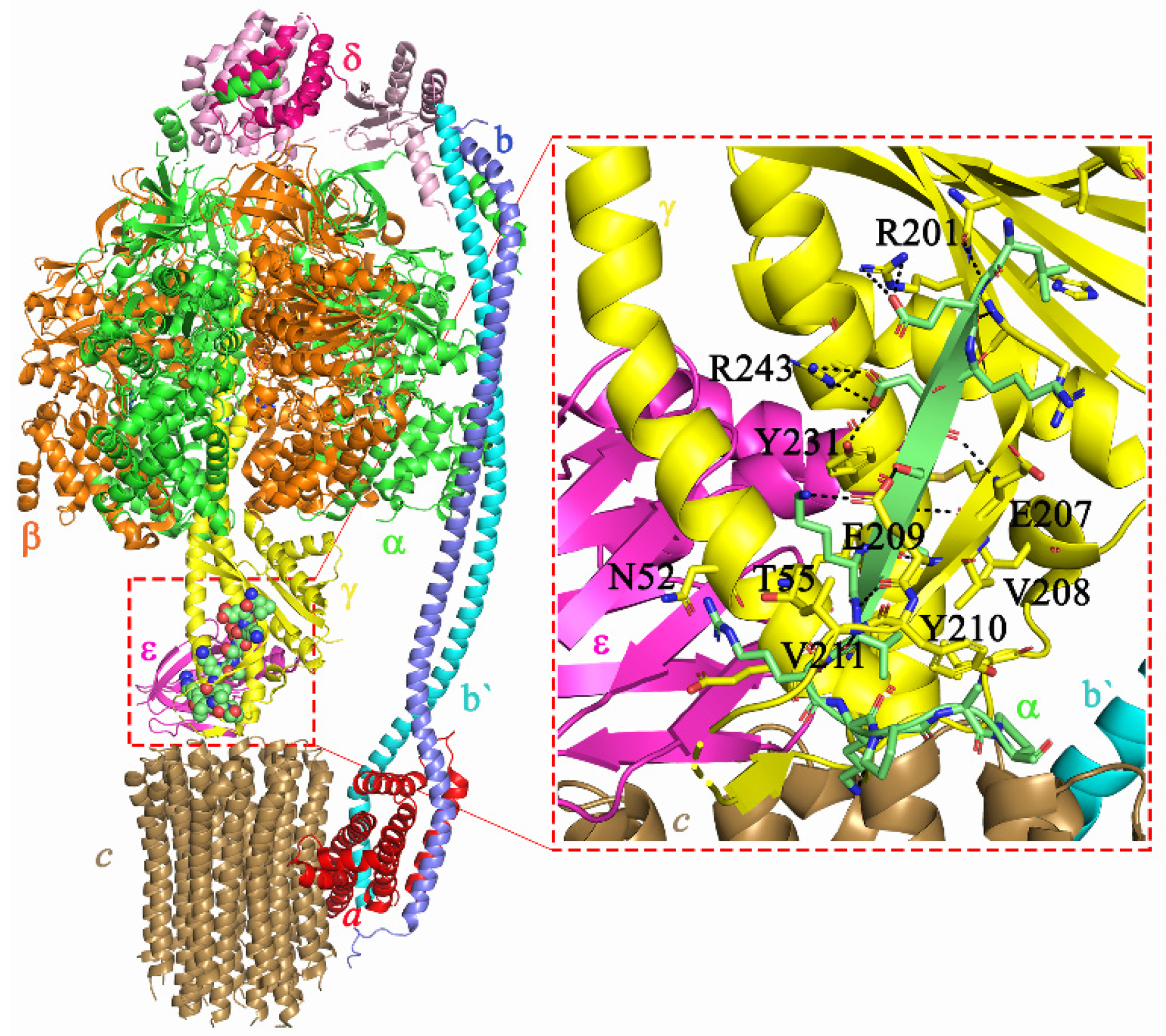
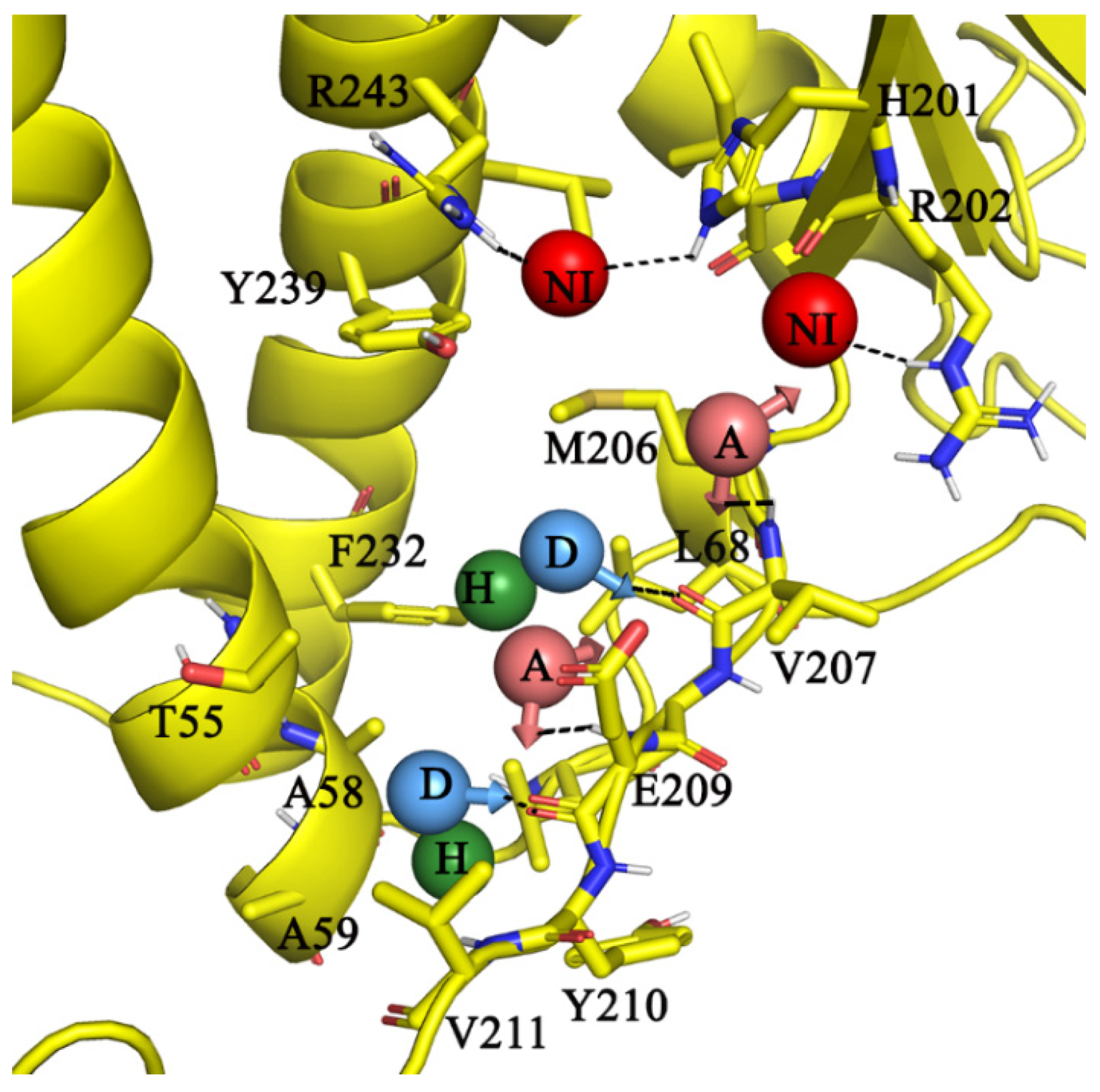
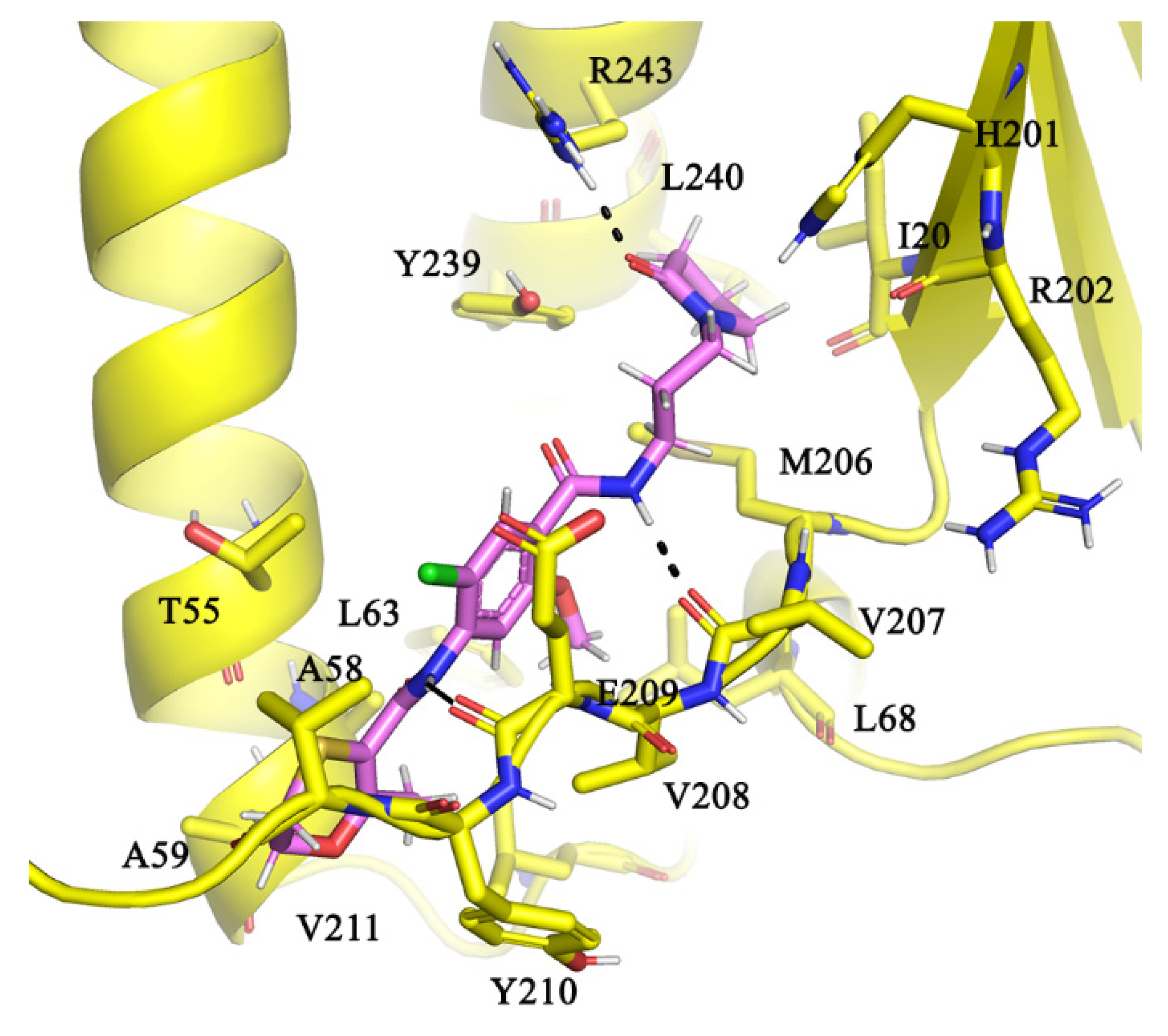
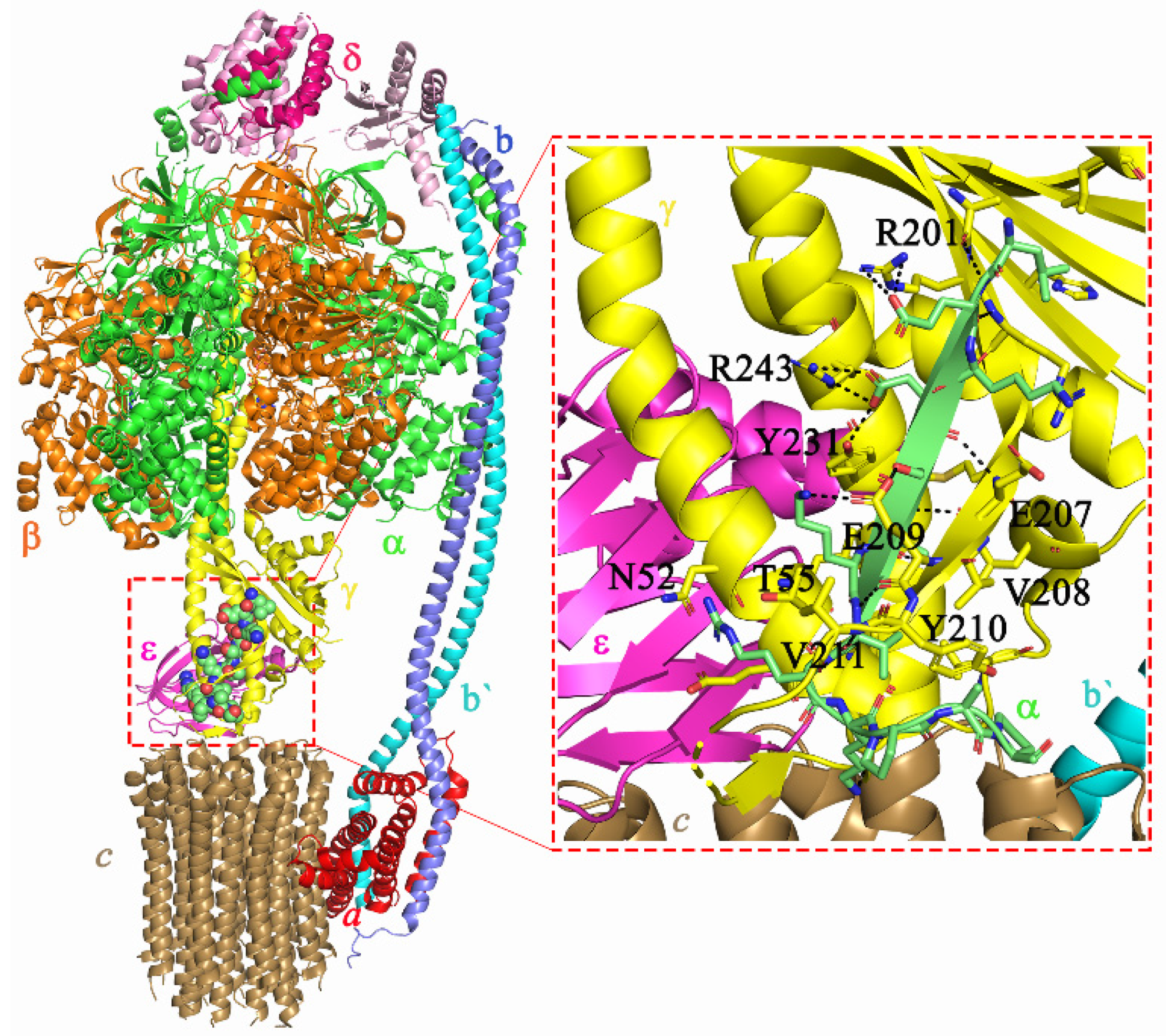
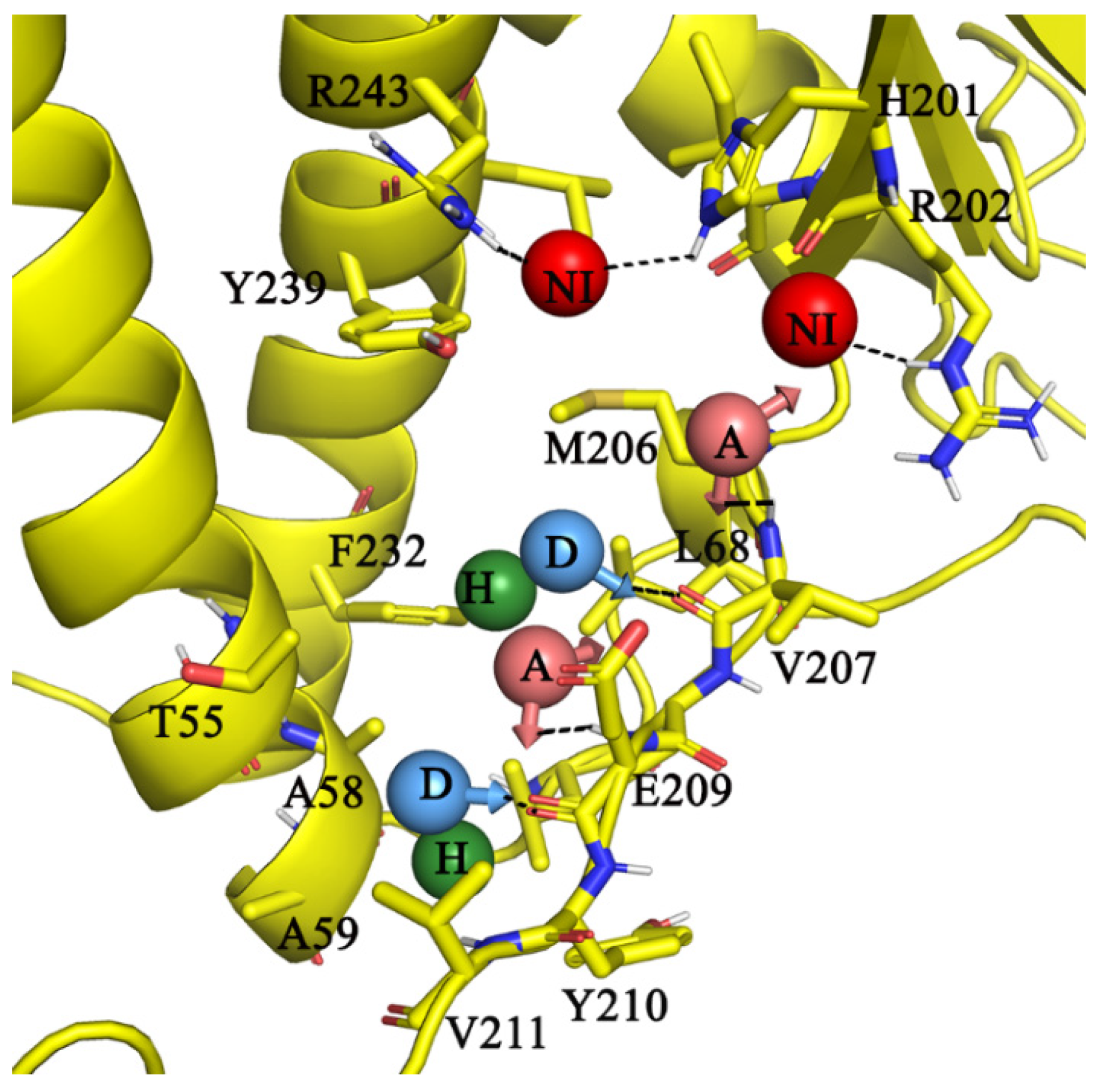
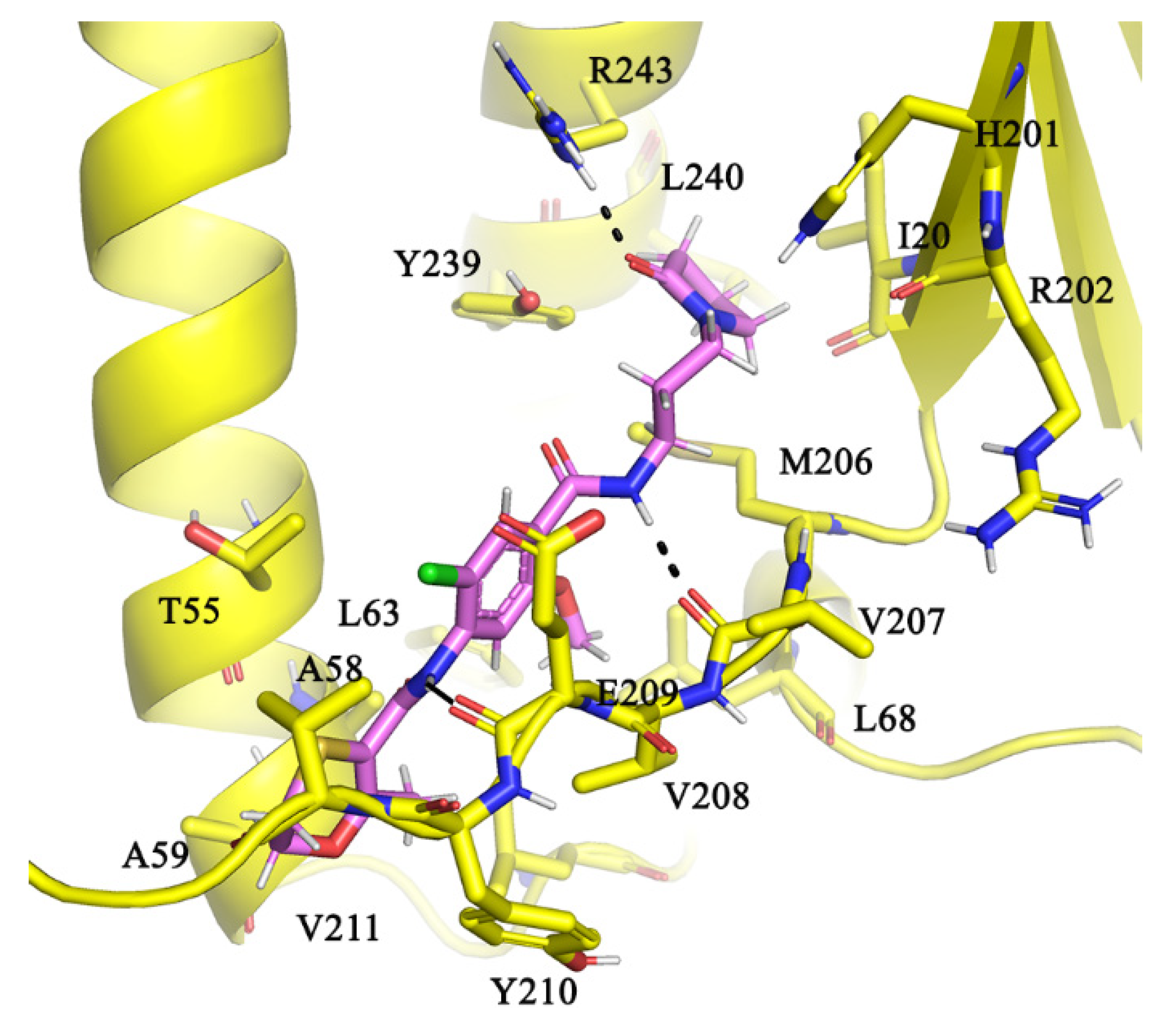
2.5. Molecular Interactions of AlMF1 with Mycobacterial Subunit γ
3. Conclusions
4. Materials and Methods
4.1. Modeling Simulations
4.2. Preparation of M. smegmatis and E. coli Inverted Membrane Vesicles (IMVs)
4.3. ATP Synthesis Assay Using IMVs
4.4. Reconstitution and ATP Synthesis of Recombinant M. smegmatis F-ATP Synthase
4.5. Bacterial Growth in Absence and Presence of AlMF1
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Global Tuberculosis Report 2019; WHO/CDS/TB/2019.15; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- History of TB drugs (Time Line). Available online: https://www.tbfacts.org/history-of-tb-drugs/ (accessed on 11 July 2021).
- FDA. FDA Approves New Drug for Treatment-Resistant Forms of Tuberculosis that Affects the Lungs. 2019. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-drug-treatment-resistant-forms-tuberculosis-affects-lungs (accessed on 29 June 2021).
- FDA. SIRTURO™ (bedaquiline). 2012. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/204384s002lbl.pdf (accessed on 15 October 2019).
- Diacon, A.H.; Donald, P.R.; Pym, A.; Grobusch, M.; Patientia, R.F.; Mahanyele, R.; Bantubani, N.; Narasimooloo, R.; De Marez, T.; van Heeswijk, R.; et al. Randomized pilot trial of eight weeks of bedaquiline (TMC207) treatment for multidrug-resistant tuberculosis: Long-term outcome, tolerability, and effect on emergence of drug resistance. Antimicrob. Agents Chemother. 2012, 56, 3271–3276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, M.; Zhou, W.; Patel, H.; Srivastava, A.P.; Symersky, J.; Bonar, M.M.; Faraldo-Gómez, J.D.; Liao, M.; Mueller, D.M. Bedaquiline inhibits the yeast and human mitochondrial ATP synthases. Commun. Biol. 2020, 3, 452. [Google Scholar] [CrossRef] [PubMed]
- Hotra, A.; Ragunathan, P.; Ng, P.S.; Seankongsuk, P.; Harikishore, A.; Sarathy, J.P.; Saw, W.-G.; Lakshmanan, U.; Sae-Lao, P.; Kalia, N.P.; et al. Discovery of a Novel Mycobacterial F-ATP Synthase Inhibitor and its Potency in Combination with Diarylquinolines. Angew. Chem. Int. Ed. 2020, 59, 13295–13304. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.F.; Lau, A.M.; Harikishore, A.; Saw, W.G.; Shin, J.; Ragunathan, P.; Bhushan, S.; Ngan, S.C.; Sze, S.K.; Bates, R.W.; et al. A systematic assessment of mycobacterial F(1)-ATPase subunit ε‘s role in latent ATPase hydrolysis. FEBS J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Saw, W.G.; Wu, M.L.; Ragunathan, P.; Biukovic, G.; Lau, A.M.; Shin, J.; Harikishore, A.; Cheung, C.Y.; Hards, K.; Sarathy, J.P.; et al. Disrupting coupling within mycobacterial F-ATP synthases subunit epsilon causes dysregulated energy production and cell wall biosynthesis. Sci. Rep. 2019, 9, 16759. [Google Scholar] [CrossRef] [PubMed]
- Kamariah, N.; Ragunathan, P.; Shin, J.; Saw, W.-G.; Wong, C.-F.; Dick, T.; Grüber, G. Unique structural and mechanistic properties of mycobacterial F-ATP synthases: Implications for drug design. Prog. Biophys. Mol. Biol. 2020, 152, 64–73. [Google Scholar] [CrossRef]
- Guo, H.; Courbon, G.M.; Bueler, S.A.; Mai, J.; Liu, J.; Rubinstein, J.L. Structure of mycobacterial ATP synthase bound to the tuberculosis drug bedaquiline. Nature 2021, 589, 143–147. [Google Scholar] [CrossRef]
- Haagsma, A.C.; Driessen, N.N.; Hahn, M.M.; Lill, H.; Bald, D. ATP synthase in slow- and fast-growing mycobacteria is active in ATP synthesis and blocked in ATP hydrolysis direction. FEMS Microbiol. Lett. 2010, 313, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Hotra, A.; Suter, M.; Biukovic, G.; Ragunathan, P.; Kundu, S.; Dick, T.; Grüber, G. Deletion of a unique loop in the mycobacterial F-ATP synthase gamma subunit sheds light on its inhibitory role in ATP hydrolysis-driven H(+) pumping. FEBS J. 2016, 283, 1947–1961. [Google Scholar] [CrossRef] [Green Version]
- Ragunathan, P.; Sielaff, H.; Sundararaman, L.; Biuković, G.; Subramanian Manimekalai, M.S.; Singh, D.; Kundu, S.; Wohland, T.; Frasch, W.; Dick, T.; et al. The uniqueness of subunit alpha of mycobacterial F-ATP synthases: An evolutionary variant for niche adaptation. J. Biol. Chem. 2017, 292, 11262–11279. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.F.; Grüber, G. The Unique C-Terminal Extension of Mycobacterial F-ATP Synthase Subunit α Is the Major Contributor to Its Latent ATP Hydrolysis Activity. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput.-Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef] [PubMed]
- Rebollo-Lopez, M.J.; Lelièvre, J.; Alvarez-Gomez, D.; Castro-Pichel, J.; Martínez-Jiménez, F.; Papadatos, G.; Kumar, V.; Colmenarejo, G.; Mugumbate, G.; Hurle, M.; et al. Release of 50 new, drug-like compounds and their computational target predictions for open source anti-tubercular drug discovery. PLoS ONE 2015, 10, e0142293. [Google Scholar] [CrossRef] [PubMed]
- ChemDiv 10320 Camino Santa Fe, Suite B, San Diego CA 92121-3103 USA. Available online: https://www.chemdiv.com/screening-libraries11/ (accessed on 29 June 2021).
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, C.E.; Boshoff, H.I.; Dartois, V.; Dick, T.; Ehrt, S.; Flynn, J.; Schnappinger, D.; Wilkinson, R.J.; Young, D. The spectrum of latent tuberculosis: Rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 2009, 7, 845–855. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-4: Maestro, Force Fields, MacroModel, Prime, Protein Preparation Wizard, Ligprep, ConfGen, Phase, QikProp, Glide; Schrödinger, LLC: New York, NY, USA, 2019.
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Motulsky, H.; Christopoulos, A. Fitting Models to Biological Data Using Linear and Nonlinear Regression: A Practical Guide to Curve Fitting; GraphPad Software Inc.: San Diego, CA, USA, 2003; Available online: www.graphpad.com (accessed on 21 September 2021).
- Saw, W.-G.; Wong, C.-F.; Dick, T.; Grüber, G. Overexpression, purification, enzymatic and microscopic characterization of recombinant mycobacterial F-ATP synthase. Biochem. Biophys. Res. Commun. 2019. [Google Scholar] [CrossRef]
- Knol, J.; Sjollema, K.; Poolman, B. Detergent-Mediated Reconstitution of Membrane Proteins. Biochemistry 1998, 37, 16410–16415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harikishore, A.; Wong, C.-F.; Ragunathan, P.; Litty, D.; Müller, V.; Grüber, G. Targeting Mycobacterial F-ATP Synthase C-Terminal α Subunit Interaction Motif on Rotary Subunit γ. Antibiotics 2021, 10, 1456. https://doi.org/10.3390/antibiotics10121456
Harikishore A, Wong C-F, Ragunathan P, Litty D, Müller V, Grüber G. Targeting Mycobacterial F-ATP Synthase C-Terminal α Subunit Interaction Motif on Rotary Subunit γ. Antibiotics. 2021; 10(12):1456. https://doi.org/10.3390/antibiotics10121456
Chicago/Turabian StyleHarikishore, Amaravadhi, Chui-Fann Wong, Priya Ragunathan, Dennis Litty, Volker Müller, and Gerhard Grüber. 2021. "Targeting Mycobacterial F-ATP Synthase C-Terminal α Subunit Interaction Motif on Rotary Subunit γ" Antibiotics 10, no. 12: 1456. https://doi.org/10.3390/antibiotics10121456
APA StyleHarikishore, A., Wong, C.-F., Ragunathan, P., Litty, D., Müller, V., & Grüber, G. (2021). Targeting Mycobacterial F-ATP Synthase C-Terminal α Subunit Interaction Motif on Rotary Subunit γ. Antibiotics, 10(12), 1456. https://doi.org/10.3390/antibiotics10121456