Improving the Accuracy of Single-Nucleotide Variant Diagnosis Using On–Off Discriminating Primers


Abstract
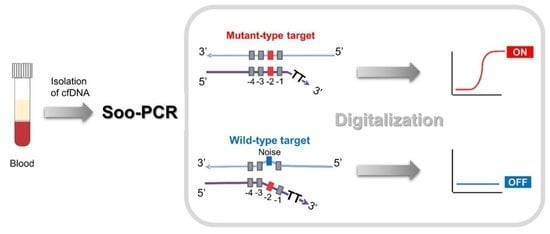
1. Introduction
2. Materials and Methods
2.1. Design and Synthesis of Oligonucleotides for Soo-PCR
2.2. Screening Various Taq DNA Polymerases for Soo-PCR Primers with a 3′-End Tailing Structure
2.3. Detection of Low-Abundance SNVs (KRAS G12D, EGFR L858R, and EGFR T790M)
3. Results and Discussion
3.1. Schematic Overview of Soo-PCR
3.2. Screening Taq DNA Polymerases for High Specificity
3.3. Optimization of 3′-End Tailing Length of Soo-PCR Primer for Major Cancer Markers
3.4. Validation of Soo-PCR Sensitivity and Specificity for Low-Abundance SNVs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kuang, Y.; Rogers, A.; Yeap, B.Y.; Wang, L.; Makrigiorgos, M.; Vetrand, K.; Thiede, S.; Distel, R.J.; Jänne, P.A. Noninvasive detection of EGFR T790M in gefitinib or erlotinib resistant non-small cell lung cancer. Clin. Cancer Res. 2009, 15, 2630–2636. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Murtaz, M.; Dawson, S.J.; Tsui, D.W.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.F.; Kingsbury, Z.; Wong, A.S.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Thierry, A.R.; Mouliere, F.; El Messaoudi, S.; Mollevi, C.; Lopez-Crapez, E.; Rolet, F.; Gillet, B.; Gongora, C.; Dechelotte, P.; Robert, B.; et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat. Med. 2014, 20, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Dawson, S.J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef]
- Diaz, L.A., Jr.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Hoon, D.S.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Milbury, C.A.; Li, J.; Makrigiorgos, G.M. PCR-based methods for the enrichment of minority alleles and mutations. Clin. Chem. 2009, 55, 632–640. [Google Scholar] [CrossRef]
- Sidransky, D.; Von Eschenbach, A.; Tsai, Y.C.; Jones, P.I.; Summerhayes, F.; Marshall, M.; Paul, P.; Green, S.R.; Hamilton, P.; Frost, B.; et al. Identification of p53 gene mutations in bladder cancers and urine samples. Science 1991, 252, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Tada, M.; Omata, M.; Kawai, S.; Saisho, H.; Ohto, M.; Saiki, R.K.; Sninsky, J.J. Detection of ras gene Mutations in Pancreatic Juice and peripheral Blood of Patients with Pancreatic adenocarcinoma. Cancer Res. 1993, 53, 2472–2474. [Google Scholar] [PubMed]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.; Kaper, F.; Dawson, S.J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl. Med. 2012, 4, 136ra68. [Google Scholar] [CrossRef]
- Leary, R.J.; Sausen, M.; Kinde, I.; Papadopoulos, N.; Carpten, J.D.; Craig, D.; O’Shaughnessy, J.; Kinzler, K.W.; Parmigiani, G.; Vogelstein, B.; et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci. Transl. Med. 2012, 4, 162ra154. [Google Scholar] [CrossRef] [PubMed]
- Kinde, I.; Wu, J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 9530–9535. [Google Scholar] [CrossRef]
- Tie, J.; Kinde, I.; Wang, Y.; Wong, H.L.; Roebert, J.; Christie, M.; Tacey, M.; Wong, R.; Singh, M.; Karapetis, C.S.; et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann. Oncol. 2015, 26, 1715–1722. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, E.Y.; Kim, T.; Chang, Y.S. Compared to plasma, bronchial washing fluid shows higher diagnostic yields for detecting EGFR-TKI sensitizing mutations by ddPCR in lung cancer. Respir. Res. 2020, 21, 1–8. [Google Scholar] [CrossRef]
- Olmedillas-López, S.; Olivera-Salazar, R.; García-Arranz, M.; García-Olmo, D. Current and emerging applications of droplet digital PCR in oncology: An updated review. Mol. Diagn. Ther. 2022, 26, 61–87. [Google Scholar] [CrossRef]
- Soverini, S.; De Santis, S.; Martelli, M.; Monaldi, C.; Castagnetti, F.; Gugliotta, G.; Papayannidis, C.; Mancini, M.; Bruno, S.; Venturi, C. Droplet digital PCR for the detection of second-generation tyrosine kinase inhibitor-resistant BCR::ABL1 kinase domain mutations in chronic myeloid leukemia. Leukemia 2022, 36, 2250–2260. [Google Scholar] [CrossRef] [PubMed]
- Wolter, M.; Felsberg, J.; Malzkorn, B.; Kaulich, K.; Reifenberger, G. Droplet digital PCR-based analyses for robust, rapid, and sensitive molecular diagnostics of gliomas. Acta Neuropathol. Commun. 2022, 10, 42. [Google Scholar] [CrossRef] [PubMed]
- Callens, C.; Bidard, F.-C.; Curto-Taribo, A.; Trabelsi-Grati, O.; Melaabi, S.; Delaloge, S.; Hardy-Bessard, A.-C.; Bachelot, T.; Clatot, F.; De La Motte Rouge, T. Real-time detection of ESR1 mutation in blood by droplet digital PCR in the PADA-1 trial: Feasibility and cross-validation with NGS. Anal. Chem. 2022, 94, 6297–6303. [Google Scholar] [CrossRef]
- Visser, E.; de Kock, R.; Genet, S.; van den Borne, B.; Youssef-El Soud, M.; Belderbos, H.; Stege, G.; de Saegher, M.; van’t Westeinde, S.; Broeren, M. Up-front mutation detection in circulating tumor DNA by droplet digital PCR has added diagnostic value in lung cancer. Transl. Oncol. 2023, 27, 101589. [Google Scholar] [CrossRef]
- Lefever, S.; Rihani, A.; Van der Meulen, J.; Pattyn, F.; Van Maerken, T.; Van Dorpe, J.; Hellemans, J.; Vandesompele, J. Cost-effective and robust genotyping using double-mismatch allele-specific quantitative PCR. Sci. Rep. 2019, 9, 2150. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-K.; Fan, L.; Wang, T.-Y.; Wu, P.-S. A new primer construction technique that effectively increases amplification of rare mutant templates in samples. BMC Biotechnol. 2019, 19, 62. [Google Scholar]
- Narang, A.; Marras, S.A.E.; Kurepina, N.; Chauhan, V.; Shashkina, E.; Kreiswirth, B.; Varma-Basil, M.; Vinnard, C.; Subbian, S. Ultrasensitive Detection of Multidrug-Resistant Mycobacterium tuberculosis Using SuperSelective Primer-Based Real-Time PCR Assays. Int. J. Mol. Sci. 2022, 23, 15752. [Google Scholar] [CrossRef]
- Kwak, J.Y.; Kim, E.K.; Kim, J.K.; Han, J.H.; Hong, S.W.; Park, T.S.; Choi, J.R. Dual priming oligonucleotide-based multiplex PCR analysis for detection of BRAFV600E mutation in FNAB samples of thyroid nodules in BRAFV600E mutation-prevalent area. Head Neck 2010, 32, 490–498. [Google Scholar]
- Lee, S.T.; Kim, S.W.; Ki, C.S.; Jang, J.H.; Shin, J.H.; Oh, Y.L.; Kim, J.W.; Chung, J.H. Clinical implication of highly sensitive detection of the BRAF V600E mutation in fine-needle aspirations of thyroid nodules: A comparative analysis of three molecular assays in 4585 consecutive cases in a BRAF V600E mutation-prevalent area. J. Clin. Endocrinol. Metab. 2012, 97, 2299–2306. [Google Scholar] [CrossRef]
- Guha, M.; Castellanos-Rizaldos, E.; Makrigiorgos, G.M. DISSECT method using PNA-LNA clamp improves detection of EGFR T790m mutation. PLoS ONE 2013, 8, e67782. [Google Scholar] [CrossRef]
- Nagai, Y.; Miyazawa, H.; Tanaka, T.; Udagawa, K.; Kato, M.; Fukuyama, S.; Yokote, A.; Kobayashi, K.; Kanazawa, M.; Hagiwara, K. Genetic heterogeneity of the epidermal growth factor receptor in non–small cell lung cancer cell lines revealed by a rapid and sensitive detection system, the peptide nucleic acid-locked nucleic acid PCR clamp. Cancer Res. 2005, 65, 7276–7282. [Google Scholar] [CrossRef] [PubMed]
- Pinzani, P.; Salvianti, F.; Cascella, R.; Massi, D.; De Giorgi, V.; Pazzagli, M.; Orlando, C. Allele specific TaqMan-based real-time PCR assay to quantify circulating BRAFV600E mutated DNA in plasma of melanoma patients. Clin. Chim. Acta 2010, 411, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.S.; Zhang, D.Y. Simulation-guided DNA probe design for consistently ultraspecific hybridization. Nat. Chem. 2015, 7, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Shin, S.; Kim, B.; Lee, K.-A. Selecting short length nucleic acids localized in exosomes improves plasma EGFR mutation detection in NSCLC patients. Cancer Cell Int. 2019, 19, 1–9. [Google Scholar] [CrossRef]
- Galli, A.; Gale, D.; Lawson, A.R.J.; Howarth, K.; Madi, M.; Durham, B.; Smalley, S.; Calaway, J.; Blais, S.; Jones, G.; et al. Development of a highly sensitive liquid biopsy platform to detect clinically-relevant cancer mutations at low allele fractions in cell-free DNA. PLoS ONE 2018, 13, e0194630. [Google Scholar] [CrossRef]
- Johnson, S.J.; Beese, L.S. Structures of mismatch replication errors observed in a DNA polymerase. Cell 2004, 116, 803–816. [Google Scholar] [CrossRef]
- Beard, W.A.; Shock, D.D.; Wilson, S.H. Influence of DNA structure on DNA polymerase β active site function: Extension of mutagenic DNA intermediates. J. Biol. Chem. 2004, 279, 31921–31929. [Google Scholar] [CrossRef]
- Stadhouders, R.; Pas, S.D.; Anber, J.; Voermans, J.; Mes, T.H.M.; Schutten, M. The Effect of Primer-Template Mismatches on the Detection and Quantification of Nucleic Acids Using the 5′ Nuclease Assay. J. Mol. Diagn. 2010, 12, 109–117. [Google Scholar] [CrossRef]
- Zhang, D.Y.; Seelig, G. Dynamic DNA nanotechnology using strand-displacement reactions. Nat. Chem. 2011, 3, 103–113. [Google Scholar] [CrossRef]
- Zhang, D.Y.; Chen, S.X.; Yin, P. Optimizing the specificity of nucleic acid hybridization. Nat. Chem. 2012, 4, 208–214. [Google Scholar] [CrossRef]
- Alcaide, M.; Cheung, M.; Hillman, J.; Rassekh, S.R.; Deyell, R.J.; Batist, G.; Karsan, A.; Wyatt, A.W.; Johnson, N.; Scott, D.W.; et al. Evaluating the quantity, quality and size distribution of cell-free DNA by multiplex droplet digital PCR. Sci. Rep. 2020, 10, 12564. [Google Scholar] [CrossRef] [PubMed]
- Meddeb, R.; Dache, Z.A.A.; Thezenas, S.; Otandault, A.; Tanos, R.; Pastor, B.; Sanchez, C.; Azzi, J.; Tousch, G.; Azan, S.; et al. Quantifying circulating cell-free DNA in humans. Sci. Rep. 2019, 9, 5220. [Google Scholar] [CrossRef] [PubMed]
- Volik, S.; Alcaide, M.; Morin, R.D.; Collins, C. Cell-free DNA (cfDNA): Clinical significance and utility in cancer shaped by emerging technologies. Mol. Cancer Res. 2016, 14, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Akarapipad, P.; Bertelson, E.; Pessell, A.; Wang, T.-H.; Hsieh, K. Emerging Multiplex Nucleic Acid Diagnostic Tests for Combating COVID-19. Biosensors 2022, 12, 978. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, S.; Visweswar Kambhampati, N.S.; Ganesh, K.M.; Mahesh, S.P.; Srinivasan, V.; Ramamurthy, S.S. Metal-Free, Graphene Oxide-Based Tunable Soliton and Plasmon Engineering for Biosensing Applications. ACS Appl. Mater. Interfaces 2021, 13, 17046–17061. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taq DNA Polymerase | PCR Cycling Conditions (°C/Times) | ||||
|---|---|---|---|---|---|
| Initial Denaturation | 70 Cycles | Final Extension | |||
| Denaturation | Annealing | Extension | |||
| Ampli Taq | 95/10 min | 95/30 s | 56/30 s | 72/15 s | 72/7 min |
| Hot Start Taq | 95/30 s | 95/30 s | 56/30 s | 68/15 s | 68/5 min |
| EzWay | 95/15 min | 94/30 s | 56/30 s | 72/15 s | 72/10 min |
| Dream Taq | 95/3 min | 95/30 s | 61/30 s | 72/1 min | 72/7 min |
| Go Taq | 95/2 min | 95/30 s | 56/30 s | 72/15 s | 72/5 min |
| Taq DNA Polymerase | ΔCtMP-PMP | ||||
|---|---|---|---|---|---|
| Length of 3′-End Tailing Structure (nt) | |||||
| 0 | 1 | 2 | 3 | 4 | |
| Ampli Taq | 0.6 | 9.4 | −45 ** | * | * |
| Hot Start Taq | 0.3 | 9.3 | −53.4 ** | * | * |
| EzWay | 0.5 | 9.1 | * | * | * |
| DreamTaq | 0.6 | 7.1 | 17.4 | * | * |
| GoTaq | 0.4 | 8.7 | * | * | * |
| Target | ΔCtMP-PMP | ||||
|---|---|---|---|---|---|
| Length of 3′-End Tailing Structure (nt) | |||||
| 0 | 1 | 2 | 3 | 4 | |
| KRAS G12D | 0.4 | 9.1 | −41.3 ** | * | * |
| EGFR L858R | 4.9 | 12.2 | −42.6 ** | * | * |
| EGFR T790M | 2.5 | 12.3 | −57.6 ** | * | * |
| Horizon cfDNA (HD780) | Droplet Digital PCR | Soo-PCR | ||||||
|---|---|---|---|---|---|---|---|---|
| Gene | Variant | Copy Number | Expected AF (%) | Actual AF (%) | On/Off | Ct Value | ||
| WT | MT | Total | ||||||
| 100% Wild Type | ||||||||
| KRAS G12D | 14,995 | 5 | 15,000 | 0.00 | 0.02 | Off | No amplification | |
| EGFR L858R | 15,000 | 0 | 15,000 | 0.00 | 0.00 | Off | No amplification | |
| EGFR T790M | 14,993 | 7 | 15,000 | 0.00 | 0.05 | Off | No amplification | |
| 0.1% Allele Frequency | ||||||||
| KRAS G12D | 14,980 | 20 | 15,000 | 0.13 | 0.13 | On | 65.3 | |
| EGFR L858R | 14,983 | 17 | 15,000 | 0.10 | 0.11 | On | 68.9 | |
| EGFR T790M | 14,980 | 20 | 15,000 | 0.10 | 0.12 | On | 65.7 | |
| 1% Allele Frequency | ||||||||
| KRAS G12D | 14,822 | 178 | 15,000 | 1.30 | 1.17 | On | 63.2 | |
| EGFR L858R | 14,870 | 130 | 15,000 | 1.00 | 0.87 | On | 52.8 | |
| EGFR T790M | 14,878 | 122 | 15,000 | 1.00 | 0.81 | On | 61.1 | |
| 5% Allele Frequency | ||||||||
| KRAS G12D | 14,094 | 906 | 15,000 | 6.30 | 6.03 | On | 53.4 | |
| EGFR L858R | 14,337 | 663 | 15,000 | 5.00 | 4.43 | On | 47.9 | |
| EGFR T790M | 14,364 | 636 | 15,000 | 5.00 | 4.24 | On | 47.3 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, J.; Jung, C. Improving the Accuracy of Single-Nucleotide Variant Diagnosis Using On–Off Discriminating Primers. Biosensors 2023, 13, 380. https://doi.org/10.3390/bios13030380
Shin J, Jung C. Improving the Accuracy of Single-Nucleotide Variant Diagnosis Using On–Off Discriminating Primers. Biosensors. 2023; 13(3):380. https://doi.org/10.3390/bios13030380
Chicago/Turabian StyleShin, Juny, and Cheulhee Jung. 2023. "Improving the Accuracy of Single-Nucleotide Variant Diagnosis Using On–Off Discriminating Primers" Biosensors 13, no. 3: 380. https://doi.org/10.3390/bios13030380
APA StyleShin, J., & Jung, C. (2023). Improving the Accuracy of Single-Nucleotide Variant Diagnosis Using On–Off Discriminating Primers. Biosensors, 13(3), 380. https://doi.org/10.3390/bios13030380