Red-Shifted FRET Biosensors for High-Throughput Fluorescence Lifetime Screening

 and
and 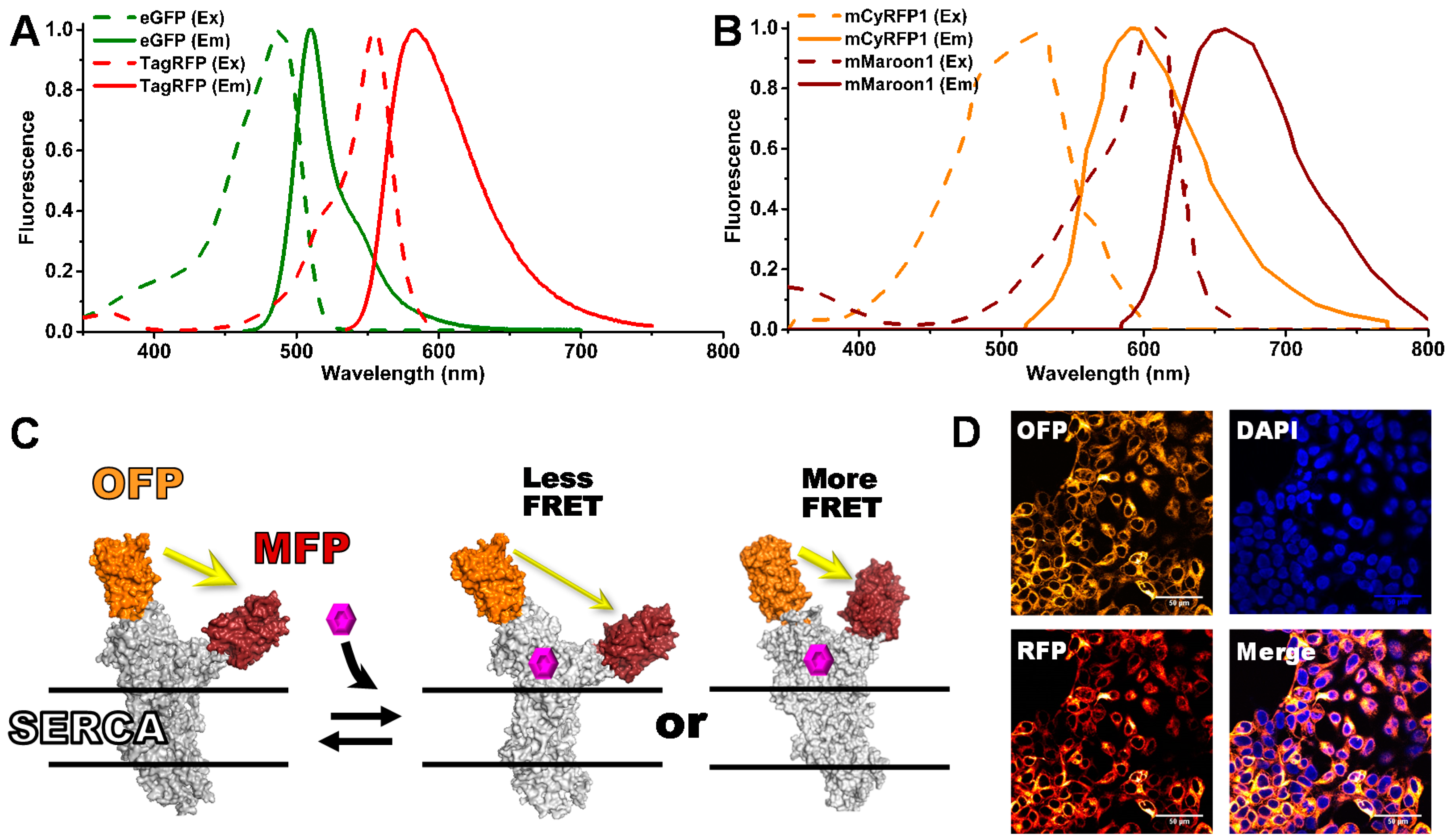{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Stable Clone Generation
2.2. Liquid Dispensing
2.3. FLT Detection and Data Analysis
2.4. Enzymatic SERCA Activity Assays of FRET Hits
3. Results
3.1. Red-Shifted FRET Pairs
3.2. Evaluation of FLT Detection Using 532 nm Pulsed-Laser Excitation
3.3. FLT Biosensor Dynamic Range and HTS Assay Quality (Z′)
3.4. Pilot Small-Molecule Library HTS Comparison
3.5. Structural (FRET) and Functional (ATPase Activity) Relationship of LOPAC Hits
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A


References
- Hou, Z.; Hu, Z.; Blackwell, D.J.; Miller, T.D.; Thomas, D.D.; Robia, S.L. 2-Color calcium pump reveals closure of the cytoplasmic headpiece with calcium binding. PLoS ONE 2012, 7, e40369. [Google Scholar] [CrossRef] [PubMed]
- Gruber, S.J.; Cornea, R.L.; Li, J.; Peterson, K.C.; Schaaf, T.M.; Gillispie, G.D.; Dahl, R.; Zsebo, K.M.; Robia, S.L.; Thomas, D.D. Discovery of enzyme modulators via high-throughput time-resolved FRET in living cells. J. Biomol. Screen. 2014, 19, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Stryer, L. Fluorescence energy transfer as a spectroscopic ruler. Annu. Rev. Biochem. 1978, 47, 819–846. [Google Scholar] [CrossRef] [PubMed]
- Muretta, J.M.; Kyrychenko, A.; Ladokhin, A.S.; Kast, D.; Gillispie, G.E.; Thomas, D.D. High-performance time-resolved fluorescence by direct waveform recording. Rev. Sci. Instrum. 2010, 81, 103101–103108. [Google Scholar] [CrossRef] [PubMed]
- Muretta, J.M.; Petersen, K.J.; Thomas, D.D. Direct real-time detection of the actin-activated power stroke within the myosin catalytic domain. Proc. Natl. Acad. Sci. USA 2013, 110, 7211–7216. [Google Scholar] [CrossRef] [PubMed]
- Cornea, R.L.; Gruber, S.J.; Lockamy, E.L.; Muretta, J.M.; Jin, D.; Chen, J.; Dahl, R.; Bartfai, T.; Zsebo, K.M.; Gillispie, G.D.; et al. High-Throughput FRET Assay Yields Allosteric SERCA Activators. J. Biomol. Screen. 2013, 18, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.J.; Peterson, K.C.; Muretta, J.M.; Higgins, S.E.; Gillispie, G.D.; Thomas, D.D. Fluorescence lifetime plate reader: Resolution and precision meet high-throughput. Rev. Sci. Instrum. 2014, 85, 113101. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.H.; Vunnam, N.; Lewis, A.K.; Chiu, T.L.; Brummel, B.E.; Schaaf, T.M.; Grant, B.D.; Bawaskar, P.; Thomas, D.D.; Sachs, J.N. An Innovative High-Throughput Screening Approach for Discovery of Small Molecules That Inhibit TNF Receptors. SLAS Discov. 2017, 22, 950–961. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, T.M.; Peterson, K.C.; Grant, B.D.; Bawaskar, P.; Yuen, S.; Li, J.; Muretta, J.M.; Gillispie, G.D.; Thomas, D.D. High-Throughput Spectral and Lifetime-Based FRET Screening in Living Cells to Identify Small-Molecule Effectors of SERCA. SLAS Discov. 2017, 22, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, T.M.; Peterson, K.C.; Grant, B.D.; Thomas, D.D.; Gillispie, G.D. Spectral Unmixing Plate Reader: High-Throughput, High-Precision FRET Assays in Living Cells. SLAS Discov. 2017, 22, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Waldeck-Weiermair, M.; Bischof, H.; Blass, S.; Deak, A.T.; Klec, C.; Graier, T.; Roller, C.; Rost, R.; Eroglu, E.; Gottschalk, B.; et al. Generation of Red-Shifted Cameleons for Imaging Ca2+ Dynamics of the Endoplasmic Reticulum. Sensors 2015, 15, 13052–13068. [Google Scholar] [CrossRef] [PubMed]
- Cranfill, P.J.; Sell, B.R.; Baird, M.A.; Allen, J.R.; Lavagnino, Z.; de Gruiter, H.M.; Kremers, G.J.; Davidson, M.W.; Ustione, A.; Piston, D.W. Quantitative assessment of fluorescent proteins. Nat. Methods 2016, 13, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Bajar, B.T.; Lam, A.J.; Badiee, R.K.; Oh, Y.H.; Chu, J.; Zhou, X.X.; Kim, N.; Kim, B.B.; Chung, M.; Yablonovitch, A.L.; et al. Fluorescent indicators for simultaneous reporting of all four cell cycle phases. Nat. Methods 2016, 13, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Bajar, B.T.; Wang, E.S.; Zhang, S.; Lin, M.Z.; Chu, J. A Guide to Fluorescent Protein FRET Pairs. Sensors 2016, 16, 1488. [Google Scholar] [CrossRef] [PubMed]
- Jung, G.; Brockhinke, A.; Gensch, T.; Hötzer, B.; Schwedler, S.; Veettil, S.K. Fluorescence Lifetime of Fluorescent Proteins. In Fluorescent Proteins I: From Understanding to Design; Springer: Berlin/Heidelberg, Germany, 2011; Volume 11, pp. 69–97. [Google Scholar]
- Rohde, J.A.; Thomas, D.D.; Muretta, J.M. Heart failure drug changes the mechanoenzymology of the cardiac myosin powerstroke. Proc. Natl. Acad. Sci. USA 2017, 114, E1796–E1804. [Google Scholar] [CrossRef] [PubMed]
- Muretta, J.M.; Rohde, J.A.; Johnsrud, D.O.; Cornea, S.; Thomas, D.D. Direct real-time detection of the structural and biochemical events in the myosin power stroke. Proc. Natl. Acad. Sci. USA 2015, 112, 14272–14277. [Google Scholar] [CrossRef] [PubMed]
- Laviv, T.; Kim, B.B.; Chu, J.; Lam, A.J.; Lin, M.Z.; Yasuda, R. Simultaneous dual-color fluorescence lifetime imaging with novel red-shifted fluorescent proteins. Nat. Methods 2016, 13, 989–992. [Google Scholar] [CrossRef] [PubMed]
- Pallikkuth, S.; Blackwell, D.J.; Hu, Z.; Hou, Z.; Zieman, D.T.; Svensson, B.; Thomas, D.D.; Robia, S.L. Phosphorylated Phospholamban Stabilizes a Compact Conformation of the Cardiac Calcium-ATPase. Biophys. J. 2013, 105, 1812–1821. [Google Scholar] [CrossRef] [PubMed]
- Smolin, N.; Robia, S.L. A structural mechanism for calcium transporter headpiece closure. J. Phys. Chem. B 2015, 119, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Suhling, K.; Siegel, J.; Phillips, D.; French, P.M.; Leveque-Fort, S.; Webb, S.E.; Davis, D.M. Imaging the environment of green fluorescent protein. Biophys. J. 2002, 83, 3589–3595. [Google Scholar] [CrossRef]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Mueller, B.; Karim, C.B.; Negrashov, I.V.; Kutchai, H.; Thomas, D.D. Direct detection of phospholamban and sarcoplasmic reticulum Ca-ATPase interaction in membranes using fluorescence resonance energy transfer. Biochemistry 2004, 43, 8754–8765. [Google Scholar] [CrossRef] [PubMed]
- Svensson, B.; Autry, J.M.; Thomas, D.D. Molecular Modeling of Fluorescent SERCA Biosensors. Methods Mol. Biol. 2016, 1377, 503–522. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.M.; Javadzadeh-Tabatabaie, M.; Benton, D.C.; Ganellin, C.R.; Haylett, D.G. Enhancement of hippocampal pyramidal cell excitability by the novel selective slow-afterhyperpolarization channel blocker 3-(triphenylmethylaminomethyl)pyridine (UCL2077). Mol. Pharmacol. 2006, 70, 1494–1502. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Rampe, D.; Triggle, D.J. Pharmacological, radioligand binding, and electrophysiological characteristics of FPL 64176, a novel nondihydropyridine Ca2+ channel activator, in cardiac and vascular preparations. Mol. Pharmacol. 1991, 40, 734–741. [Google Scholar] [PubMed]
- Goc, A.; Al-Azayzih, A.; Abdalla, M.; Al-Husein, B.; Kavuri, S.; Lee, J.; Moses, K.; Somanath, P.R. P21 activated kinase-1 (Pak1) promotes prostate tumor growth and microinvasion via inhibition of transforming growth factor beta expression and enhanced matrix metalloproteinase 9 secretion. J. Biol. Chem. 2013, 288, 3025–3035. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Tenorio, M.; Niggli, E. Stabilization of Ca2+ signaling in cardiac muscle by stimulation of SERCA. J. Mol. Cell. Cardiol. 2018, 119, 87–95. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schaaf, T.M.; Li, A.; Grant, B.D.; Peterson, K.; Yuen, S.; Bawaskar, P.; Kleinboehl, E.; Li, J.; Thomas, D.D.; Gillispie, G.D. Red-Shifted FRET Biosensors for High-Throughput Fluorescence Lifetime Screening. Biosensors 2018, 8, 99. https://doi.org/10.3390/bios8040099
Schaaf TM, Li A, Grant BD, Peterson K, Yuen S, Bawaskar P, Kleinboehl E, Li J, Thomas DD, Gillispie GD. Red-Shifted FRET Biosensors for High-Throughput Fluorescence Lifetime Screening. Biosensors. 2018; 8(4):99. https://doi.org/10.3390/bios8040099
Chicago/Turabian StyleSchaaf, Tory M., Ang Li, Benjamin D. Grant, Kurt Peterson, Samantha Yuen, Prachi Bawaskar, Evan Kleinboehl, Ji Li, David D. Thomas, and Gregory D. Gillispie. 2018. "Red-Shifted FRET Biosensors for High-Throughput Fluorescence Lifetime Screening" Biosensors 8, no. 4: 99. https://doi.org/10.3390/bios8040099
APA StyleSchaaf, T. M., Li, A., Grant, B. D., Peterson, K., Yuen, S., Bawaskar, P., Kleinboehl, E., Li, J., Thomas, D. D., & Gillispie, G. D. (2018). Red-Shifted FRET Biosensors for High-Throughput Fluorescence Lifetime Screening. Biosensors, 8(4), 99. https://doi.org/10.3390/bios8040099