Biofunctionalization of Multiplexed Silicon Photonic Biosensors

Abstract

1. Introduction
2. Bioreceptors
2.1. Antibodies
2.2. Aptamers
2.3. Nucleic Acid Probes (Hybridization-Based Sensing)
2.4. Molecularly Imprinted Polymers (MIPs)
2.5. Peptides and Protein-Catalyzed Capture Agents
2.6. Glycans and Lectins
2.7. Other
2.7.1. High Contrast Cleavage Detection (i.e., CRISPR Cleavage Detection)
2.7.2. CRISPR-dCas9-Mediated Sensing
2.7.3. Lipid Nanodiscs
2.8. Summary and Future Directions
3. Bioreceptor Immobilization Strategies
3.1. Passive Adsorption
3.2. Bioaffinity-Based Immobilization
3.3. Covalent Immobilization
3.3.1. Silane-Mediated Immobilization
3.3.2. Organophosphonate-Mediated Immobilization
3.3.3. Click Chemistry
3.3.4. UV-Crosslinking
3.4. Summary and Future Directions
4. Patterning Techniques
4.1. Microcontact Printing
4.2. Pin and Pipette Spotting
4.3. Microfluidic Patterning in Channels
4.4. Inkjet Printing
4.5. Microfluidic Probes
4.6. Summary and Future Directions
5. Critical Comparative Analysis of Solutions and Discussion of the Interplay between the Three Aspects of Biofunctionalization
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luan, E.; Shoman, H.; Ratner, D.M.; Cheung, K.C.; Chrostowski, L. Silicon Photonic Biosensors Using Label-Free Detection. Sensors 2018, 18, 3519. [Google Scholar] [CrossRef] [PubMed]
- Stratis-Cullum, D.N.; Finch, A.S. Current trends in ubiquitous biosensing. J. Anal. Bioanal. Tech. 2013, S7, 9. [Google Scholar] [CrossRef]
- Blevins, M.G.; Fernandez-Galiana, A.; Hooper, M.J.; Boriskina, S.V. Roadmap on Universal Photonic Biosensors for Real-Time Detection of Emerging Pathogens. Photonics 2021, 8, 342. [Google Scholar] [CrossRef]
- Chrostowski, L.; Grist, S.; Flueckiger, J.; Shi, W.; Wang, X.; Ouellet, E.; Yun, H.; Webb, M.; Nie, B.; Liang, Z.; et al. Silicon photonic resonator sensors and devices. In Laser Resonators, Microresonators, and Beam Control XIV; Kudryashov, A.V., Paxton, A.H., Ilchenko, V.S., Eds.; SPIE Proceedings; SPIE: Bellingham, WA, USA, 2012; Volume 8236, p. 823620. [Google Scholar]
- Pohanka, M.; Skládal, P. Electrochemical biosensors—Principles and applications. J. Appl. Biomed. 2008, 6, 57–64. [Google Scholar] [CrossRef]
- Pohanka, M. Overview of piezoelectric biosensors, immunosensors and DNA sensors and their applications. Materials 2018, 11, 448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.X.J.; Hoshino, K. Mechanical transducers: Cantilevers, acoustic wave sensors, and thermal sensors. In Molecular Sensors and Nanodevices; Elsevier: Amsterdam, The Netherlands, 2019; pp. 311–412. ISBN 9780128148624. [Google Scholar]
- Ciminelli, C.; Dell’Olio, F.; Conteduca, D.; Armenise, M.N. Silicon photonic biosensors. IET Optoelectronics 2018, 13, 48–54. [Google Scholar] [CrossRef]
- Soref, R. The past, present, and future of silicon photonics. IEEE J. Select. Topics Quantum Electron. 2006, 12, 1678–1687. [Google Scholar] [CrossRef]
- Bailey, R.C.; Washburn, A.L.; Qavi, A.J.; Iqbal, M.; Gleeson, M.; Tybor, F.; Gunn, L.C. A robust silicon photonic platform for multiparameter biological analysis. In Silicon Photonics IV; Kubby, J.A., Reed, G.T., Eds.; SPIE Proceedings; SPIE: Bellingham, WA, USA, 2009; Volume 7220, p. 72200N. [Google Scholar]
- Chrostowski, L.; Hochberg, M. Silicon Photonics Design; Cambridge University Press: Cambridge, UK, 2015; ISBN 9781316084168. [Google Scholar]
- Steglich, P.; Lecci, G.; Mai, A. Surface plasmon resonance (SPR) spectroscopy and photonic integrated circuit (PIC) biosensors: A comparative review. Sensors 2022, 22, 2901. [Google Scholar] [CrossRef] [PubMed]
- Chrostowski, L.; Leanne, D.; Matthew, M.; Connor, M.; Luan, E.; Al-Qadasi, M.; Avineet, R.; Mojaver, H.R.; Lyall, E.; Gervais, A.; et al. A silicon photonic evanescent-field sensor architecture using a fixed-wavelength laser. In Optical Interconnects XXI; Schröder, H., Chen, R.T., Eds.; SPIE: Bellingham, WA, USA, 2021; Volume 11692. [Google Scholar]
- Steglich, P.; Hülsemann, M.; Dietzel, B.; Mai, A. Optical Biosensors Based on Silicon-On-Insulator Ring Resonators: A Review. Molecules 2019, 24, 519. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.; Gleeson, M.A.; Spaugh, B.; Tybor, F.; Gunn, W.G.; Hochberg, M.; Baehr-Jones, T.; Bailey, R.C.; Gunn, L.C. Label-Free Biosensor Arrays Based on Silicon Ring Resonators and High-Speed Optical Scanning Instrumentation. IEEE J. Select. Topics Quantum Electron. 2010, 16, 654–661. [Google Scholar] [CrossRef]
- Puumala, L.S.; Grist, S.M.; Wickremasinghe, K.; Al-Qadasi, M.A.; Chowdhury, S.J.; Liu, Y.; Mitchell, M.; Chrostowski, L.; Shekhar, S.; Cheung, K.C. An Optimization Framework for Silicon Photonic Evanescent-Field Biosensors Using Sub-Wavelength Gratings. Biosensors 2022, 12, 840. [Google Scholar] [CrossRef] [PubMed]
- Valera, E.; Shia, W.W.; Bailey, R.C. Development and validation of an immunosensor for monocyte chemotactic protein 1 using a silicon photonic microring resonator biosensing platform. Clin. Biochem. 2016, 49, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Luchansky, M.S.; Bailey, R.C. Silicon photonic microring resonators for quantitative cytokine detection and T-cell secretion analysis. Anal. Chem. 2010, 82, 1975–1981. [Google Scholar] [CrossRef] [PubMed]
- Arshavsky-Graham, S.; Massad-Ivanir, N.; Segal, E.; Weiss, S. Porous Silicon-Based Photonic Biosensors: Current Status and Emerging Applications. Anal. Chem. 2019, 91, 441–467. [Google Scholar] [CrossRef] [PubMed]
- Dincer, C.; Bruch, R.; Kling, A.; Dittrich, P.S.; Urban, G.A. Multiplexed Point-of-Care Testing—xPOCT. Trends Biotechnol. 2017, 35, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Jarockyte, G.; Karabanovas, V.; Rotomskis, R.; Mobasheri, A. Multiplexed nanobiosensors: Current trends in early diagnostics. Sensors 2020, 20, 6890. [Google Scholar] [CrossRef] [PubMed]
- Washburn, A.L.; Luchansky, M.S.; Bowman, A.L.; Bailey, R.C. Quantitative, label-free detection of five protein biomarkers using multiplexed arrays of silicon photonic microring resonators. Anal. Chem. 2010, 82, 69–72. [Google Scholar] [CrossRef]
- Shin, Y.; Perera, A.P.; Kee, J.S.; Song, J.; Fang, Q.; Lo, G.-Q.; Park, M.K. Label-free methylation specific sensor based on silicon microring resonators for detection and quantification of DNA methylation biomarkers in bladder cancer. Sens. Actuators B Chem. 2013, 177, 404–411. [Google Scholar] [CrossRef]
- Park, M.K.; Kee, J.S.; Quah, J.Y.; Netto, V.; Song, J.; Fang, Q.; La Fosse, E.M.; Lo, G.-Q. Label-free aptamer sensor based on silicon microring resonators. Sens. Actuators B Chem. 2013, 176, 552–559. [Google Scholar] [CrossRef]
- Chalyan, T.; Pasquardini, L.; Gandolfi, D.; Guider, R.; Samusenko, A.; Zanetti, M.; Pucker, G.; Pederzolli, C.; Pavesi, L. Aptamer- and Fab’- Functionalized Microring Resonators for Aflatoxin M1 Detection. IEEE J. Select. Topics Quantum Electron. 2017, 23, 350–357. [Google Scholar] [CrossRef]
- Adamopoulos, C.; Buchbinder, S.; Zarkos, P.; Bhargava, P.; Gharia, A.; Niknejad, A.; Anwar, M.; Stojanovic, V. Fully Integrated Electronic–Photonic Biosensor for Label-Free Real-Time Molecular Sensing in Advanced Zero-Change CMOS-SOI Process. IEEE Solid-State Circuits Lett. 2021, 4, 198–201. [Google Scholar] [CrossRef]
- Robison, H.M.; Bailey, R.C. A Guide to Quantitative Biomarker Assay Development using Whispering Gallery Mode Biosensors. Curr. Protoc. Chem. Biol. 2017, 9, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, N.; Jolly, P.; Formisano, N.; Estrela, P. Introduction to biosensors. Essays Biochem. 2016, 60, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Soler, M.; Lechuga, L.M. Biochemistry strategies for label-free optical sensor biofunctionalization: Advances towards real applicability. Anal. Bioanal. Chem. 2021, 414, 5071–5085. [Google Scholar] [CrossRef] [PubMed]
- Morales, M.A.; Halpern, J.M. Guide to selecting a biorecognition element for biosensors. Bioconjug. Chem. 2018, 29, 3231–3239. [Google Scholar] [CrossRef] [PubMed]
- Bañuls, M.-J.; Puchades, R.; Maquieira, Á. Chemical surface modifications for the development of silicon-based label-free integrated optical (IO) biosensors: A review. Anal. Chim. Acta 2013, 777, 1–16. [Google Scholar] [CrossRef]
- Delamarche, E.; Pereiro, I.; Kashyap, A.; Kaigala, G.V. Biopatterning: The art of patterning biomolecules on surfaces. Langmuir 2021, 37, 9637–9651. [Google Scholar] [CrossRef]
- Nicu, L.; Leïchlé, T. Micro- and Nanoelectromechanical Biosensors; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2013; pp. 65–91. ISBN 9781118760857. [Google Scholar]
- Baker, J.E.; Sriram, R.; Miller, B.L. Two-dimensional photonic crystals for sensitive microscale chemical and biochemical sensing. Lab Chip 2015, 15, 971–990. [Google Scholar] [CrossRef]
- Byrne, B.; Stack, E.; Gilmartin, N.; O’Kennedy, R. Antibody-based sensors: Principles, problems and potential for detection of pathogens and associated toxins. Sensors 2009, 9, 4407–4445. [Google Scholar] [CrossRef]
- Sajid, M.; Kawde, A.-N.; Daud, M. Designs, formats and applications of lateral flow assay: A literature review. Journal of Saudi Chemical Society 2015, 19, 689–705. [Google Scholar] [CrossRef]
- Arya, S.K.; Estrela, P. Recent Advances in Enhancement Strategies for Electrochemical ELISA-Based Immunoassays for Cancer Biomarker Detection. Sensors 2018, 18, 2010. [Google Scholar] [CrossRef]
- Narita, F.; Wang, Z.; Kurita, H.; Li, Z.; Shi, Y.; Jia, Y.; Soutis, C. A Review of Piezoelectric and Magnetostrictive Biosensor Materials for Detection of COVID-19 and Other Viruses. Adv. Mater. 2021, 33, e2005448. [Google Scholar] [CrossRef]
- Huertas, C.S.; Calvo-Lozano, O.; Mitchell, A.; Lechuga, L.M. Advanced Evanescent-Wave Optical Biosensors for the Detection of Nucleic Acids: An Analytic Perspective. Front. Chem. 2019, 7, 724. [Google Scholar] [CrossRef] [PubMed]
- Fard, S.T.; Donzella, V.; Schmidt, S.A.; Flueckiger, J.; Grist, S.M.; Talebi Fard, P.; Wu, Y.; Bojko, R.J.; Kwok, E.; Jaeger, N.A.F.; et al. Performance of ultra-thin SOI-based resonators for sensing applications. Opt. Express 2014, 22, 14166–14179. [Google Scholar] [CrossRef] [PubMed]
- Oliverio, M.; Perotto, S.; Messina, G.C.; Lovato, L.; De Angelis, F. Chemical functionalization of plasmonic surface biosensors: A tutorial review on issues, strategies, and costs. ACS Appl. Mater. Interfaces 2017, 9, 29394–29411. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, J.; Yang, Z.; Wilkinson, J.S.; Zhou, X. Optical biosensors based on refractometric sensing schemes: A review. Biosens. Bioelectron. 2019, 144, 111693. [Google Scholar] [CrossRef] [PubMed]
- Wijaya, E.; Lenaerts, C.; Maricot, S.; Hastanin, J.; Habraken, S.; Vilcot, J.-P.; Boukherroub, R.; Szunerits, S. Surface plasmon resonance-based biosensors: From the development of different SPR structures to novel surface functionalization strategies. Curr. Opin. Solid State Mater. Sci. 2011, 15, 208–224. [Google Scholar] [CrossRef]
- Luan, E. Improving the Performance of Silicon Photonic Optical Resonator-based Sensors for Biomedical Applications. Doctoral Dissertation, University of British Columbia, Vancouver, BC, Canada, 2020. [Google Scholar]
- Donzella, V.; Sherwali, A.; Flueckiger, J.; Talebi Fard, S.; Grist, S.M.; Chrostowski, L. Sub-wavelength grating components for integrated optics applications on SOI chips. Opt. Express 2014, 22, 21037–21050. [Google Scholar] [CrossRef] [PubMed]
- Donzella, V.; Sherwali, A.; Flueckiger, J.; Grist, S.M.; Fard, S.T.; Chrostowski, L. Design and fabrication of SOI micro-ring resonators based on sub-wavelength grating waveguides. Opt. Express 2015, 23, 4791–4803. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, L.; Zhang, H.; Li, Y.; Liu, L.; Chen, Y.; Qiu, X.; Yu, D. Development of a portable SPR sensor for nucleic acid detection. Micromachines 2020, 11, 526. [Google Scholar] [CrossRef]
- Harpaz, D.; Koh, B.; Marks, R.S.; Seet, R.C.S.; Abdulhalim, I.; Tok, A.I.Y. Point-of-Care Surface Plasmon Resonance Biosensor for Stroke Biomarkers NT-proBNP and S100β Using a Functionalized Gold Chip with Specific Antibody. Sensors 2019, 19, 2533. [Google Scholar] [CrossRef] [PubMed]
- Masson, J.-F. Portable and field-deployed surface plasmon resonance and plasmonic sensors. Analyst 2020, 145, 3776–3800. [Google Scholar] [CrossRef]
- Ouellet, E.; Lausted, C.; Lin, T.; Yang, C.W.T.; Hood, L.; Lagally, E.T. Parallel microfluidic surface plasmon resonance imaging arrays. Lab Chip 2010, 10, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Yu, F.; Zare, R.N. Microfluidic device for immunoassays based on surface plasmon resonance imaging. Lab Chip 2008, 8, 694–700. [Google Scholar] [CrossRef]
- Endo, T.; Kerman, K.; Nagatani, N.; Hiepa, H.M.; Kim, D.-K.; Yonezawa, Y.; Nakano, K.; Tamiya, E. Multiple label-free detection of antigen-antibody reaction using localized surface plasmon resonance-based core-shell structured nanoparticle layer nanochip. Anal. Chem. 2006, 78, 6465–6475. [Google Scholar] [CrossRef] [PubMed]
- Tokel, O.; Inci, F.; Demirci, U. Advances in plasmonic technologies for point of care applications. Chem. Rev. 2014, 114, 5728–5752. [Google Scholar] [CrossRef] [PubMed]
- Breault-Turcot, J.; Masson, J.-F. Nanostructured substrates for portable and miniature SPR biosensors. Anal. Bioanal. Chem. 2012, 403, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Zhang, J.; Jiang, X.; Gan, H.; Zhu, Y.; Peng, Q.; Fang, X.; Guo, Y.; Wang, L. SPR/SERS dual-mode plasmonic biosensor via catalytic hairpin assembly-induced AuNP network. Biosens. Bioelectron. 2021, 190, 113376. [Google Scholar] [CrossRef] [PubMed]
- Barchiesi, D.; Grosges, T.; Colas, F.; de la Chapelle, M.L. Combined SPR and SERS: Otto and Kretschmann configurations. J. Opt. 2015, 17, 114009. [Google Scholar] [CrossRef]
- Meyer, S.A.; Le Ru, E.C.; Etchegoin, P.G. Combining surface plasmon resonance (SPR) spectroscopy with surface-enhanced Raman scattering (SERS). Anal. Chem. 2011, 83, 2337–2344. [Google Scholar] [CrossRef]
- Bontempi, N.; Vassalini, I.; Danesi, S.; Ferroni, M.; Donarelli, M.; Colombi, P.; Alessandri, I. Non-Plasmonic SERS with Silicon: Is It Really Safe? New Insights into the Optothermal Properties of Core/Shell Microbeads. J. Phys. Chem. Lett. 2018, 9, 2127–2132. [Google Scholar] [CrossRef] [PubMed]
- Juan-Colás, J.; Parkin, A.; Dunn, K.E.; Scullion, M.G.; Krauss, T.F.; Johnson, S.D. The electrophotonic silicon biosensor. Nat. Commun. 2016, 7, 12769. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.; Mujawar, M.A. Point of care sensing devices: Better care for everyone. Sensors 2018, 18, 4303. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.M.; Cho, P.; Bickford, J.R.; Pellegrino, P.M.; Leake, G.; Fanto, M.L. Development of army relevant wearable Photonic Integrated Circuit (PIC) biosensors. In Chemical, Biological, Radiological, Nuclear, and Explosives (CBRNE) Sensing XXII; Guicheteau, J.A., Howle, C.R., Eds.; SPIE: Bellingham, WA, USA, 2021; p. 15. [Google Scholar]
- Zan, G.; Wu, T.; Zhu, F.; He, P.; Cheng, Y.; Chai, S.; Wang, Y.; Huang, X.; Zhang, W.; Wan, Y.; et al. A biomimetic conductive super-foldable material. Matter 2021, 4, 3232–3247. [Google Scholar] [CrossRef]
- Steglich, P.; Bondarenko, S.; Mai, C.; Paul, M.; Weller, M.G.; Mai, A. CMOS-Compatible Silicon Photonic Sensor for Refractive Index Sensing Using Local Back-Side Release. IEEE Photon. Technol. Lett. 2020, 32, 1241–1244. [Google Scholar] [CrossRef]
- Steglich, P.; Paul, M.; Mai, C.; Böhme, A.; Bondarenko, S.; Weller, M.G.; Mai, A. A monolithically integrated micro fluidic channel in a silicon-based photonic-integrated-circuit technology for biochemical sensing. In Optical Sensors 2021; Lieberman, R.A., Baldini, F., Homola, J., Eds.; SPIE: Bellingham, WA, USA, 2021; p. 3. [Google Scholar]
- Zhou, Z.; Yin, B.; Michel, J. On-chip light sources for silicon photonics. Light Sci. Appl. 2015, 4, e358. [Google Scholar] [CrossRef]
- Laplatine, L.; Luan, E.; Cheung, K.; Ratner, D.M.; Dattner, Y.; Chrostowski, L. System-level integration of active silicon photonic biosensors using Fan-Out Wafer-Level-Packaging for low cost and multiplexed point-of-care diagnostic testing. Sens. Actuators B Chem. 2018, 273, 1610–1617. [Google Scholar] [CrossRef]
- Steglich, P.; Rabus, D.G.; Sada, C.; Paul, M.; Weller, M.G.; Mai, C.; Mai, A. Silicon Photonic Micro-Ring Resonators for Chemical and Biological Sensing: A Tutorial. IEEE Sens. J. 2021, 22, 10089–10105. [Google Scholar] [CrossRef]
- Gao, S.; Guisán, J.M.; Rocha-Martin, J. Oriented immobilization of antibodies onto sensing platforms—A critical review. Anal. Chim. Acta 2022, 1189, 338907. [Google Scholar] [CrossRef]
- Welch, N.G.; Scoble, J.A.; Muir, B.W.; Pigram, P.J. Orientation and characterization of immobilized antibodies for improved immunoassays (Review). Biointerphases 2017, 12, 02D301. [Google Scholar] [CrossRef]
- Teles, F.; Fonseca, L. Trends in DNA biosensors. Talanta 2008, 77, 606–623. [Google Scholar] [CrossRef]
- Nimse, S.B.; Song, K.; Sonawane, M.D.; Sayyed, D.R.; Kim, T. Immobilization techniques for microarray: Challenges and applications. Sensors 2014, 14, 22208–22229. [Google Scholar] [CrossRef] [PubMed]
- Chiappini, A.; Pasquardini, L.; Bossi, A.M. Molecular imprinted polymers coupled to photonic structures in biosensors: The state of art. Sensors 2020, 20, 5069. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.P.; Arulanandam, B.P.; Matta, L.L.; Weis, A.; Valdes, J.J. Biosensor recognition elements. Curr. Issues Mol. Biol. 2008, 10, 1–12. [Google Scholar] [CrossRef]
- Vashist, S.K.; Lam, E.; Hrapovic, S.; Male, K.B.; Luong, J.H.T. Immobilization of antibodies and enzymes on 3-aminopropyltriethoxysilane-functionalized bioanalytical platforms for biosensors and diagnostics. Chem. Rev. 2014, 114, 11083–11130. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Rusling, J.; Dixit, C.K. Site-selective orientated immobilization of antibodies and conjugates for immunodiagnostics development. Methods 2017, 116, 95–111. [Google Scholar] [CrossRef]
- Mauriz, E.; García-Fernández, M.C.; Lechuga, L.M. Towards the design of universal immunosurfaces for SPR-based assays: A review. TrAC Trends in Analytical Chemistry 2016, 79, 191–198. [Google Scholar] [CrossRef]
- Barbulovic-Nad, I.; Lucente, M.; Sun, Y.; Zhang, M.; Wheeler, A.R.; Bussmann, M. Bio-microarray fabrication techniques--a review. Crit. Rev. Biotechnol. 2006, 26, 237–259. [Google Scholar] [CrossRef]
- Nicu, L.; Leïchlé, T. Biosensors and tools for surface functionalization from the macro- to the nanoscale: The way forward. J. Appl. Phys. 2008, 104, 111101. [Google Scholar] [CrossRef]
- Amirjani, A.; Rahbarimehr, E. Recent advances in functionalization of plasmonic nanostructures for optical sensing. Mikrochim. Acta 2021, 188, 57. [Google Scholar] [CrossRef]
- Sassolas, A.; Leca-Bouvier, B.D.; Blum, L.J. DNA biosensors and microarrays. Chem. Rev. 2008, 108, 109–139. [Google Scholar] [CrossRef] [PubMed]
- Ravina; Kumar, D.; Prasad, M.; Mohan, H. Biological recognition elements. In Electrochemical Sensors; Elsevier: Amsterdam, The Netherlands, 2022; pp. 213–239. ISBN 9780128231487. [Google Scholar]
- Goode, J.A.; Rushworth, J.V.H.; Millner, P.A. Biosensor regeneration: A review of common techniques and outcomes. Langmuir 2015, 31, 6267–6276. [Google Scholar] [CrossRef] [PubMed]
- Autebert, J.; Cors, J.F.; Taylor, D.P.; Kaigala, G.V. Convection-Enhanced Biopatterning with Recirculation of Hydrodynamically Confined Nanoliter Volumes of Reagents. Anal. Chem. 2016, 88, 3235–3242. [Google Scholar] [CrossRef]
- Abbas MBBS, A.K.; Lichtman MD PhD, A.H.; Pillai MBBS PhD, S. Cellular and Molecular Immunology, 10th ed.; Elsevier: Amsterdam, The Netherlands, 2021; p. 618. ISBN 978-0-323-75748-5. [Google Scholar]
- Nimjee, S.M.; Rusconi, C.P.; Sullenger, B.A. Aptamers: An emerging class of therapeutics. Annu. Rev. Med. 2005, 56, 555–583. [Google Scholar] [CrossRef] [PubMed]
- Landry, J.P.; Ke, Y.; Yu, G.-L.; Zhu, X.D. Measuring affinity constants of 1450 monoclonal antibodies to peptide targets with a microarray-based label-free assay platform. J. Immunol. Methods 2015, 417, 86–96. [Google Scholar] [CrossRef]
- Kaur, H. Overview of monoclonal antibodies. In Monoclonal Antibodies; Elsevier: Amsterdam, The Netherlands, 2021; pp. 1–29. ISBN 9780128223185. [Google Scholar]
- Singh, A.; Mishra, A.; Verma, A. Antibodies: Monoclonal and polyclonal. In Animal Biotechnology; Elsevier: Amsterdam, The Netherlands, 2020; pp. 327–352. ISBN 9780128117101. [Google Scholar]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef]
- Lipman, N.S.; Jackson, L.R.; Trudel, L.J.; Weis-Garcia, F. Monoclonal versus polyclonal antibodies: Distinguishing characteristics, applications, and information resources. ILAR J. 2005, 46, 258–268. [Google Scholar] [CrossRef]
- Layouni, R.; Cao, T.; Coppock, M.B.; Laibinis, P.E.; Weiss, S.M. Peptide-Based Capture of Chikungunya Virus E2 Protein Using Porous Silicon Biosensor. Sensors 2021, 21, 8248. [Google Scholar] [CrossRef]
- Edfors, F.; Hober, A.; Linderbäck, K.; Maddalo, G.; Azimi, A.; Sivertsson, Å.; Tegel, H.; Hober, S.; Szigyarto, C.A.-K.; Fagerberg, L.; et al. Enhanced validation of antibodies for research applications. Nat. Commun. 2018, 9, 4130. [Google Scholar] [CrossRef]
- Antibodypedia. Available online: https://www.antibodypedia.com/ (accessed on 29 March 2022).
- Global Industry Analysts Research Antibodies. Available online: https://www.marketresearch.com/Global-Industry-Analysts-v1039/Research-Antibodies-32538641/ (accessed on 2 December 2022).
- Grand View Research Research Antibodies Market Size, Share & Trends Analysis Report By Product (Primary, Secondary), By Type, By Technology, By Source, By Application, By End-use, By Region, And Segment Forecasts, 2022–2030. Available online: https://www.grandviewresearch.com/industry-analysis/research-antibodies-market (accessed on 2 December 2022).
- Balamurugan, S.; Obubuafo, A.; Soper, S.A.; Spivak, D.A. Surface immobilization methods for aptamer diagnostic applications. Anal. Bioanal. Chem. 2008, 390, 1009–1021. [Google Scholar] [CrossRef]
- Available online: https://www.sigmaaldrich.com/CA/en/search/antibodies?facet=facet_clonality%3Amonoclonal&focus=products&page=1&perpage=30&sort=relevance&term=antibodies&type=product_namewww.sigmaaldrich.com/CA/en/search/antibodies (accessed on 19 July 2022).
- Primary Antibodies|Abcam. Available online: https://www.abcam.com/nav/primary-antibodies (accessed on 19 July 2022).
- Primary Antibodies|Thermo Fisher Scientific—CA. Available online: https://www.thermofisher.com/ca/en/home/life-science/antibodies/primary-antibodies.html?icid=ab-search-primary-icons (accessed on 19 July 2022).
- Tombelli, S.; Minunni, M.; Mascini, M. Analytical applications of aptamers. Biosens. Bioelectron. 2005, 20, 2424–2434. [Google Scholar] [CrossRef]
- Sun, H.; Zu, Y. A highlight of recent advances in aptamer technology and its application. Molecules 2015, 20, 11959–11980. [Google Scholar] [CrossRef]
- Lakhin, A.V.; Tarantul, V.Z.; Gening, L.V. Aptamers: Problems, solutions and prospects. Acta Naturae 2013, 5, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Grand View Research DNA Synthesis Market Size, Share & Trends Analysis Report By Service Type (Gene Synthesis, Oligonucleotide Synthesis), By Application (Research And Development, Therapeutics), By End-use, By Region, And Segment Forecasts, 2023–2030. Available online: https://www.grandviewresearch.com/industry-analysis/dna-synthesis-market-report (accessed on 2 December 2022).
- LP Information Inc. Global DNA Synthesis Service Market Growth (Status and Outlook) 2022–2028. Available online: https://www.marketresearch.com/LP-Information-Inc-v4134/Global-DNA-Synthesis-Service-Growth-32230582/ (accessed on 2 December 2022).
- Yoo, H.; Jo, H.; Oh, S.S. Detection and beyond: Challenges and advances in aptamer-based biosensors. Mater. Adv. 2020, 1, 2663–2687. [Google Scholar] [CrossRef]
- Large Scale DNA Synthesis Services–Bio-Synthesis. Available online: https://www.biosyn.com/large-scale-dna-synthesis.aspx (accessed on 19 October 2022).
- Bielec, K.; Sozanski, K.; Seynen, M.; Dziekan, Z.; Ten Wolde, P.R.; Holyst, R. Kinetics and equilibrium constants of oligonucleotides at low concentrations. Hybridization and melting study. Phys. Chem. Chem. Phys. 2019, 21, 10798–10807. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Fu, G.; Kong, J.; Zhou, S.; Scafa, N.; Zhang, X. Advancement of nucleic acid biosensors based on morpholino. Am. J. Biomed. Sci. 2015, 7, 40–51. [Google Scholar] [CrossRef]
- Qavi, A.J.; Kindt, J.T.; Gleeson, M.A.; Bailey, R.C. Anti-DNA:RNA antibodies and silicon photonic microring resonators: Increased sensitivity for multiplexed microRNA detection. Anal. Chem. 2011, 83, 5949–5956. [Google Scholar] [CrossRef] [PubMed]
- Yousuf, S.; Kim, J.; Orozaliev, A.; Dahlem, M.S.; Song, Y.-A.; Viegas, J. Label-Free Detection of Morpholino-DNA Hybridization Using a Silicon Photonics Suspended Slab Micro-Ring Resonator. IEEE Photonics J. 2021, 13, 1–9. [Google Scholar] [CrossRef]
- Wages, J.M. NUCLEIC ACIDS | immunoassays. In Encyclopedia of Analytical Science; Elsevier: Amsterdam, The Netherlands, 2005; pp. 408–417. ISBN 9780123693976. [Google Scholar]
- Kosuri, S.; Church, G.M. Large-scale de novo DNA synthesis: Technologies and applications. Nat. Methods 2014, 11, 499–507. [Google Scholar] [CrossRef]
- DNA and RNA Molecular Weights and Conversions|Thermo Fisher Scientific—CA. Available online: https://www.thermofisher.com/ca/en/home/references/ambion-tech-support/rna-tools-and-calculators/dna-and-rna-molecular-weights-and-conversions.html (accessed on 22 October 2022).
- Mandelkern, M.; Elias, J.G.; Eden, D.; Crothers, D.M. The dimensions of DNA in solution. J. Mol. Biol. 1981, 152, 153–161. [Google Scholar] [CrossRef]
- PNA OLIGONUCLEOTIDES. Available online: https://www.biomers.net/Media/Preislisten/biomers_net_pna_pricelist_euro.pdf (accessed on 20 July 2022).
- Affinity Plus DNA & RNA Oligonucleotides. Available online: https://www.idtdna.com/pages/products/custom-dna-rna/dna-oligos/affinity-plus-dna-rna-oligonucleotides (accessed on 20 July 2022).
- GENE TOOLS PRICE LIST. Available online: https://www.gene-tools.com/sites/default/files/Price_list_01_Jul_2022.pdf (accessed on 19 July 2022).
- Zamora-Gálvez, A.; Morales-Narváez, E.; Mayorga-Martinez, C.C.; Merkoçi, A. Nanomaterials connected to antibodies and molecularly imprinted polymers as bio/receptors for bio/sensor applications. Appl. Mater. Today 2017, 9, 387–401. [Google Scholar] [CrossRef]
- BelBruno, J.J. Molecularly Imprinted Polymers. Chem. Rev. 2019, 119, 94–119. [Google Scholar] [CrossRef] [PubMed]
- Pavan, S.; Berti, F. Short peptides as biosensor transducers. Anal. Bioanal. Chem. 2012, 402, 3055–3070. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.L.S. Lectin biosensors in cancer glycan biomarker detection. Adv. Clin. Chem. 2019, 93, 1–61. [Google Scholar] [CrossRef]
- Van Breedam, W.; Pöhlmann, S.; Favoreel, H.W.; de Groot, R.J.; Nauwynck, H.J. Bitter-sweet symphony: Glycan-lectin interactions in virus biology. FEMS Microbiol. Rev. 2014, 38, 598–632. [Google Scholar] [CrossRef]
- Ward, E.M.; Kizer, M.E.; Imperiali, B. Strategies and Tactics for the Development of Selective Glycan-Binding Proteins. ACS Chem. Biol. 2021, 16, 1795–1813. [Google Scholar] [CrossRef]
- Hideshima, S.; Hayashi, H.; Hinou, H.; Nambuya, S.; Kuroiwa, S.; Nakanishi, T.; Momma, T.; Nishimura, S.-I.; Sakoda, Y.; Osaka, T. Glycan-immobilized dual-channel field effect transistor biosensor for the rapid identification of pandemic influenza viral particles. Sci. Rep. 2019, 9, 11616. [Google Scholar] [CrossRef]
- Lim, S.Y.; Ng, B.H.; Li, S.F.Y. Glycans in blood as biomarkers for forensic applications. TrAC Trends in Analytical Chemistry 2020, 133, 116084. [Google Scholar] [CrossRef]
- Shang, J.; Cheng, F.; Dubey, M.; Kaplan, J.M.; Rawal, M.; Jiang, X.; Newburg, D.S.; Sullivan, P.A.; Andrade, R.B.; Ratner, D.M. An organophosphonate strategy for functionalizing silicon photonic biosensors. Langmuir 2012, 28, 3338–3344. [Google Scholar] [CrossRef]
- Seeberger, P.H.; Overkleeft, H.S. Chapter 53: Chemical synthesis of glycans and glycoconjugates. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015. [Google Scholar]
- Mulloy, B.; Dell, A.; Stanley, P.; Prestegard, J.H. Structural analysis of glycans. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015. [Google Scholar]
- Zhang, Q.; Li, Z.; Song, X. Preparation of complex glycans from natural sources for functional study. Front. Chem. 2020, 8, 508. [Google Scholar] [CrossRef]
- Lam, S.K.; Ng, T.B. Lectins: Production and practical applications. Appl. Microbiol. Biotechnol. 2011, 89, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Yaghoubi, M.; Rahimi, F.; Negahdari, B.; Rezayan, A.H.; Shafiekhani, A. A lectin-coupled porous silicon-based biosensor: Label-free optical detection of bacteria in a real-time mode. Sci. Rep. 2020, 10, 16017. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, F.; Eftekhar, A.A.; Gottfried, D.S.; Song, X.; Cummings, R.D.; Adibi, A. Self-referenced silicon nitride array microring biosensor for toxin detection using glycans at visible wavelength. In Nanoscale Imaging, Sensing, and Actuation for Biomedical Applications X; Cartwright, A.N., Nicolau, D.V., Eds.; SPIE Proceedings; SPIE: Bellingham, WA, USA, 2013; Volume 8594. [Google Scholar]
- Ghasemi, F.; Hosseini, E.S.; Song, X.; Gottfried, D.S.; Chamanzar, M.; Raeiszadeh, M.; Cummings, R.D.; Eftekhar, A.A.; Adibi, A. Multiplexed detection of lectins using integrated glycan-coated microring resonators. Biosens. Bioelectron. 2016, 80, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Lectins and Other Carbohydrate-Binding Proteins—Section 7.7|Thermo Fisher Scientific—CA. Available online: https://www.thermofisher.com/ca/en/home/references/molecular-probes-the-handbook/antibodies-avidins-lectins-and-related-products/lectins-and-other-carbohydrate-binding-proteins.html (accessed on 21 July 2022).
- Glycan|Sigma-Aldrich. Available online: https://www.sigmaaldrich.com/CA/en/search/glycan?focus=products&page=1&perpage=30&sort=relevance&term=glycan&type=product_namewww.sigmaaldrich.com/CA/en/search/lectin. (accessed on 24 May 2022).
- Available online: https://www.sigmaaldrich.com/CA/en/search/lectin?focus=products&page=1&perpage=30&sort=relevance&term=lectin&type=product_name (accessed on 22 October 2022).
- Layouni, R.; Dubrovsky, M.; Bao, M.; Chung, H.; Du, K.; Boriskina, S.V.; Weiss, S.M.; Vermeulen, D. High contrast cleavage detection for enhancing porous silicon sensor sensitivity. Opt. Express 2021, 29, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dubrovsky, M.; Blevins, M.; Boriskina, S.V.; Vermeulen, D. High contrast cleavage detection. Opt. Lett. 2021, 46, 2593–2596. [Google Scholar] [CrossRef]
- Liu, L.; Dubrovsky, M.; Gundavarapu, S.; Vermeulen, D.; Du, K. Viral nucleic acid detection with CRISPR-Cas12a using high contrast cleavage detection on micro-ring resonator biosensors. In Frontiers in Biological Detection: From Nanosensors to Systems XIII; Miller, B.L., Weiss, S.M., Danielli, A., Eds.; SPIE: Bellingham, WA, USA, 2021; p. 8. [Google Scholar]
- Alt-R CRISPR-Cas12a (Cpf1) Genome Editing. Available online: https://www.idtdna.com/pages/products/crispr-genome-editing/alt-r-crispr-cpf1-genome-editing (accessed on 19 October 2022).
- Gold Nanoparticles: Properties and Applications. Available online: https://www.sigmaaldrich.com/CA/en/technical-documents/technical-article/materials-science-and-engineering/biosensors-and-imaging/gold-nanoparticles (accessed on 7 December 2022).
- Quantum Dots. Available online: https://www.sigmaaldrich.com/CA/en/technical-documents/technical-article/materials-science-and-engineering/biosensors-and-imaging/quantum-dots (accessed on 7 December 2022).
- Koo, B.; Kim, D.-E.; Kweon, J.; Jin, C.E.; Kim, S.-H.; Kim, Y.; Shin, Y. CRISPR/dCas9-mediated biosensor for detection of tick-borne diseases. Sens. Actuators B Chem. 2018, 273, 316–321. [Google Scholar] [CrossRef]
- Start Genome Editing with CRISPR-Cas9|IDT. Available online: https://www.idtdna.com/pages/products/crispr-genome-editing/alt-r-crispr-cas9-system (accessed on 19 October 2022).
- Kaminski, M.M.; Abudayyeh, O.O.; Gootenberg, J.S.; Zhang, F.; Collins, J.J. CRISPR-based diagnostics. Nat. Biomed. Eng. 2021, 5, 643–656. [Google Scholar] [CrossRef]
- Rosser, A.; Rollinson, D.; Forrest, M.; Webster, B.L. Isothermal Recombinase Polymerase amplification (RPA) of Schistosoma haematobium DNA and oligochromatographic lateral flow detection. Parasit. Vectors 2015, 8, 446. [Google Scholar] [CrossRef]
- Daher, R.K.; Stewart, G.; Boissinot, M.; Bergeron, M.G. Recombinase polymerase amplification for diagnostic applications. Clin. Chem. 2016, 62, 947–958. [Google Scholar] [CrossRef]
- Medfisch, S.M.; Muehl, E.M.; Morrissey, J.H.; Bailey, R.C. Phosphatidylethanolamine-phosphatidylserine binding synergy of seven coagulation factors revealed using Nanodisc arrays on silicon photonic sensors. Sci. Rep. 2020, 10, 17407. [Google Scholar] [CrossRef]
- Muehl, E.M.; Gajsiewicz, J.M.; Medfisch, S.M.; Wiersma, Z.S.B.; Morrissey, J.H.; Bailey, R.C. Multiplexed silicon photonic sensor arrays enable facile characterization of coagulation protein binding to nanodiscs with variable lipid content. J. Biol. Chem. 2017, 292, 16249–16256. [Google Scholar] [CrossRef] [PubMed]
- Sloan, C.D.K.; Marty, M.T.; Sligar, S.G.; Bailey, R.C. Interfacing lipid bilayer nanodiscs and silicon photonic sensor arrays for multiplexed protein-lipid and protein-membrane protein interaction screening. Anal. Chem. 2013, 85, 2970–2976. [Google Scholar] [CrossRef] [PubMed]
- Denisov, I.G.; Sligar, S.G. Nanodiscs for structural and functional studies of membrane proteins. Nat. Struct. Mol. Biol. 2016, 23, 481–486. [Google Scholar] [CrossRef]
- Nanodisc Products Protein Research|Cube Biotech. Available online: https://cube-biotech.com/us/products/nanodisc-products/ (accessed on 19 October 2022).
- Borrebaeck, C.A. Antibodies in diagnostics—From immunoassays to protein chips. Immunol. Today 2000, 21, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr.; Travers, P.; Walport, M.; Shlomchik, M.J. The structure of a typical antibody molecule. In Immunobiology: The Immune System in Health and Disease; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Antibody production|Abcam. Available online: https://www.abcam.com/protocols/antibody-production (accessed on 7 November 2022).
- Marx, V. Finding the right antibody for the job. Nat. Methods 2013, 10, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Frenzel, A.; Hust, M.; Schirrmann, T. Expression of recombinant antibodies. Front. Immunol. 2013, 4, 217. [Google Scholar] [CrossRef]
- Grand View Research Antibody Fragments Market Size, Share & Trends Analysis Report by Specificity (Monoclonal Antibodies, Polyclonal Antibodies), by Type, by Therapy, by Application, by Region, and Segment Forecasts, 2022–2030. Available online: https://www.grandviewresearch.com/industry-analysis/antibody-fragments-market-report (accessed on 6 December 2022).
- Transparency Market Research Antibody Fragments Market Trend Shows a Rapid Growth by 2024 according to New Research Report|BioSpace. Available online: https://www.biospace.com/article/antibody-fragments-market-trend-shows-a-rapid-growth-by-2024-according-to-new-research-report/ (accessed on 5 December 2022).
- Cox, K.L.; Devanarayan, V.; Kriauciunas, A.; Manetta, J.; Montrose, C.; Sittampalam, S. Immunoassay Methods. In Assay Guidance Manual; Sittampalam, G.S., Coussens, N.P., Nelson, H., Arkin, M., Auld, D., Austin, C., Bejcek, B., Glicksman, M., Inglese, J., Iversen, P.W., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Washburn, A.L.; Gunn, L.C.; Bailey, R.C. Label-free quantitation of a cancer biomarker in complex media using silicon photonic microring resonators. Anal. Chem. 2009, 81, 9499–9506. [Google Scholar] [CrossRef]
- Kindt, J.T.; Luchansky, M.S.; Qavi, A.J.; Lee, S.-H.; Bailey, R.C. Subpicogram per milliliter detection of interleukins using silicon photonic microring resonators and an enzymatic signal enhancement strategy. Anal. Chem. 2013, 85, 10653–10657. [Google Scholar] [CrossRef]
- Washburn, A.L.; Shia, W.W.; Lenkeit, K.A.; Lee, S.-H.; Bailey, R.C. Multiplexed cancer biomarker detection using chip-integrated silicon photonic sensor arrays. Analyst 2016, 141, 5358–5365. [Google Scholar] [CrossRef]
- Christenson, C.; Baryeh, K.; Ahadian, S.; Nasiri, R.; Dokmeci, M.R.; Goudie, M.; Khademhosseini, A.; Ye, J.Y. Enhancement of label-free biosensing of cardiac troponin I. In Label-Free Biomedical Imaging and Sensing (LBIS); SPIE: Bellingham, WA, USA, 2020; Volume 11251. [Google Scholar] [CrossRef]
- Zhang, B.; Tamez-Vela, J.M.; Solis, S.; Bustamante, G.; Peterson, R.; Rahman, S.; Morales, A.; Tang, L.; Ye, J.Y. Detection of Myoglobin with an Open-Cavity-Based Label-Free Photonic Crystal Biosensor. J. Med. Eng. 2013, 2013, 808056. [Google Scholar] [CrossRef]
- Arnfinnsdottir, N.B.; Chapman, C.A.; Bailey, R.C.; Aksnes, A.; Stokke, B.T. Impact of silanization parameters and antibody immobilization strategy on binding capacity of photonic ring resonators. Sensors 2020, 20, 3163. [Google Scholar] [CrossRef] [PubMed]
- Cognetti, J.S.; Miller, B.L. Monitoring Serum Spike Protein with Disposable Photonic Biosensors Following SARS-CoV-2 Vaccination. Sensors 2021, 21, 5857. [Google Scholar] [CrossRef] [PubMed]
- Shia, W.W.; Bailey, R.C. Single domain antibodies for the detection of ricin using silicon photonic microring resonator arrays. Anal. Chem. 2013, 85, 805–810. [Google Scholar] [CrossRef] [PubMed]
- McClellan, M.S.; Domier, L.L.; Bailey, R.C. Label-free virus detection using silicon photonic microring resonators. Biosens. Bioelectron. 2012, 31, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Yadav, A.R.; Lifson, M.A.; Baker, J.E.; Fauchet, P.M.; Miller, B.L. Selective virus detection in complex sample matrices with photonic crystal optical cavities. Biosens. Bioelectron. 2013, 44, 229–234. [Google Scholar] [CrossRef]
- Griol, A.; Peransi, S.; Rodrigo, M.; Hurtado, J.; Bellieres, L.; Ivanova, T.; Zurita, D.; Sánchez, C.; Recuero, S.; Hernández, A.; et al. Design and development of photonic biosensors for swine viral diseases detection. Sensors 2019, 19, 3985. [Google Scholar] [CrossRef]
- Ramachandran, A.; Wang, S.; Clarke, J.; Ja, S.J.; Goad, D.; Wald, L.; Flood, E.M.; Knobbe, E.; Hryniewicz, J.V.; Chu, S.T.; et al. A universal biosensing platform based on optical micro-ring resonators. Biosens. Bioelectron. 2008, 23, 939–944. [Google Scholar] [CrossRef]
- Yoshimoto, K.; Nishio, M.; Sugasawa, H.; Nagasaki, Y. Direct observation of adsorption-induced inactivation of antibody fragments surrounded by mixed-PEG layer on a gold surface. J. Am. Chem. Soc. 2010, 132, 7982–7989. [Google Scholar] [CrossRef]
- Byeon, J.-Y.; Bailey, R.C. Multiplexed evaluation of capture agent binding kinetics using arrays of silicon photonic microring resonators. Analyst 2011, 136, 3430–3433. [Google Scholar] [CrossRef][Green Version]
- Xu, J.; Suarez, D.; Gottfried, D.S. Detection of avian influenza virus using an interferometric biosensor. Anal. Bioanal. Chem. 2007, 389, 1193–1199. [Google Scholar] [CrossRef]
- Chhasatia, R.; Sweetman, M.J.; Harding, F.J.; Waibel, M.; Kay, T.; Thomas, H.; Loudovaris, T.; Voelcker, N.H. Non-invasive, in vitro analysis of islet insulin production enabled by an optical porous silicon biosensor. Biosens. Bioelectron. 2017, 91, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Barrios, C.A.; Bañuls, M.J.; González-Pedro, V.; Gylfason, K.B.; Sánchez, B.; Griol, A.; Maquieira, A.; Sohlström, H.; Holgado, M.; Casquel, R. Label-free optical biosensing with slot-waveguides. Opt. Lett. 2008, 33, 708–710. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Bandrowski, A.; Carr, S.; Edwards, A.; Ellenberg, J.; Lundberg, E.; Rimm, D.L.; Rodriguez, H.; Hiltke, T.; Snyder, M.; et al. A proposal for validation of antibodies. Nat. Methods 2016, 13, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Bunka, D.H.J.; Stockley, P.G. Aptamers come of age—At last. Nat. Rev. Microbiol. 2006, 4, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Kohlberger, M.; Gadermaier, G. SELEX: Critical factors and optimization strategies for successful aptamer selection. Biotechnol. Appl. Biochem. 2022, 69, 1771–1792. [Google Scholar] [CrossRef] [PubMed]
- Cambio—Excellence in Molecular Biology—Aptamers. Available online: https://www.cambio.co.uk/products/apps/?id=28 (accessed on 27 May 2022).
- Custom Oligos. Available online: https://www.idtdna.com/pages/products/custom-dna-rna (accessed on 27 May 2022).
- Bernhardt, H.S.; Tate, W.P. Primordial soup or vinaigrette: Did the RNA world evolve at acidic pH? Biol. Direct 2012, 7, 4. [Google Scholar] [CrossRef]
- Schasfoort, R.B.M. Chapter 1. introduction to surface plasmon resonance. In Handbook of Surface Plasmon Resonance; Schasfoort, R.B.M., Ed.; Royal Society of Chemistry: Cambridge, UK, 2017; pp. 1–26. ISBN 978-1-78262-730-2. [Google Scholar]
- Guider, R.; Gandolfi, D.; Chalyan, T.; Pasquardini, L.; Samusenko, A.; Pederzolli, C.; Pucker, G.; Pavesi, L. Sensitivity and Limit of Detection of biosensors based on ring resonators. Sens. Bio-Sens. Res. 2015, 6, 99–102. [Google Scholar] [CrossRef]
- Kusser, W. Chemically modified nucleic acid aptamers for in vitro selections: Evolving evolution. Rev. Mol. Biotechnol. 2000, 74, 27–38. [Google Scholar] [CrossRef]
- Wang, T.; Chen, C.; Larcher, L.M.; Barrero, R.A.; Veedu, R.N. Three decades of nucleic acid aptamer technologies: Lessons learned, progress and opportunities on aptamer development. Biotechnol. Adv. 2019, 37, 28–50. [Google Scholar] [CrossRef]
- Paniel, N.; Baudart, J.; Hayat, A.; Barthelmebs, L. Aptasensor and genosensor methods for detection of microbes in real world samples. Methods 2013, 64, 229–240. [Google Scholar] [CrossRef]
- Wang, S.; Kool, E.T. Origins of the large differences in stability of DNA and RNA helices: C-5 methyl and 2′-hydroxyl effects. Biochemistry 1995, 34, 4125–4132. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.A.; Ellington, A.D. Synthetic DNA synthesis and assembly: Putting the synthetic in synthetic biology. Cold Spring Harb. Perspect. Biol. 2017, 9, a023812. [Google Scholar] [CrossRef] [PubMed]
- Braasch, D.A.; Corey, D.R. Locked nucleic acid (LNA): Fine-tuning the recognition of DNA and RNA. Chem. Biol. 2001, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Tu, X.; Kim, K.W.; Kee, J.S.; Shin, Y.; Han, K.; Yoon, Y.-J.; Lo, G.-Q.; Park, M.K. Highly sensitive Mach–Zehnder interferometer biosensor based on silicon nitride slot waveguide. Sens. Actuators B Chem. 2013, 188, 681–688. [Google Scholar] [CrossRef]
- Lai, Q.; Chen, W.; Zhang, Y.; Liu, Z. Application strategies of peptide nucleic acids toward electrochemical nucleic acid sensors. Analyst 2021, 146, 5822–5835. [Google Scholar] [CrossRef]
- Qavi, A.J.; Bailey, R.C. Multiplexed detection and label-free quantitation of microRNAs using arrays of silicon photonic microring resonators. Angew. Chem. Int. Ed. 2010, 49, 4608–4611. [Google Scholar] [CrossRef]
- Graybill, R.M.; Cardenosa-Rubio, M.C.; Yang, H.; Johnson, M.D.; Bailey, R.C. Multiplexed microRNA Expression Profiling by Combined Asymmetric PCR and Label-Free Detection using Silicon Photonic Sensor Arrays. Anal. Methods 2018, 10, 1618–1623. [Google Scholar] [CrossRef]
- Kindt, J.T.; Bailey, R.C. Chaperone probes and bead-based enhancement to improve the direct detection of mRNA using silicon photonic sensor arrays. Anal. Chem. 2012, 84, 8067–8074. [Google Scholar] [CrossRef]
- Shin, Y.; Perera, A.P.; Park, M.K. Label-free DNA sensor for detection of bladder cancer biomarkers in urine. Sens. Actuators B Chem. 2013, 178, 200–206. [Google Scholar] [CrossRef]
- Scheler, O.; Kindt, J.T.; Qavi, A.J.; Kaplinski, L.; Glynn, B.; Barry, T.; Kurg, A.; Bailey, R.C. Label-free, multiplexed detection of bacterial tmRNA using silicon photonic microring resonators. Biosens. Bioelectron. 2012, 36, 56–61. [Google Scholar] [CrossRef]
- Liu, Q.; Lim, B.K.L.; Lim, S.Y.; Tang, W.Y.; Gu, Z.; Chung, J.; Park, M.K.; Barkham, T. Label-free, real-time and multiplex detection of Mycobacterium tuberculosis based on silicon photonic microring sensors and asymmetric isothermal amplification technique (SPMS-AIA). Sens. Actuators B Chem. 2017, 255, 1595–1603. [Google Scholar] [CrossRef]
- Sepúlveda, B.; del Río, J.S.; Moreno, M.; Blanco, F.J.; Mayora, K.; Domínguez, C.; Lechuga, L.M. Optical biosensor microsystems based on the integration of highly sensitive Mach–Zehnder interferometer devices. J. Opt. A Pure Appl. Opt. 2006, 8, S561–S566. [Google Scholar] [CrossRef]
- Hu, S.; Zhao, Y.; Qin, K.; Retterer, S.T.; Kravchenko, I.I.; Weiss, S.M. Enhancing the Sensitivity of Label-Free Silicon Photonic Biosensors through Increased Probe Molecule Density. ACS Photonics 2014, 1, 590–597. [Google Scholar] [CrossRef]
- Peserico, N.; Castagna, R.; Bellieres, L.; Rodrigo, M.; Melloni, A. Tip-mould microcontact printing for functionalisation of optical microring resonator. IET Nanobiotechnol. 2018, 12, 87–91. [Google Scholar] [CrossRef]
- Toccafondo, V.; García-Rupérez, J.; Bañuls, M.J.; Griol, A.; Castelló, J.G.; Peransi-Llopis, S.; Maquieira, A. Single-strand DNA detection using a planar photonic-crystal-waveguide-based sensor. Opt. Lett. 2010, 35, 3673–3675. [Google Scholar] [CrossRef] [PubMed]
- Rashid, J.I.A.; Yusof, N.A. The strategies of DNA immobilization and hybridization detection mechanism in the construction of electrochemical DNA sensor: A review. Sens. Bio-Sens. Res. 2017, 16, 19–31. [Google Scholar] [CrossRef]
- Zhu, B.; Travas-Sejdic, J. PNA versus DNA in electrochemical gene sensing based on conducting polymers: Study of charge and surface blocking effects on the sensor signal. Analyst 2018, 143, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Kaisti, M.; Kerko, A.; Aarikka, E.; Saviranta, P.; Boeva, Z.; Soukka, T.; Lehmusvuori, A. Real-time wash-free detection of unlabeled PNA-DNA hybridization using discrete FET sensor. Sci. Rep. 2017, 7, 15734. [Google Scholar] [CrossRef] [PubMed]
- El-Schich, Z.; Zhang, Y.; Feith, M.; Beyer, S.; Sternbæk, L.; Ohlsson, L.; Stollenwerk, M.; Wingren, A.G. Molecularly imprinted polymers in biological applications. BioTechniques 2020, 69, 406–419. [Google Scholar] [CrossRef] [PubMed]
- Ertürk, G.; Mattiasson, B. Molecular imprinting techniques used for the preparation of biosensors. Sensors 2017, 17, 288. [Google Scholar] [CrossRef]
- Ahmad, O.S.; Bedwell, T.S.; Esen, C.; Garcia-Cruz, A.; Piletsky, S.A. Molecularly imprinted polymers in electrochemical and optical sensors. Trends Biotechnol. 2019, 37, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Wackerlig, J.; Schirhagl, R. Applications of Molecularly Imprinted Polymer Nanoparticles and Their Advances toward Industrial Use: A Review. Anal. Chem. 2016, 88, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Hammond, G.D.; Vojta, A.L.; Grant, S.A.; Hunt, H.K. Integrating Nanostructured Artificial Receptors with Whispering Gallery Mode Optical Microresonators via Inorganic Molecular Imprinting Techniques. Biosensors 2016, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Y.; Shen, X.; Chang, Z.; Tang, L.; Dong, W.-F.; Li, M.; He, J.-J. Ultrasensitive Detection of Testosterone Using Microring Resonator with Molecularly Imprinted Polymers. Sensors 2015, 15, 31558–31565. [Google Scholar] [CrossRef]
- Eisner, L.; Wilhelm, I.; Flachenecker, G.; Hürttlen, J.; Schade, W. Molecularly Imprinted Sol-Gel for TNT Detection with Optical Micro-Ring Resonator Sensor Chips. Sensors 2019, 19, 3909. [Google Scholar] [CrossRef] [PubMed]
- Farrell, M.E.; Coppock, M.B.; Holthoff, E.L.; Pellegrino, P.M.; Bickford, J.R.; Cho, P.S. Towards Army Relevant Sensing with Integrated Molecularly Imprinted Polymer Photonic (IMIPP) Devices. In Proceedings of the 2018 IEEE Research and Applications of Photonics In Defense Conference (RAPID), Miramar Beach, FL, USA, 22–24 August 2018; pp. 1–4. [Google Scholar]
- Farrell, M.E.; Holthoff, E.L.; Bickford, J.; Cho, P.S.; Pellegrino, P.M. Development of army relevant integrated photonics MIP platform. In Smart Biomedical and Physiological Sensor Technology XVI; Cullum, B.M., McLamore, E.S., Kiehl, D., Eds.; SPIE: Bellingham, WA, USA, 2019; p. 13. [Google Scholar]
- Lorenzo, R.A.; Carro, A.M.; Alvarez-Lorenzo, C.; Concheiro, A. To remove or not to remove? The challenge of extracting the template to make the cavities available in Molecularly Imprinted Polymers (MIPs). Int. J. Mol. Sci. 2011, 12, 4327–4347. [Google Scholar] [CrossRef] [PubMed]
- Han, X.-Y.; Wu, Z.-L.; Yang, S.-C.; Shen, F.-F.; Liang, Y.-X.; Wang, L.-H.; Wang, J.-Y.; Ren, J.; Jia, L.-Y.; Zhang, H.; et al. Recent progress of imprinted polymer photonic waveguide devices and applications. Polymers 2018, 10, 603. [Google Scholar] [CrossRef]
- Refaat, D.; Aggour, M.G.; Farghali, A.A.; Mahajan, R.; Wiklander, J.G.; Nicholls, I.A.; Piletsky, S.A. Strategies for Molecular Imprinting and the Evolution of MIP Nanoparticles as Plastic Antibodies-Synthesis and Applications. Int. J. Mol. Sci. 2019, 20, 6304. [Google Scholar] [CrossRef]
- Xie, Z.; Cao, Z.; Liu, Y.; Zhang, Q.; Zou, J.; Shao, L.; Wang, Y.; He, J.; Li, M. Highly-sensitive optical biosensor based on equal FSR cascaded microring resonator with intensity interrogation for detection of progesterone molecules. Opt. Express 2017, 25, 33193. [Google Scholar] [CrossRef]
- Marfà, J.; Pupin, R.R.; Sotomayor, M.P.T.; Pividori, M.I. Magnetic-molecularly imprinted polymers in electrochemical sensors and biosensors. Anal. Bioanal. Chem. 2021, 413, 6141–6157. [Google Scholar] [CrossRef]
- Kupai, J.; Razali, M.; Buyuktiryaki, S.; Kecili, R.; Szekely, G. Long-term stability and reusability of molecularly imprinted polymers. Polym. Chem. 2017, 8, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, I.A.; Andersson, H.S.; Charlton, C.; Henschel, H.; Karlsson, B.C.G.; Karlsson, J.G.; O’Mahony, J.; Rosengren, A.M.; Rosengren, K.J.; Wikman, S. Theoretical and computational strategies for rational molecularly imprinted polymer design. Biosens. Bioelectron. 2009, 25, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Umpleby, R.J.; Baxter, S.C.; Chen, Y.; Shah, R.N.; Shimizu, K.D. Characterization of Molecularly Imprinted Polymers with the Langmuir−Freundlich Isotherm. Anal. Chem. 2001, 73, 4584–4591. [Google Scholar] [CrossRef] [PubMed]
- Ndunda, E.N. Molecularly imprinted polymers-A closer look at the control polymer used in determining the imprinting effect: A mini review. J. Mol. Recognit. 2020, 33, e2855. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.M.; Hawkins, D.M.; Phan, Q.T.; Stevenson, D.; Warriner, K. Protein detection using hydrogel-based molecularly imprinted polymers integrated with dual polarisation interferometry. Sens. Actuators B Chem. 2013, 176, 190–197. [Google Scholar] [CrossRef]
- Harris, L.J.; Skaletsky, E.; McPherson, A. Crystallographic structure of an intact IgG1 monoclonal antibody. J. Mol. Biol. 1998, 275, 861–872. [Google Scholar] [CrossRef]
- Carothers, J.M.; Davis, J.H.; Chou, J.J.; Szostak, J.W. Solution structure of an informationally complex high-affinity RNA aptamer to GTP. RNA 2006, 12, 567–579. [Google Scholar] [CrossRef]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, E.A.; Abbott, W.M. Expression of recombinant proteins in insect and mammalian cells. Methods 2018, 147, 40–49. [Google Scholar] [CrossRef]
- Chandrudu, S.; Simerska, P.; Toth, I. Chemical methods for peptide and protein production. Molecules 2013, 18, 4373–4388. [Google Scholar] [CrossRef]
- Merrifield, R.B. Solid phase synthesis (nobel lecture). Angew. Chem. Int. Ed. Engl. 1985, 24, 799–810. [Google Scholar] [CrossRef]
- Tian, J.; Li, Y.; Ma, B.; Tan, Z.; Shang, S. Automated peptide synthesizers and glycoprotein synthesis. Front. Chem. 2022, 10, 896098. [Google Scholar] [CrossRef]
- Monty, O.B.C.; Simmons, N.; Chamakuri, S.; Matzuk, M.M.; Young, D.W. Solution-Phase Fmoc-Based Peptide Synthesis for DNA-Encoded Chemical Libraries: Reaction Conditions, Protecting Group Strategies, and Pitfalls. ACS Comb. Sci. 2020, 22, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Agnew, H.D.; Coppock, M.B.; Idso, M.N.; Lai, B.T.; Liang, J.; McCarthy-Torrens, A.M.; Warren, C.M.; Heath, J.R. Protein-Catalyzed Capture Agents. Chem. Rev. 2019, 119, 9950–9970. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Ó’Fágáin, C.; O’Kennedy, R. Antibody stability: A key to performance—Analysis, influences and improvement. Biochimie 2020, 177, 213–225. [Google Scholar] [CrossRef]
- Martínez-Ceron, M.C.; Giudicessi, S.L.; Saavedra, S.L.; Gurevich-Messina, J.M.; Erra-Balsells, R.; Albericio, F.; Cascone, O.; Camperi, S.A. Latest advances in OBOC peptide libraries. improvements in screening strategies and enlarging the family from linear to cyclic libraries. Curr. Pharm. Biotechnol. 2016, 17, 449–457. [Google Scholar] [CrossRef]
- Bozovičar, K.; Bratkovič, T. Evolving a peptide: Library platforms and diversification strategies. Int. J. Mol. Sci. 2019, 21, 215. [Google Scholar] [CrossRef]
- Angelopoulou, M.; Makarona, E.; Salapatas, A.; Misiakos, K.; Synolaki, E.; Ioannidis, A.; Chatzipanagiotou, S.; Ritvos, M.A.; Pasternack, A.; Ritvos, O.; et al. Directly immersible silicon photonic probes: Application to rapid SARS-CoV-2 serological testing. Biosens. Bioelectron. 2022, 215, 114570. [Google Scholar] [CrossRef]
- Martucci, N.M.; Rea, I.; Ruggiero, I.; Terracciano, M.; De Stefano, L.; Migliaccio, N.; Palmieri, C.; Scala, G.; Arcari, P.; Rendina, I.; et al. A new strategy for label-free detection of lymphoma cancer cells. Biomed. Opt. Express 2015, 6, 1353–1362. [Google Scholar] [CrossRef]
- Cao, T.; Layouni, R.; Coppock, M.B.; Laibinis, P.E.; Weiss, S.M. Use of peptide capture agents in porous silicon biosensors. In Frontiers in Biological Detection: From Nanosensors to Systems XII; Miller, B.L., Weiss, S.M., Danielli, A., Eds.; SPIE: Bellingham, WA, USA, 2020; p. 21. [Google Scholar]
- Lamberti, A.; Sanges, C.; Migliaccio, N.; De Stefano, L.; Rea, I.; Orabona, E.; Scala, G.; Rendina, I.; Arcari, P. Silicon-Based Technology for Ligand-Receptor Molecular Identification. J. At. Mol. Opt. Phys. 2012, 2012, 948390. [Google Scholar] [CrossRef]
- Chen, S.; Liu, L.; Zhou, J.; Jiang, S. Controlling Antibody Orientation on Charged Self-Assembled Monolayers. Langmuir 2003, 19, 2859–2864. [Google Scholar] [CrossRef]
- Coppock, M.B.; Warner, C.R.; Dorsey, B.; Orlicki, J.A.; Sarkes, D.A.; Lai, B.T.; Pitram, S.M.; Rohde, R.D.; Malette, J.; Wilson, J.A.; et al. Protein catalyzed capture agents with tailored performance for in vitro and in vivo applications. Biopolymers 2017, 108, e22934. [Google Scholar] [CrossRef] [PubMed]
- Belický, Š.; Katrlík, J.; Tkáč, J. Glycan and lectin biosensors. Essays Biochem. 2016, 60, 37–47. [Google Scholar] [CrossRef]
- Overkleeft, H.S.; Seeberger, P.H. Chapter 54: Chemoenzymatic synthesis of glycans and glycoconjugates. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015. [Google Scholar]
- Kirk, J.T.; Fridley, G.E.; Chamberlain, J.W.; Christensen, E.D.; Hochberg, M.; Ratner, D.M. Multiplexed inkjet functionalization of silicon photonic biosensors. Lab Chip 2011, 11, 1372–1377. [Google Scholar] [CrossRef] [PubMed]
- Van Landuyt, L.; Lonigro, C.; Meuris, L.; Callewaert, N. Customized protein glycosylation to improve biopharmaceutical function and targeting. Curr. Opin. Biotechnol. 2019, 60, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Li, Y.; Xu, F.; Yang, X. Powerful CRISPR-Based Biosensing Techniques and Their Integration With Microfluidic Platforms. Front. Bioeng. Biotechnol. 2022, 10, 851712. [Google Scholar] [CrossRef]
- Li, Y.; Li, S.; Wang, J.; Liu, G. CRISPR/Cas Systems towards Next-Generation Biosensing. Trends Biotechnol. 2019, 37, 730–743. [Google Scholar] [CrossRef] [PubMed]
- Leung, R.K.-K.; Cheng, Q.-X.; Wu, Z.-L.; Khan, G.; Liu, Y.; Xia, H.-Y.; Wang, J. CRISPR-Cas12-based nucleic acids detection systems. Methods 2021, 203, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Park, J.; Boriskina, S.V. Inverse-designed waveguide-based biosensor for high-sensitivity, single-frequency detection of biomolecules. Nanophotonics 2022, 11, 1427–1442. [Google Scholar] [CrossRef]
- Lobato, I.M.; O’Sullivan, C.K. Recombinase polymerase amplification: Basics, applications and recent advances. Trends Analyt. Chem. 2018, 98, 19–35. [Google Scholar] [CrossRef]
- Bruch, R.; Urban, G.A.; Dincer, C. Crispr/cas powered multiplexed biosensing. Trends Biotechnol. 2019, 37, 791–792. [Google Scholar] [CrossRef] [PubMed]
- McCarty, N.S.; Graham, A.E.; Studená, L.; Ledesma-Amaro, R. Multiplexed CRISPR technologies for gene editing and transcriptional regulation. Nat. Commun. 2020, 11, 1281. [Google Scholar] [CrossRef] [PubMed]
- Vatankhah, M.; Azizi, A.; Sanajouyan Langeroudi, A.; Ataei Azimi, S.; Khorsand, I.; Kerachian, M.A.; Motaei, J. CRISPR-based biosensing systems: A way to rapidly diagnose COVID-19. Crit. Rev. Clin. Lab. Sci. 2021, 58, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.A.; Denisov, I.G.; Sligar, S.G. Nanodiscs as a new tool to examine lipid-protein interactions. Methods Mol. Biol. 2013, 974, 415–433. [Google Scholar] [CrossRef]
- Morita, M.; Ohmi, T.; Hasegawa, E.; Kawakami, M.; Ohwada, M. Growth of native oxide on a silicon surface. J. Appl. Phys. 1990, 68, 1272–1281. [Google Scholar] [CrossRef]
- Aissaoui, N.; Bergaoui, L.; Landoulsi, J.; Lambert, J.-F.; Boujday, S. Silane layers on silicon surfaces: Mechanism of interaction, stability, and influence on protein adsorption. Langmuir 2012, 28, 656–665. [Google Scholar] [CrossRef]
- Coen, M.C.; Lehmann, R.; Gröning, P.; Bielmann, M.; Galli, C.; Schlapbach, L. Adsorption and bioactivity of protein A on silicon surfaces studied by AFM and XPS. J. Colloid Interface Sci. 2001, 233, 180–189. [Google Scholar] [CrossRef]
- Cuddy, M.F.; Poda, A.R.; Brantley, L.N. Determination of isoelectric points and the role of pH for common quartz crystal microbalance sensors. ACS Appl. Mater. Interfaces 2013, 5, 3514–3518. [Google Scholar] [CrossRef]
- Jönsson, U.; Malmqvist, M.; Rönnberg, I. Immobilization of immunoglobulins on silica surfaces. Stability. Biochem. J. 1985, 227, 363–371. [Google Scholar] [CrossRef]
- Lin, J.N.; Andrade, J.D.; Chang, I.N. The influence of adsorption of native and modified antibodies on their activity. J. Immunol. Methods 1989, 125, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Vashist, S.K.; Dixit, C.K.; MacCraith, B.D.; O’Kennedy, R. Effect of antibody immobilization strategies on the analytical performance of a surface plasmon resonance-based immunoassay. Analyst 2011, 136, 4431–4436. [Google Scholar] [CrossRef] [PubMed]
- Flueckiger, J.; Schmidt, S.; Donzella, V.; Sherwali, A.; Ratner, D.M.; Chrostowski, L.; Cheung, K.C. Sub-wavelength grating for enhanced ring resonator biosensor. Opt. Express 2016, 24, 15672–15686. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, G.T. Avidin–Biotin Systems. In Bioconjugate Techniques; Elsevier: Amsterdam, The Netherlands, 2008; pp. 900–923. ISBN 9780123705013. [Google Scholar]
- Yalcin, A.; Popat, K.C.; Aldridge, J.C.; Desai, T.A.; Hryniewicz, J.; Chbouki, N.; Little, B.E.; King, O.; Van, V.; Chu, S.; et al. Optical sensing of biomolecules using microring resonators. IEEE J. Select. Topics Quantum Electron. 2006, 12, 148–155. [Google Scholar] [CrossRef]
- Yadav, A.R.; Sriram, R.; Carter, J.A.; Miller, B.L. Comparative study of solution-phase and vapor-phase deposition of aminosilanes on silicon dioxide surfaces. Mater. Sci. Eng. C Mater. Biol. Appl. 2014, 35, 283–290. [Google Scholar] [CrossRef]
- Zhu, M.; Lerum, M.Z.; Chen, W. How to prepare reproducible, homogeneous, and hydrolytically stable aminosilane-derived layers on silica. Langmuir 2012, 28, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Hunt, H.K.; Soteropulos, C.; Armani, A.M. Bioconjugation strategies for microtoroidal optical resonators. Sensors 2010, 10, 9317–9336. [Google Scholar] [CrossRef]
- Cattani-Scholz, A.; Pedone, D.; Dubey, M.; Neppl, S.; Nickel, B.; Feulner, P.; Schwartz, J.; Abstreiter, G.; Tornow, M. Organophosphonate-based PNA-functionalization of silicon nanowires for label-free DNA detection. ACS Nano 2008, 2, 1653–1660. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Chen, R.; Surman, C.; Potyrailo, R.; Pris, A.; Holwitt, E.A.; Sorola, V.K.; Kiel, J.L. Immobilization of aptamers onto unmodified glass surfaces for affordable biosensors. In Photonic Microdevices/Microstructures for Sensing III; Xiao, H., Fan, X., Wang, A., Eds.; SPIE Proceedings; SPIE: Bellingham, WA, USA, 2011; Volume 8034, p. 803405. [Google Scholar]
- Gudnason, H.; Dufva, M.; Duong Bang, D.; Wolff, A. An inexpensive and simple method for thermally stable immobilization of DNA on an unmodified glass surface: UV linking of poly(T)10-poly(C)10-tagged DNA probes. BioTechniques 2008, 45, 261–271. [Google Scholar] [CrossRef]
- Antoniou, M.; Tsounidi, D.; Petrou, P.S.; Beltsios, K.G.; Kakabakos, S.E. Functionalization of silicon dioxide and silicon nitride surfaces with aminosilanes for optical biosensing applications. Med. Devices Sens. 2020, 3, e10072. [Google Scholar] [CrossRef]
- Anderson, T.H.; Min, Y.; Weirich, K.L.; Zeng, H.; Fygenson, D.; Israelachvili, J.N. Formation of supported bilayers on silica substrates. Langmuir 2009, 25, 6997–7005. [Google Scholar] [CrossRef]
- Goluch, E.D.; Shaw, A.W.; Sligar, S.G.; Liu, C. Microfluidic patterning of nanodisc lipid bilayers and multiplexed analysis of protein interaction. Lab Chip 2008, 8, 1723–1728. [Google Scholar] [CrossRef]
- Seo, J.; Lee, S.; Poulter, C.D. Regioselective covalent immobilization of recombinant antibody-binding proteins A, G, and L for construction of antibody arrays. J. Am. Chem. Soc. 2013, 135, 8973–8980. [Google Scholar] [CrossRef]
- Ikeda, T.; Hata, Y.; Ninomiya, K.-I.; Ikura, Y.; Takeguchi, K.; Aoyagi, S.; Hirota, R.; Kuroda, A. Oriented immobilization of antibodies on a silicon wafer using Si-tagged protein A. Anal. Biochem. 2009, 385, 132–137. [Google Scholar] [CrossRef]
- Choe, W.; Durgannavar, T.A.; Chung, S.J. Fc-Binding Ligands of Immunoglobulin G: An Overview of High Affinity Proteins and Peptides. Materials 2016, 9, 994. [Google Scholar] [CrossRef]
- Åkerström, B.; Björck, L. Protein L: An Immunoglobulin Light Chain-binding Bacterial Protein. J. Biol. Chem. 1989, 264, 19740–19746. [Google Scholar] [CrossRef]
- Taniguchi, K.; Nomura, K.; Hata, Y.; Nishimura, T.; Asami, Y.; Kuroda, A. The Si-tag for immobilizing proteins on a silica surface. Biotechnol. Bioeng. 2007, 96, 1023–1029. [Google Scholar] [CrossRef]
- Fukuyama, M.; Nishida, M.; Abe, Y.; Amemiya, Y.; Ikeda, T.; Kuroda, A.; Yokoyama, S. Detection of Antigen–Antibody Reaction Using Si Ring Optical Resonators Functionalized with an Immobilized Antibody-Binding Protein. Jpn. J. Appl. Phys. 2011, 50, 04DL07. [Google Scholar] [CrossRef]
- Anderson, G.P.; Jacoby, M.A.; Ligler, F.S.; King, K.D. Effectiveness of protein A for antibody immobilization for a fiber optic biosensor. Biosens. Bioelectron. 1997, 12, 329–336. [Google Scholar] [CrossRef]
- Knoglinger, C.; Zich, A.; Traxler, L.; Poslední, K.; Friedl, G.; Ruttmann, B.; Schorpp, A.; Müller, K.; Zimmermann, M.; Gruber, H.J. Regenerative biosensor for use with biotinylated bait molecules. Biosens. Bioelectron. 2018, 99, 684–690. [Google Scholar] [CrossRef]
- Choi, H.W.; Takahashi, H.; Ooya, T.; Takeuchi, T. Label-free detection of glycoproteins using reflectometric interference spectroscopy-based sensing system with upright episcopic illumination. Anal. Methods 2011, 3, 1366. [Google Scholar] [CrossRef]
- Lü, H.; Zhao, Y.; Ma, J.; Li, W.; Lu, Z. Characterization of DNA hybridization on the optical fiber surface. Colloids Surf. A Physicochem. Eng. Asp. 2000, 175, 147–152. [Google Scholar] [CrossRef]
- Holmberg, A.; Blomstergren, A.; Nord, O.; Lukacs, M.; Lundeberg, J.; Uhlén, M. The biotin-streptavidin interaction can be reversibly broken using water at elevated temperatures. Electrophoresis 2005, 26, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Bonanno, L.M.; DeLouise, L.A. Whole blood optical biosensor. Biosens. Bioelectron. 2007, 23, 444–448. [Google Scholar] [CrossRef]
- Bonanno, L.M.; Delouise, L.A. Steric crowding effects on target detection in an affinity biosensor. Langmuir 2007, 23, 5817–5823. [Google Scholar] [CrossRef]
- Hermanson, G.T. Silane Coupling Agents. In Bioconjugate Techniques; Elsevier: Amsterdam, The Netherlands, 2008; pp. 562–581. ISBN 9780123705013. [Google Scholar]
- Escorihuela, J.; Bañuls, M.J.; García Castelló, J.; Toccafondo, V.; García-Rupérez, J.; Puchades, R.; Maquieira, Á. Chemical silicon surface modification and bioreceptor attachment to develop competitive integrated photonic biosensors. Anal. Bioanal. Chem. 2012, 404, 2831–2840. [Google Scholar] [CrossRef]
- De Vos, K.; Bartolozzi, I.; Schacht, E.; Bienstman, P.; Baets, R. Silicon-on-Insulator microring resonator for sensitive and label-free biosensing. Opt. Express 2007, 15, 7610–7615. [Google Scholar] [CrossRef]
- Mudumba, S.; de Alba, S.; Romero, R.; Cherwien, C.; Wu, A.; Wang, J.; Gleeson, M.A.; Iqbal, M.; Burlingame, R.W. Photonic ring resonance is a versatile platform for performing multiplex immunoassays in real time. J. Immunol. Methods 2017, 448, 34–43. [Google Scholar] [CrossRef]
- Ksendzov, A.; Lin, Y. Integrated optics ring-resonator sensors for protein detection. Opt. Lett. 2005, 30, 3344–3346. [Google Scholar] [CrossRef]
- Byeon, J.-Y.; Limpoco, F.T.; Bailey, R.C. Efficient bioconjugation of protein capture agents to biosensor surfaces using aniline-catalyzed hydrazone ligation. Langmuir 2010, 26, 15430–15435. [Google Scholar] [CrossRef]
- Arkles, B. Silane Coupling Agents: Connecting Across Boundaries, 3rd ed.; Gelest Inc.: Morrisville, PA, USA, 2014. [Google Scholar]
- Zhang, F.; Srinivasan, M.P. Self-assembled molecular films of aminosilanes and their immobilization capacities. Langmuir 2004, 20, 2309–2314. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.A.; Chen, W. How to prevent the loss of surface functionality derived from aminosilanes. Langmuir 2008, 24, 12405–12409. [Google Scholar] [CrossRef] [PubMed]
- Bañuls, M.-J.; González-Pedro, V.; Barrios, C.A.; Puchades, R.; Maquieira, A. Selective chemical modification of silicon nitride/silicon oxide nanostructures to develop label-free biosensors. Biosens. Bioelectron. 2010, 25, 1460–1466. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, G.T. Homobifunctional Crosslinkers. In Bioconjugate Techniques; Elsevier: Amsterdam, The Netherlands, 2013; pp. 275–298. ISBN 9780123822390. [Google Scholar]
- Sheng, J.C.-C.; De La Franier, B.; Thompson, M. Assembling Surface Linker Chemistry with Minimization of Non-Specific Adsorption on Biosensor Materials. Materials 2021, 14, 472. [Google Scholar] [CrossRef]
- Xia, B.; Xiao, S.-J.; Guo, D.-J.; Wang, J.; Chao, J.; Liu, H.-B.; Pei, J.; Chen, Y.-Q.; Tang, Y.-C.; Liu, J.-N. Biofunctionalisation of porous silicon (PS) surfaces by using homobifunctional cross-linkers. J. Mater. Chem 2006, 16, 570–578. [Google Scholar] [CrossRef]
- Qavi, A.J.; Mysz, T.M.; Bailey, R.C. Isothermal discrimination of single-nucleotide polymorphisms via real-time kinetic desorption and label-free detection of DNA using silicon photonic microring resonator arrays. Anal. Chem. 2011, 83, 6827–6833. [Google Scholar] [CrossRef]
- Qavi, A.J.; Meserve, K.; Aman, M.J.; Vu, H.; Zeitlin, L.; Dye, J.M.; Froude, J.W.; Leung, D.W.; Yang, L.; Holtsberg, F.W.; et al. Rapid detection of an Ebola biomarker with optical microring resonators. Cell Rep. Methods 2022, 2, 100234. [Google Scholar] [CrossRef]
- Cardenosa-Rubio, M.C.; Graybill, R.M.; Bailey, R.C. Combining asymmetric PCR-based enzymatic amplification with silicon photonic microring resonators for the detection of lncRNAs from low input human RNA samples. Analyst 2018, 143, 1210–1216. [Google Scholar] [CrossRef]
- Karyakin, A.A.; Presnova, G.V.; Rubtsova, M.Y.; Egorov, A.M. Oriented immobilization of antibodies onto the gold surfaces via their native thiol groups. Anal. Chem. 2000, 72, 3805–3811. [Google Scholar] [CrossRef]
- Hanson, E.L.; Schwartz, J.; Nickel, B.; Koch, N.; Danisman, M.F. Bonding self-assembled, compact organophosphonate monolayers to the native oxide surface of silicon. J. Am. Chem. Soc. 2003, 125, 16074–16080. [Google Scholar] [CrossRef]
- Hermanson, G.T. Chemoselective ligation: Bioorthogonal reagents. In Bioconjugate Techniques; Elsevier: Amsterdam, The Netherlands, 2013; pp. 757–785. ISBN 9780123822390. [Google Scholar]
- SuSoS Functional Coatings for Glass, Polymeric, Metallic Surfaces. Available online: https://susos.com/products/ (accessed on 27 October 2022).
- Andreatta, G.A.L.; Hendricks, N.R.; Grivel, A.; Billod, M. Grafted-to Polymeric Layers Enabling Highly Adhesive Copper Films Deposited by Electroless Plating on Ultra-Smooth Three-Dimensional-Printed Surfaces. ACS Appl. Electron. Mater. 2022, 4, 1864–1874. [Google Scholar] [CrossRef]
- Weydert, S.; Zürcher, S.; Tanner, S.; Zhang, N.; Ritter, R.; Peter, T.; Aebersold, M.J.; Thompson-Steckel, G.; Forró, C.; Rottmar, M.; et al. Easy to Apply Polyoxazoline-Based Coating for Precise and Long-Term Control of Neural Patterns. Langmuir 2017, 33, 8594–8605. [Google Scholar] [CrossRef] [PubMed]
- Garg, H.; Nair, P.R. Stochastic modeling of steric hindrance effects in biosensors. In Proceedings of the 2018 IEEE SENSORS, New Delhi, India, 28–31 October 2018; pp. 1–4. [Google Scholar]
- González-Guerrero, A.B.; Alvarez, M.; García Castaño, A.; Domínguez, C.; Lechuga, L.M. A comparative study of in-flow and micro-patterning biofunctionalization protocols for nanophotonic silicon-based biosensors. J. Colloid Interface Sci. 2013, 393, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Arshak, K.; Korostynska, O.; Cunniffe, C. Nanopatterning using the bioforce nanoenabler. In Functionalized Nanoscale Materials, Devices and Systems; Vaseashta, A., Mihailescu, I.N., Eds.; NATO Science for Peace and Security Series B: Physics and Biophysics; Springer: Dordrecht, The Netherlands, 2008; pp. 299–304. ISBN 978-1-4020-8902-2. [Google Scholar]
- ChipMaker 3 Microarray Printing Pins (90–100 Micron Features). Available online: https://shop.arrayit.com/chipmaker3microspottingpin90-100micronfeatures.aspx (accessed on 6 October 2022).
- Folch, A. Introduction to BioMEMS; CRC Press: Boca Raton, FL, USA, 2016; ISBN 9780429194047. [Google Scholar]
- Kaigala, G.V.; Lovchik, R.D.; Drechsler, U.; Delamarche, E. A vertical microfluidic probe. Langmuir 2011, 27, 5686–5693. [Google Scholar] [CrossRef] [PubMed]
- Ness, S.J.; Kim, S.; Woolley, A.T.; Nordin, G.P. Single-sided inkjet functionalization of silicon photonic microcantilevers. Sensors and Actuators B: Chemical 2012, 161, 80–87. [Google Scholar] [CrossRef]
- Laplatine, L.; Fournier, M.; Gaignebet, N.; Hou, Y.; Mathey, R.; Herrier, C.; Liu, J.; Descloux, D.; Gautheron, B.; Livache, T. Silicon photonic olfactory sensor based on an array of 64 biofunctionalized Mach-Zehnder interferometers. Opt. Express 2022, 30, 33955–33968. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Whitesides, G.M. Soft Lithography. Angew. Chem. Int. Ed. 1998, 37, 550–575. [Google Scholar] [CrossRef]
- Kane, R.S.; Takayama, S.; Ostuni, E.; Ingber, D.E.; Whitesides, G.M. Patterning proteins and cells using soft lithography. Biomaterials 1999, 20, 2363–2376. [Google Scholar] [CrossRef]
- Graber, D.J.; Zieziulewicz, T.J.; Lawrence, D.A.; Shain, W.; Turner, J.N. Antigen binding specificity of antibodies patterned by microcontact printing. Langmuir 2003, 19, 5431–5434. [Google Scholar] [CrossRef]
- Lange, S.A.; Benes, V.; Kern, D.P.; Hörber, J.K.H.; Bernard, A. Microcontact printing of DNA molecules. Anal. Chem. 2004, 76, 1641–1647. [Google Scholar] [CrossRef] [PubMed]
- Thibault, C.; Le Berre, V.; Casimirius, S.; Trévisiol, E.; François, J.; Vieu, C. Direct microcontact printing of oligonucleotides for biochip applications. J. Nanobiotechnol. 2005, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Goh, S.H.; Bi, X.; Yang, K.-L. Replication of DNA submicron patterns by combining nanoimprint lithography and contact printing. J. Colloid Interface Sci. 2009, 333, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Idil, N.; Hedström, M.; Denizli, A.; Mattiasson, B. Whole cell based microcontact imprinted capacitive biosensor for the detection of Escherichia coli. Biosens. Bioelectron. 2017, 87, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Buhl, M.; Traboni, S.; Körsgen, M.; Lamping, S.; Arlinghaus, H.F.; Ravoo, B.J. On surface O-glycosylation by catalytic microcontact printing. Chem. Commun. 2017, 53, 6203–6206. [Google Scholar] [CrossRef]
- Qiu, S.; Ji, J.; Sun, W.; Pei, J.; He, J.; Li, Y.; Li, J.J.; Wang, G. Recent advances in surface manipulation using micro-contact printing for biomedical applications. Smart Mater. Med. 2021, 2, 65–73. [Google Scholar] [CrossRef]
- Romanov, V.; Davidoff, S.N.; Miles, A.R.; Grainger, D.W.; Gale, B.K.; Brooks, B.D. A critical comparison of protein microarray fabrication technologies. Analyst 2014, 139, 1303–1326. [Google Scholar] [CrossRef]
- Ong, J.K.Y.; Moore, D.; Kane, J.; Saraf, R.F. Negative printing by soft lithography. ACS Appl. Mater. Interfaces 2014, 6, 14278–14285. [Google Scholar] [CrossRef]
- Yunus, S.; de Looringhe, C.D.C.; Poleunis, C.; Delcorte, A. Diffusion of oligomers from polydimethylsiloxane stamps in microcontact printing: Surface analysis and possible application. Surf. Interface Anal. 2007, 39, 922–925. [Google Scholar] [CrossRef]
- Yang, L.; Shirahata, N.; Saini, G.; Zhang, F.; Pei, L.; Asplund, M.C.; Kurth, D.G.; Ariga, K.; Sautter, K.; Nakanishi, T.; et al. Effect of surface free energy on PDMS transfer in microcontact printing and its application to ToF-SIMS to probe surface energies. Langmuir 2009, 25, 5674–5683. [Google Scholar] [CrossRef]
- Fluent®Automated Workstation-Tecan. Available online: https://www.tecan.com/fluent-automated-workstation?utm_term=robotic%20pipette&utm_campaign=SO-Liquid+Handling&utm_source=adwords&utm_medium=ppc&hsa_net=adwords&hsa_tgt=kwd-1223309053168&hsa_ad=605529119671&hsa_acc=9279258943&hsa_grp=121576188140&hsa_mt=p&hsa_cam=12736641036&hsa_kw=robotic%20pipette&hsa_ver=3&hsa_src=g&gclid=Cj0KCQjw1vSZBhDuARIsAKZlijS14MpBw3DcUBTV9B0GbtZIDM8IKOPaTo58M70vwGtNy3tyXpkfbdsaAp9FEALw_wcB (accessed on 5 October 2022).
- Pipetting Robot Using Electronic Pipettes for Liquid Handling-Andrew+ -. Available online: https://www.andrewalliance.com/pipetting-robot/ (accessed on 27 October 2022).
- Pipetting Robots|INTEGRA. Available online: https://www.integra-biosciences.com/canada/en/pipetting-robots (accessed on 27 October 2022).
- Pipetting Robot—Automated Pipettor Robots for Liquid Handling. Available online: https://hudsonrobotics.com/products/liquid-handling/solo-liquid-handling/ (accessed on 27 October 2022).
- Austin, J.; Holway, A.H. Contact printing of protein microarrays. Methods Mol. Biol. 2011, 785, 379–394. [Google Scholar] [CrossRef]
- Wu, D.; Song, L.; Chen, K.; Liu, F. Modelling and hydrostatic analysis of contact printing microarrays by quill pins. International Journal of Mechanical Sciences 2012, 54, 206–212. [Google Scholar] [CrossRef]
- Arrayit 946 Microarray Pins—Printing Spotting Robot Automation Microfluidic Chip RT-PCR PCR DNA Sequencing. Available online: https://shop.arrayit.com/microarray_printing_spotting_pins.aspx (accessed on 6 October 2022).
- Arrayit Stealth Microarray Pins and Printheads—Printing Spotting Robot Automation Microfluidic Chip RT-PCR PCR DNA Sequencing. Available online: https://shop.arrayit.com/microarray_pins_stealth.aspx (accessed on 6 October 2022).
- Arrayit Green Microarray Pins and Printheads—Printing Spotting Robot Automation Microfluidic Chip RT-PCR PCR DNA Sequencing. Available online: https://shop.arrayit.com/greenmicroarraypins.aspx (accessed on 6 October 2022).
- Delamarche, E.; Bernard, A.; Schmid, H.; Michel, B.; Biebuyck, H. Patterned delivery of immunoglobulins to surfaces using microfluidic networks. Science 1997, 276, 779–781. [Google Scholar] [CrossRef] [PubMed]
- Silzel, J.W.; Dodson, C.; Obremski, R.J.; Tsay, T. Mass-Sensing Multianalyte Micro-Array Based Immunoassay by Direct Nir Fluorescence and Quantitative Image Analysis. In Micro Total Analysis Systems ’98; Harrison, D.J., van den Berg, A., Eds.; Springer: Dordrecht, The Netherlands, 1998; pp. 65–68. ISBN 978-94-010-6225-1. [Google Scholar]
- SCIENION|Global Leader in Precision Dispensing|SCIENION. Available online: https://www.scienion.com/ (accessed on 31 October 2022).
- Micro-Dispensing and Inkjet Printing Applications—Microdrop Technologies GmbH. Available online: https://www.microdrop.de/ (accessed on 31 October 2022).
- Delehanty, J.B.; Ligler, F.S. Method for printing functional protein microarrays. BioTechniques 2003, 34, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Melnik, E.; Strasser, F.; Muellner, P.; Heer, R.; Mutinati, G.C.; Koppitsch, G.; Lieberzeit, P.; Laemmerhofer, M.; Hainberger, R. Surface modification of integrated optical MZI sensor arrays using inkjet printing technology. Procedia Eng. 2016, 168, 337–340. [Google Scholar] [CrossRef]
- Juncker, D.; Schmid, H.; Delamarche, E. Multipurpose microfluidic probe. Nat. Mater. 2005, 4, 622–628. [Google Scholar] [CrossRef]
- Queval, A.; Ghattamaneni, N.R.; Perrault, C.M.; Gill, R.; Mirzaei, M.; McKinney, R.A.; Juncker, D. Chamber and microfluidic probe for microperfusion of organotypic brain slices. Lab Chip 2010, 10, 326–334. [Google Scholar] [CrossRef]
- Seo, J.; Poulter, C.D. Sandwich antibody arrays using recombinant antibody-binding protein L. Langmuir 2014, 30, 6629–6635. [Google Scholar] [CrossRef]
- Randhawa, A. Exploring the Microfluidic Organ-On-Chip Platform for Aerosol Exposure Study. Master’s Thesis, University of British Columbia, Vancouver, BC, Canada, 2022. [Google Scholar]
- Squires, T.M.; Messinger, R.J.; Manalis, S.R. Making it stick: Convection, reaction and diffusion in surface-based biosensors. Nat. Biotechnol. 2008, 26, 417–426. [Google Scholar] [CrossRef]
- Luan, E.; Yun, H.; Laplatine, L.; Dattner, Y.; Ratner, D.M.; Cheung, K.C.; Chrostowski, L. Enhanced Sensitivity of Subwavelength Multibox Waveguide Microring Resonator Label-Free Biosensors. IEEE J. Select. Topics Quantum Electron. 2019, 25, 7300211. [Google Scholar] [CrossRef]
- Mai, A.; Mai, C.; Steglich, P. From Lab-on-chip to Lab-in-App: Challenges towards silicon photonic biosensors product developments. Results Opt. 2022, 9, 100317. [Google Scholar] [CrossRef]
- Koos, C.; Freude, W.; Lindenmann, N.; Leuthold, J. Photonic wire bonds, 2013.
- Kappert, E.J.; Raaijmakers, M.J.T.; Tempelman, K.; Cuperus, F.P.; Ogieglo, W.; Benes, N.E. Swelling of 9 polymers commonly employed for solvent-resistant nanofiltration membranes: A comprehensive dataset. J. Memb. Sci. 2019, 569, 177–199. [Google Scholar] [CrossRef]























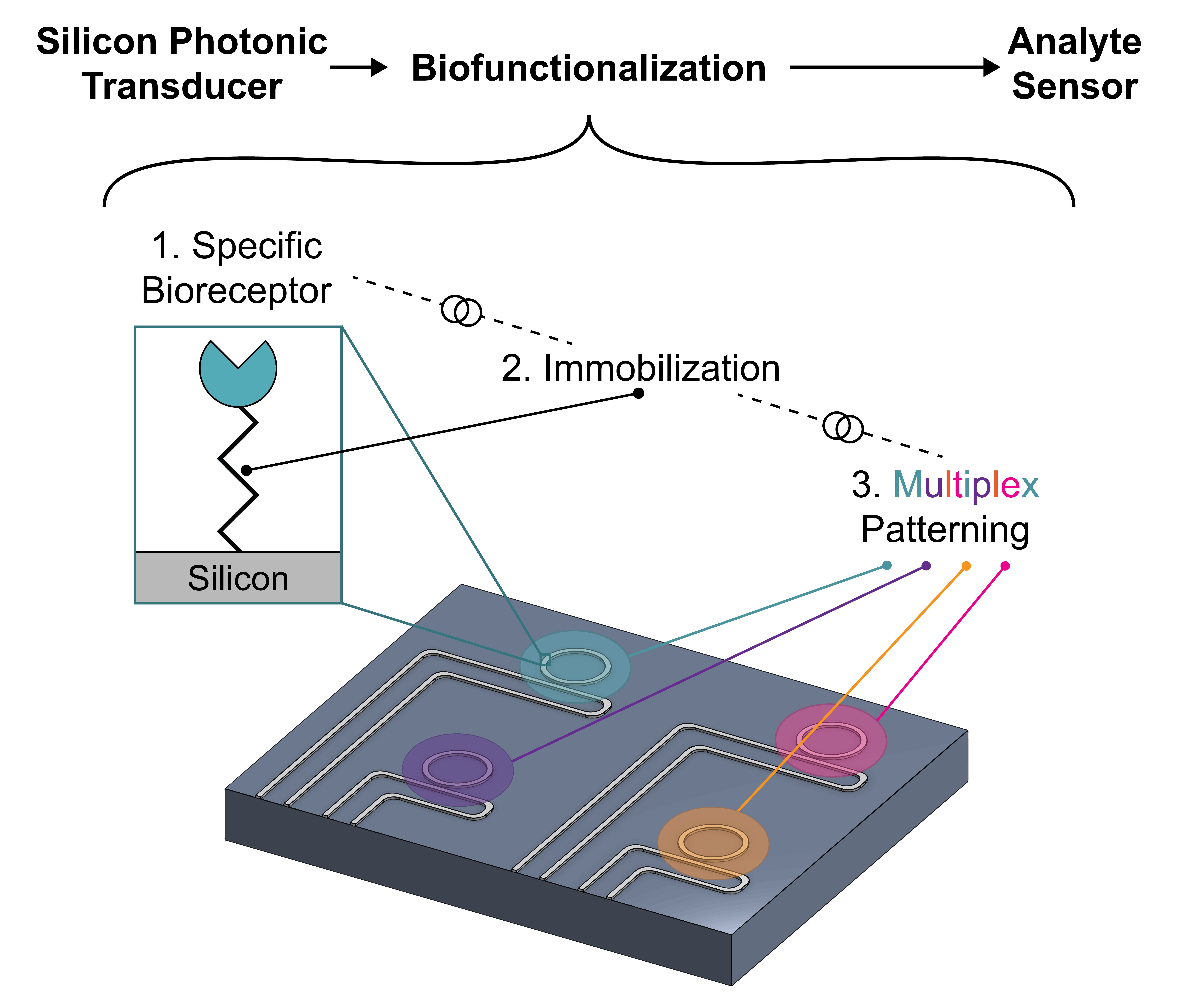{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
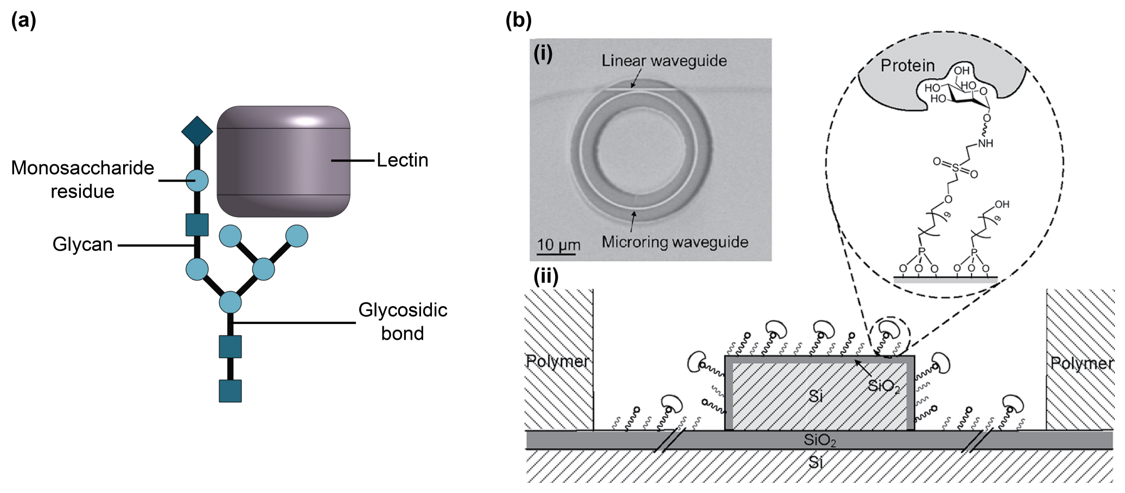{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SiP | SPR | |
|---|---|---|
| Surface [11,12,42] | Si or Si3N4, typically coated with native SiO2 film | Au |
| Approx. evanescent field decay distance [4,12,43,44,45,46] | ~40–200 nm; depends on waveguide geometry and polarization | Typically ~200 nm; up to 600 nm when using an infrared laser or long-range surface plasmons |
| Miniaturization [12,13,47,48,49] | Very compact: chip-level integration with microfluidics, electronics, and optical inputs/outputs possible | Moderate: portable instrumentation demonstrated with dimensions ~10–20 cm |
| Sensor size [4,50,51,52] | Total sensor chip dimensions ~1–10 mm; active sensing spot dimensions ~10–100 µm | Total sensor chip dimensions ~10 mm; active sensing spot dimensions ~100 µm–1000 µm for multiplexed SPRi and LSPR devices |
| Cost [12,53] | Low (at high volume) | High |
| Multiplexing [53] | Multiplexable | Not possible with conventional SPR; multiplexable with SPRi and LSPR |
| Bioreceptor | Immobilization Chemistry | Patterning Technique |
|---|---|---|
|
|
|
| Bioreceptor | Targets | Affinity | Specificity | Stability | Availability | Reproducibility of Production | Regenerability | Size | Cost * |
|---|---|---|---|---|---|---|---|---|---|
| Antibody | Antigens: diverse range of small molecules, complex macromolecules, viruses, and bacteria; exclude many toxins and non-immunogenic targets [29,33,84,85] | Very high; dissociation constant (KD) values typically in the low-nM to pM range [85,86] | Very high [87,88,89,90] | Poor [24,91] | High [92,93] | Poor to moderate [87,88,89] | Moderate; may experience activity loss [96] |
| High; ~USD 500/100 µg [89,97,98,99] |
| Aptamer | Very wide range including ions, small inorganic molecules, peptides, proteins, toxins, viral particles, and cells [85,89,100] | Very high; KD values typically in the low-nM to pM range [85,100,101] | Very high [89,100] |
[89,100,102] | Moderate
[103,104] | High due to chemical synthesis [89] | Good [96,105] |
[85,89,102] | |
| Nucleic acid probe | Nucleic acids | High to very high, depending on type of nucleic acid; KD values demonstrated in the low-nM range for DNA-DNA duplexes [107] | High to very high [29,73,81,108,109] |
[73,81,101,108,110] | Moderate-high
| High due to chemical synthesis [111,112] | Good [80] | ||
| Molecularly imprinted polymer (MIP) | Various small molecules; template molecules must be able to survive in organic solvents | Wide range; KD values reported in the 10−15–10−5 M range [72] | Moderate | High mechanical and chemical stability [118] | Low | High | Good | Thin film (sub-nanometer to micrometers) [72] | Dependent on development |
| Synthetic peptides [91,120] | A range of small molecules to proteins | Very high | High |
| High
| Very high | Good |
| Moderate; CAD 10–100 for custom synthesis dependent on amount and purity for <0.5 g |
| Native peptides [120] | A range of small molecules to proteins | Very high | High | Poor | High
| Very high | Moderate | 2–14 kDa; 89–204 Da per amino acid | High; CAD 100–1000; dependent on peptide, purity, and quantity |
| Glycan and lectin |
| Low; KD in the µM–mM range [121,122] | Low [121,123] |
|
|
|
| ||
| High contrast cleavage detection (HCCD) | Nucleic acids | High | Very high [137,138] |
| Moderate
| Moderate-high | Not regenerable |
| High |
| CRISPR-dCas9-mediated detection | Nucleic acids | High | Very high [143] |
| Moderate
| Moderate-high | Good | High | |
| Lipid nanodisc | Proteins | Moderate; KD in nM–µM range for phospholipid nanodiscs [148,149] | Variable; depends on membrane protein content [148,149,150] | Poor due to protein content | Low-moderate
| Moderate | Good [148,149] | 8–16 nm diameter [150,151] | High
|
| Affinity [86,87,88,89] | Specificity [87,88,89,90] | Availability [92,93,94,95,158,159] | Reproducibility of Production [87,88,89] | Size [30,87,89] | Cost * [89,97,98,99] | |
|---|---|---|---|---|---|---|
| Polyclonal antibody | Very high, but with significant variability within a sample | High
| High; many vendors available (key companies include Abcam plc, GenScript, Thermo Fisher Scientific Inc., etc.) † | Poor |
| High ~CAD 500/100 µg |
| Monoclonal antibody | Very high with KD values as low as 10 pM | Very high
| High; many vendors available (key companies include Abcam plc, GenScript, Thermo Fisher Scientific Inc., etc.) † | Moderate |
| High ~CAD 500/100 µg |
| Fab fragment | Very high | High to very high, depending on origin | High; can be prepared from available antibodies or purchased commercially (major antibody-fragment producing companies include AbbVie, Amgen, Novartis AG, etc.) ‡ | Poor to moderate, depending on origin | MW: ~50 kDa | High |
| Bioreceptor Description | Sensor Type | Target | Detection Performance | Assay Format | Refs. | |
|---|---|---|---|---|---|---|
| Figure of Merit | Value | |||||
| Monoclonal antibody | Si MRR | Thrombin | Min. detected concentration | 500 pM | Label-free | [174] |
| Monoclonal antibodies | Si MRR | Carcinoembryonic antigen | Min. detected concentration | 10 ng/mL | Label-free | [22] |
| Prostate-specific antigen | ||||||
| α-fetoprotein | ||||||
| Interleukin-8 | ||||||
| Tumor necrosis factor-α | ||||||
| Monoclonal antibody | Si MRR | Monocyte chemotactic protein 1 | Limit of detection (LoD) | 0.5 pg/mL | Amplification with secondary antibody and enzymatic enhancement | [17] |
| Monoclonal antibody | Si MRR | Carcinoembryonic antigen | LoD | 2 ng/mL in buffer, 25 ng/mL in serum | Label-free | [161] |
| Monoclonal antibodies | Si MRR | Interleukin-2 | LoD | 100 pg/mL | Sandwich immunoassay | [18] |
| Interleukin-8 | LoD | 100 pg/mL | ||||
| Monoclonal antibody | Si MRR | Bean pod mottle virus | LoD | 10 ng/mL | Label-free | [169] |
| Monoclonal antibody | Si MRR | C-reactive protein | - | - | Label-free | [166] |
| Monoclonal antibodies | Si MRR | α-fetoprotein | Working range | 0.3–20.6 ng/mL | Amplification with secondary antibody and protein-based multilayer signal enhancement | [163] |
| Activated leukocyte cell adhesion molecule | Working range | 1.0–43.7 ng/mL | ||||
| Cancer antigen 15-3 | Working range | 2.0–91.5 units/mL | ||||
| Cancer antigen 19-9 | Working range | 2.5–96.6 units/mL | ||||
| Cancer antigen-125 | Working range | 2.4–95.6 units/mL | ||||
| Carcinoembryonic antigen | Working range | 0.16–20.2 ng/mL | ||||
| Osteopontin | Working range | 4.3–50.3 ng/mL | ||||
| Prostate specific antigen | Working range | 0.054–4.7 ng/mL | ||||
| Monoclonal antibodies | Si MRR | Interleukin-2 | LoD | 1 pg/mL | Amplification with secondary antibody and enzymatic enhancement | [162] |
| Interleukin-6 | LoD | 1 pg/mL | ||||
| Interleukin-8 | LoD | 0.5 pg/mL | ||||
| Monoclonal antibody | Hydex MRR | E. coli O157:H7 bacterial cells | LoD | 105 CFU/mL | Label-free | [172] |
| Monoclonal antibody | Si PhC | Human Papillomavirus virus-like particles | LoD | 1.5 nM | Label-free | [170] |
| Antibody | Si PhC | Cardiac myoglobin | Min. detected concentration | 70 ng/mL | Label-free | [165] |
| Monoclonal antibodies | Si3N4 planar waveguide interferometer | Hemagglutinin (H7N2 and H7N3) | Min. detected concentration | 0.05 hemagglutination (HA) units/mL | Label-free | [175] |
| Polyclonal antibodies | Si3N4 planar waveguide interferometer | Hemagglutinin (H7N2) | Min. detected concentration | 0.0005 HA units/mL | ||
| Hemagglutinin (H7N3) | Min. detected concentration | 0.005 HA units/mL | ||||
| Polyclonal antibody | Porous Si sensor using reflectometric interference spectroscopy | Insulin | LoD | 4.3 µg/mL | Label-free | [176] |
| Antibody | Si PhC total internal reflection | Cardiac troponin I | LoD | 0.01 ng/mL | Label-free | [164] |
| Antibody | Si3N4/SiO2 slot-waveguide MRR | Bovine serum albumin | LoD | 16 pg/mm2 | Label-free | [177] |
| Antigen-binding fragment (Fab) from protease digestion of polyclonal IgG | SiOxNy MRR | Aflatoxin M1 | LoD | 5 nM | Label-free | [25] |
| Single domain antibodies | Si MRR | Ricin | LoD | 200 pM | Label-free | [168] |
| Advantages | Limitations |
|---|---|
|
|
| Affinity [89,100] | Specificity [89,100] | Stability [89,100,102,183] | Availability [89] | Reproducibility of Production [89] | Attachment Chemistry | Size * [85,89,102] | Cost † [89,106,182] | |
|---|---|---|---|---|---|---|---|---|
| DNA aptamer | High | High |
| Moderate
| High due to chemical synthesis | Typically, covalent immobilization via terminal functional groups (e.g., 5′ amines) |
| Low
|
| RNA aptamer | Very high due to more diverse 3D conformations | Very high due to more diverse 3D conformations |
| Moderate
| High due to chemical synthesis | Typically, covalent immobilization via terminal functional groups (e.g., 5′ amines) |
| Moderate
|
| Sensor Type | Target | Detection Performance | Refs. | |
|---|---|---|---|---|
| Figure of Merit | Value | |||
| Si MRR | IgE | LoD | 33 pM | [24] |
| Thrombin | LoD | 1.4 nM | ||
| Si MRR | Thrombin | Min. detected concentration | 500 pM | [174] |
| SiOxNy MRR | Aflatoxin M1 | LoD | 5 nM | [25] |
| SiOxNy MRR | Aflatoxin M1 | Min. detected concentration | 1.58 nM | [185] |
| Si PhC total internal reflection | Cardiac troponin I | LoD | 0.1 ng/mL | [164] |
| Porous Si reflectometric interference spectroscopy | Insulin | LoD | 1.9 µg/mL | [176] |
| Advantages | Limitations |
|---|---|
| Affinity [29,30,81,108] | Specificity [29,73,81,108,109] | Stability [73,81,101,108,110,183,189] | Availability | Reproducibility of Production [89] | Attachment Chemistry | Cost * [115,116,117,182] | |
|---|---|---|---|---|---|---|---|
| DNA | High | High |
| Moderate
| High due to chemical synthesis | Typically, covalent immobilization via terminal functional groups (e.g., 5′ amines) | Low
|
| RNA | High | High |
| Moderate
| High due to chemical synthesis | Typically, covalent immobilization via terminal functional groups (e.g., 5′ amines) | Medium
|
| PNA | Higher than DNA/RNA due to neutral charge | Very high | Very high physical, thermal, and enzymatic stability | Low
| High due to chemical synthesis | Typically, covalent immobilization via terminal functional groups (e.g., 5′ amines) | High
|
| LNA | Higher than DNA/RNA due to decreased configurational flexibility | Very high | Very high physical, thermal, and enzymatic stability | Low
| High due to chemical synthesis | Typically, covalent immobilization via terminal functional groups (e.g., 5′ amines) | High
|
| Morpholino | Higher than DNA/RNA due to neutral charge, but lower affinity than PNA | Very high |
| Low
| High due to chemical synthesis | Typically, covalent immobilization via terminal functional groups (e.g., 5′ amines) | High
|
| Bioreceptor | Sensor Type | Target | Detection Performance | Assay Format * | Refs. | |
|---|---|---|---|---|---|---|
| Figure of Merit | Value | |||||
| ssDNA | Si MRR | microRNA | LoD | 150 fmol (i.e., 75 µL of 2 nM microRNA solution) | Label-free | [194] |
| ssDNA | Si MRR | Complementary DNA generated from targeted microRNAs | - | - | Label-free | [195] |
| ssDNA | Si MRR | Full-length mRNA transcripts | LoD | 32 fmol for label-free detection; 512 amol with bead-based amplification | Label-free and with streptavidin-coated bead-based amplification | [196] |
| ssDNA | Si MRR | microRNA | LoD | 10 pM (i.e., 350 amol in a 35 µL sample) | Amplification with anti-DNA:RNA antibodies | [109] |
| ssDNA | Si MRR | ssDNA | LoD | 400 fmol (i.e., 16 µL of 25 nM ssDNA solution) | Label-free | [197] |
| ssDNA | Si MRR | Bacterial transfer-messenger RNA (tmRNA) | LoD | 52.4 fmol (i.e., 100 µL of 524 pM tmRNA solution) | Label-free | [198] |
| ssDNA | Si MRR | Methylated DNA | - | - | Label-free | [23] |
| ssDNA | Cascaded Si MRRs | IS6110 ssDNA biomarker | LoD | 1 fg (corresponds to 10 µL of 0.1 pg/mL ssDNA solution) | Label-free | [199] |
| IS1081 ssDNA biomarker | LoD | 10 fg (i.e., 10 µL of 1 pg/mL ssDNA solution) | ||||
| ssDNA | Si3N4 MRR | ssDNA | - | - | Label-free | [202] |
| ssDNA | N-doped Si MRR electrophotonic sensor | ssDNA | - | - | label-free | [59] |
| ssDNA | Si3N4 MZI | ssDNA | LoD | 300 pM | Label-free | [200] |
| ssDNA | Planar Si PhC waveguide | ssDNA | LoD | 19.8 nM | Label-free | [203] |
| ssDNA (directly conjugated) | Si MRR and Si PhC | ssDNA | LoD | 50 nM | Label-free | [201] |
| ssPNA | - | - | ||||
| ssDNA (synthesized in situ) | Si MRR and Silicon PhC | ssDNA | LoD | 10 nM | ||
| ssPNA | - | - | ||||
| Methylated ssDNA | Si3N4 slot waveguide MZI | Methylated ssDNA | Min. detected concentration | 1 fmol/µL (1nM) | Label-free | [192] |
| Morpholino | Suspended Si MRR | ssDNA | Min. detected concentration | 250 pM | Label-free | [110] |
| Advantages | Limitations |
|---|---|
| MIP Type | Film Constituents | Template | Film Deposition Technique | Template Extraction | Sensor Type | Refs. |
|---|---|---|---|---|---|---|
| Polymer synthesis | Methacrylic acid (functional monomer) and ethylene glycol dimethacrylate (crosslinking agent) | Testosterone | Casting, followed by thermopolymerization | Acetic acid and ethanol-based chemical extraction | Si MRR | [212] |
| Polymer synthesis | Methacrylic acid (functional monomer) and ethylene glycol dimethacrylate (crosslinking agent) | Progesterone | Coating, followed by UV photopolymerization | - | Cascaded Si MRRs | [219] |
| Sol-gel | Bis(trimethoxysilylethyl)benzene and 2-(2-pyridylethyl)trimethoxysilane, prepared in tetrahydrofuran | Carbamate (used to create trinitrotoluene binding sites) | Airspray coating or electrospray ionization | HCl and chloroform-based chemical extraction | Si MRR | [213] |
| Sol-gel | Ethanol, methyltrimethoxysilane, aminopropyltriethoxysilane, and HCl, prepared in dimethyl sulfoxide | Fluorescein isothiocyanate | Dip coating | Oxygen plasma degradation or chemical extraction with solutions of ethanol, acetic acid, and chloroform or acetonitrile. | SiO2 microsphere whispering gallery mode resonators | [211] |
| Sol-gel | Tetraethoxysilane (TEOS), water, ethanol, and HCl or methyltriethoxysilane (C1-TriEOS), ethanol, HCl, and MPTMS. | Cortisol | Spin coating | Ethanol-based chemical extraction. | Si chips | [214,215] |
| Advantages | Limitations |
|---|---|
|
| Bioreceptor | Sensor Type | Target | Detection Performance | Assay Format | Refs. | |
|---|---|---|---|---|---|---|
| Figure of Merit | Value | |||||
| MIP film | Si MRR | Testosterone | LoD | 48.7 pg/mL | Label-free | [212] |
| Sol-gel MIP | Si racetrack resonators | Trinitrotoluene vapor | Min. detected concentration | 5 ppb | Label-free | [213] |
| MIP film | Cascaded Si MRRs | Progesterone | LoD | 83.5 fg/mL | Label-free | [219] |
| MIP film | SiO2 microsphere whispering gallery mode resonator | Fluorescein isothiocyanate | - | Fluorescence intensity | - | [211] |
| MIP film | SiOxNy dual polarization interferometer | Hemoglobin | LoD | 2 µg/mL | Label-free | [225] |
| Bioreceptor | Sensor Type | Target | Detection Performance | Refs. | |
|---|---|---|---|---|---|
| Figure of Merit | Value | ||||
| Peptide | Si3N4 MZI | SARS-CoV-2 antibodies | LoD | 80 ng/mL | [238] |
| Peptide | Planar Si and porous Si microcavity | A20 lymphoma cancer cells | Coverage efficiency after 2 h incubation with 50,000 A20 cells | ~85% and ~4% for planar and porous functionalized surfaces, respectively | [239] |
| PCC | Porous Si microcavity | Chikungunya virus E2 protein | Resonance shift after 3 h incubation with 1 µM E2 protein | 1.7 ± 0.3 nm | [91] |
| PCC | Porous Si microcavity | Streptavidin | Resonance redshift after 1 h exposure to 5 µM streptavidin | 12.9 nm | [240] |
| Advantages | Limitations |
|---|---|
|
| Compatible Targets [121,122] | Affinity [121,122] | Specificity [121,123] | Stability [121,124,125,126] | Availability [127,128,129,130] | Reproducibility of Production [121,127,128] | Attachment Chemistry [244] | Size [121,128,131,132,133,134] | Cost *[135,136] | |
|---|---|---|---|---|---|---|---|---|---|
| Glycan | Lectins, toxins, and viruses | Low; KD in µM–mM range | Low | Good shelf life and stable under dry and ambient conditions | Low–moderate
| High due to chemical synthesis | Site-directed immobilization via terminal amine group | Diverse (polysaccharides containing a few monosaccharide units to thousands) | Very high; ~CAD 200–CAD 1200/10 µg for commercial products |
| Lectin | Carbohydrates, including glycans and glycoconjugates | Low; KD in µM–mM range | Low | Low due to susceptibility to permanent denaturation | Moderate–high
| Moderate due to variability introduced by cell-based synthesis techniques | Challenging to achieve oriented immobilization due to complex structure | Diverse, but typically smaller than antibodies (~10–140 kDa) | Moderate; ~CAD 1–300/mg |
| Bioreceptor | Sensor Type | Target | Detection Performance | Refs. | |
|---|---|---|---|---|---|
| Figure of Merit | Value | ||||
| Lacto-N-fucopentaose III-human serum albumin (LNFPIII-HSA) glycoprotein | Si MRR | Norovirus-like particles | LoD | 250 ng/mL | [126] |
| BSA-mannose, BSA-lactose, BSA-galactose and RNase B glycoconjugates | Si MRR | Lectins: concanavalin A, griffithsin, and ricin | - | - | [246] |
| GM1 ganglioside glycan | Si3N4 MRR | Cholera toxin subunit B | LoD | 400 ag (corresponds to 8 pg/mm2) | [132] |
| 3-fucosyl lactose glycan | Si3N4 MRR | Aleuria Aurantia Lectin | LoD | 0.5 ng/mL (7 pM) | [133] |
| α2,6-disialylated biantennary N-glycan | Si3N4 MRR | Sambucus Nigra Lectin | LoD | 12 ng/mL (86 pM) | |
| Concanavalin A lectin | Porous Si | Escherichia coli | LoD | 103 cells/mL | [131] |
| Wheat germ agglutinin lectin | Porous Si | Staphylococcus aureus | LoD | 103 cells/mL | |
| Advantages | Limitations | |
|---|---|---|
| Glycan | ||
| Lectin |
|
| Bioreceptor | Sensor Type | Target | Detection Performance | Assay Format | Ref. | |
|---|---|---|---|---|---|---|
| Figure of Merit | Value | |||||
| CRISPR-Cas12a with guide RNA complementary to target | Si MRR | SARS-CoV-2 ssDNA | Resonance shift after exposure to 1 nM of target DNA | ~8 nm | Labeled: gold nanoparticle reporters tethered to sensor surface by ssDNA were cleaved by activated Cas12a effector; amplification via collateral cleavage | [139] |
| Advantages | Limitations |
|---|---|
|
| Bioreceptor | Sensor Type | Target | Detection Performance | Assay Format | Ref. | |
|---|---|---|---|---|---|---|
| Figure of Merit | Value | |||||
| ssDNA probes | Si MRR | Scrub typhus viral DNA | LoD | 0.54 aM | Isothermal pre-amplification of targets; target-specific CRISPR-dCas9 signal amplification | [143] |
| Severe fever with thrombocytopenia viral RNA | LoD | 0.63 aM | ||||
| Advantages | Limitations |
|---|---|
|
|
| Bioreceptor | Sensor Type | Target | Detection Performance | Ref. | |
|---|---|---|---|---|---|
| Figure of Merit | Value | ||||
| Lipid nanodiscs containing PC, four binary compositions of PC and PS, and two binary combinations of PS and PA | Si MRR | Blood clotting proteins: pro-thrombin, factor X, activated factor VII, and activated protein C | - | - | [149] |
| Lipid nanodiscs containing POPC and POPC/POPS | Si MRR | Annexin V | - | - | [150] |
| Lipid nanodiscs containing GM1 | Si MRR | Cholera Toxin Subunit B | - | - | |
| Lipid nanodiscs containing biotin-DPPE | Si MRR | Streptavidin | - | - | |
| Lipid nanodiscs containing CYP3A4 | Si MRR | Anti-CYP3A4 antibody | - | - | |
| Lipid nanodiscs with 9 different compositions containing PS, PE, and PC. | Si MRR | Protein clotting factors: prothrombin, activated factor VII, factor IX, factor X, activated protein C, protein S, and protein Z | - | - | [148] |
| Advantages | Limitations |
|---|---|
|
| Immobilization Chemistry | Compatible Bioreceptors | Surface Modification | Bioreceptor Modification | Required Linkers | Stability | Thickness | Oriented Bioreceptor Immobilization? | Impact on Bioreceptor Function | Typical Replicability and Uniformity | Compatibility with System-Level Sensor Integration | Process Scalability |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Passive adsorption | |||||||||||
| Passive adsorption | All | Not required | Not required | None | Typically, poor [31,69,261] | No added thickness | Typically, no | Likely to reduce bioreceptor binding activity [29,69,262,263] | Poor | Good | Very good |
| Bioaffinity | |||||||||||
| Antibody (Ab)- binding protein | Antibodies | Ab-binding protein is typically passively adsorbed on sensor | None | None | Moderate [29,75] | 3–4 nm for adsorbed PrA [259,264] | Yes | Preserves bioreceptor binding activity | Depends on Ab-binding protein immobilization strategy | Depends on Ab-binding protein immobilization strategy | Depends on Ab-binding protein immobilization strategy |
| Biotin/(strept)avidin | Antibodies, aptamers, nucleic acid probes, peptides, PCCs, glycans/lectins | Often silanization | Biotinylation | (Strept)avidin acts as bioaffinity linker | Good [74,265] | ~6–7 nm plus thickness of chemical layer used to immobilize (strept)avidin [266] | Possible | Preserves bioreceptor binding activity [263] | Depends on (strept)avidin immobilization strategy | Depends on (strept)avidin immobilization strategy | Poor due to complexity |
| Covalent | |||||||||||
| Silane chemistry | Antibodies, aptamers, nucleic acid probes, peptides, PCCs, glycans/lectins | Silanization | Aptamers and nucleic acids require modification with terminal functional groups (e.g., amine, carboxyl, thiol). S-4FB conjugation required for SoluLink chemistry. | Often required; popular options include GA, BS3, and EDC/NHS | Good [31] | <1 nm for silane monolayer; multilayer films may exceed 10 nm [267] | Possible | Typically preserves bioreceptor binding activity; strategies requiring antibody disulfide bond reduction may reduce antibody binding activity [69,75] | Variable for solution-phase silanization; good for vapor-phase silanization [261,267,268,269] | Poor for solution-phase silanization; good for vapor-phase silanization | Fair-good, depending on reaction conditions and linker requirements |
| Organophosphonate chemistry | Antibodies, aptamers, nucleic acid probes, peptides, PCCs, glycans/lectins | UDPA deposition | Aptamers and nucleic acids require modification with terminal functional groups | Required; DVS and 3-maleimidopropionic-acid-N-hydroxysuccinimidester have been used [126,270] | Very good [126] | ~1 nm [270] | Possible | Preserves bioreceptor binding activity [126,270] | Very good | Poor due to solution-phase UDPA deposition | Fair; multistep process |
| Click chemistry | Antibodies, aptamers, nucleic acid probes, peptides, PCCs, glycans/lectins | Azide/alkyne derivatization | Modification with azide/alkyne moieties, DBCO, or tetrazine | Azide, alkyne, DBCO, or tetrazine terminations required | Good [91] | <1 nm | Possible | Preserves bioreceptor binding activity | Good; insensitive to oxygen and moisture [271] | Poor due to solution-phase chemistry and aggressive surface pre-treatment strategies, which may damage sensor [59,91,240] | Fair; multistep process |
| UV-crosslinking | Aptamers [272] and nucleic acids [273] | None required | Modification with poly(T) or poly(TC) tags | None | Good [272,273] | Thickness added by poly(T) or poly(TC) tag; depends on tag length | Yes | Preserves bioreceptor binding activity [272,273] | Good due to process simplicity [272] | Very good | Very good |
| Antibody Immobilization | ||||||
|---|---|---|---|---|---|---|
| Immobilization Strategy | Bioreceptor Subtype | Surface Pre-Treatment | Chemical Surface Modification | Linking Strategy | Sensor Type | Refs. |
| Covalent | Fab fragment | Argon plasma treatment | MPTMS silanization | Direct conjugation to silanized surface | SiOxNy MRR | [25] |
| Covalent | Monoclonal antibodies | NaOH and ethanol/water cleaning | GPTMS silanization | Direct conjugation to silanized surface | Hydex MRR | [172] |
| Covalent | Monoclonal [18,22,109,169,174,295] and single-domain antibodies [168] | Piranha treatment | HyNic silane surface modification | Conjugation of antibody with S-4FB for hydrazone bond formation with modified surface | Si MRR | [18,22,162,168,169,174,295] |
| Covalent | Monoclonal antibody | Piranha treatment | APTES silanization + S-HyNic surface modification | Conjugation of antibody with S-4FB for hydrazone bond formation with modified surface | Si MRR | [161] |
| Covalent | Monoclonal antibodies | Acetone/isopropanol cleaning [17,27] or oxygen plasma [166] | APTES silanization | BS3 crosslinker | Si MRR | [17,27,166] |
| Covalent | Monoclonal antibody | Piranha treatment | APTES silanization | N,N-diisopropylethylamine and N,N′-disuccinimidyl carbonate linker | Si MRR | [295] |
| Covalent | Monoclonal antibody | Oxygen plasma | APTES silanization | EDC/NHS activation | Si MRR | [166] |
| Covalent | Antibody | Piranha treatment | APTES silanization | EDC/NHS activation | Si PhC | [165] |
| Covalent | Monoclonal antibody | Piranha treatment | APDMES silanization | Glutaraldehyde linker | Si PhC | [170] |
| Covalent | Polyclonal antibody | Oxidization | CTES silanization | EDC/NHS activation | Si3N4 MRR | [171] |
| Covalent | Polyclonal antibody | - | Thermal hydrolyzation with undecylenic acid | EDC/NHS activation | Porous Si sensor | [176] |
| Covalent | Antibody | Piranha treatment | Native oxide removal with HF | Glutaraldehyde linker | Si3N4/SiO2 MRR | [177] |
| Covalent | Monoclonal and polyclonal antibodies | Cleaning with Micro-90 solution and chromic acid | MPTMS silanization | m-maleimidobenzoyl-N-hydroxysuccinimide ester linker | Si3N4 planar waveguide interferometer | [175] |
| Bioaffinity | Antibody | - | - | Protein A adsorbed on surface | Si MRR (sub-wavelength grating, SWG) | [264] |
| Bioaffinity | Antibody | - | - | Protein A adsorbed on surface | Si MRR (multibox SWG) | [1] |
| Bioaffinity | Antibodies | - | - | Antibody-binding fusion protein consisting of Si-tag and Protein A | Si wafer | [278] |
| Bioaffinity | Antibodies | - | - | Antibody-binding fusion protein consisting of Si-tag and Protein A | Si MRR | [282] |
| Covalent + bioaffinity | Monoclonal antibodies (oligonucleotide-conjugated) | Piranha treatment | HyNic silane surface modification | Intermediate oligonucleotides conjugated with S-4FB for hydrazone bond formation with modified surface, then used as a bioaffinity linker | Si MRR | [163] |
| Covalent + bioaffinity | Antibody | Plasma treatment | Silane-PEG-biotin surface modification | Streptavidin used as a bioaffinity linker to immobilize biotinylated Protein G | Si PhC | [164] |
| Covalent + bioaffinity | Antibodies (biotinylated) | Dry thermal oxidation | APTMS silanization | Sulfo-NHS-LC-LC-biotin linker + streptavidin used as bioaffinity linkers | Porous Si sensor | [288,289] |
| Covalent + bioaffinity | Monoclonal and polyclonal antibodies | HF treatment | MPTMS silanization | N-succinimidyl-4-malemidobutyrate crosslinker + Protein A or G | Multimode optical fibers | [283] |
| Aptamer Immobilization | ||||||
|---|---|---|---|---|---|---|
| Immobilization Strategy | Surface Pre-Treatment | Chemical Surface Modification | Linking Strategy | Targeted Aptamer Terminal Group | Sensor Type | Refs. |
| Covalent | Argon plasma [25] or piranha treatment [185] | GPTMS silanization | Direct conjugation to silanized surface | Amine | SiOxNy MRR | [25,185] |
| Covalent | Oxygen plasma | APTES silanization | Glutaraldehyde linker | Amine | Si MRR | [24] |
| Covalent | Piranha treatment | HyNic silane surface modification | Conjugation of aptamer with S-4FB for hydrazone bond formation with modified surface | Amine | Si MRR | [174] |
| Covalent | Plasma | Silane-PEG-COOH surface modification | EDC/NHS activation | Amine | Si PhC | [164] |
| Covalent | - | Thermal hydrolyzation with undecylenic acid | EDC/NHS activation | Amine | Porous Si sensor | [176] |
| Covalent | - | - | Direct UV crosslinking on surfaces | poly(T) and poly(TC) tags | Glass slides and SiO2 wafers | [272] |
| Immobilization of Nucleic Acid Probes for Hybridization Sensing | |||||||
|---|---|---|---|---|---|---|---|
| Immobilization Strategy Type | Bioreceptor Subtype | Surface Pre-Treatment | Chemical Surface Modification | Linking Strategy | Targeted Probe Terminal Group | Sensor Type | Refs. |
| Covalent | DNA | Piranha treatment | ICPTS silanization | Direct conjugation to silanized surface | Amine | Si wafers and nanostructured Si | [291] |
| Covalent | DNA | NaOH and ethanol/water cleaning | GPTMS silanization | Direct conjugation to silanized surface | Amine | Hydex MRR | [172] |
| Covalent | DNA | Piranha treatment and thermal oxidation | APTES silanization | Sulfosuccinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (Sulfo-SMCC) linker | Thiol | Si MRR and Si PhC | [201] |
| Covalent | DNA | Piranha treatment and thermal oxidation | TEOS-HBA silanization | Base-by-base in situ ssDNA probe synthesis via phosphoramidite method | - | Si MRR and Si PhC | [201] |
| Covalent | DNA | Piranha treatment | APTES silanization + S-HyNic surface modification | Conjugation of DNA probe with S-4FB for hydrazone bond formation with modified surface | Amine | Si MRR | [194] |
| Covalent | DNA | Nitric acid wash | APTES silanization | Succinic anhydride and EDC linker | Amine | Si3N4 MRR | [202] |
| Covalent | PNA | Oxygen plasma | 11-hydroxyundecylphosphonate surface modification | 3-maleimidopropionic-acid-N-hydroxysuccinimide linker | Cysteine tag | Si nanowires | [270] |
| Covalent | DNA | Ethanol and water rinse | - | Direct UV crosslinking on surfaces | No tag and poly(TC) tag | Glass slides (unmodified and GAPS II™ aminosilane coated) | [273] |
| Covalent | DNA | Piranha treatment | APTES silanization | BS3 linker | Amine | Si MRR | [195] |
| Covalent | DNA | Piranha and oxygen plasma treatment | Electrografting of alkyne- and azide-presenting diazonium salts | Cu-catalyzed azide-alkyne click reaction | Azide and alkyne | N-doped Si MRR electrophotonic sensor | [59] |
| Covalent | DNA | Piranha treatment | HyNic silane surface modification | Conjugation of DNA probe with S-4FB for hydrazone bond formation with modified surface | Amine | Si MRR | [109,196,198,303] |
| Covalent | DNA | Oxygen plasma and nitric acid treatment | MPTMS silanization | Direct conjugation via disulphide bond formation | Thiol | Si3N4 Mach-Zehnder interferometer | [200] |
| Covalent | DNA | Oxygen plasma | APTES silanization | Glutaraldehyde linker | Amine | Si MRR | [23,197,199] |
| Covalent | Morpholino | Piranha treatment | APTMS silanization | Glutaraldehyde linker | Amine | Suspended Si photonic microring resonator | [110] |
| Covalent + bioaffinity | DNA (biotinylated) | - | 3-isocyanatepropyl thriethoxysilane vapor silanization | Streptavidin conjugated to silanized surface and used as a bioaffinity linker | Biotin tag | Planar photonic crystal- waveguide-based optical sensor | [203] |
| Peptide and PCC Immobilization | |||||||
|---|---|---|---|---|---|---|---|
| Immobilization Strategy | Bioreceptor Subtype | Surface Pre-Treatment | Chemical Surface Modification | Linking Strategy | Sensor Type | Refs. | |
| Covalent | Peptide | Acetone and isopropanol cleaning | HF treatment to create secondary amines | Glutaraldehyde linker | Si3N4 Mach-Zehnder interferometer | [238] | |
| Passive adsorption | Peptide | Acetone and isopropanol cleaning + Piranha treatment | APTES silanization | - | Si MRR | [174] | |
| Covalent | Peptide | Piranha treatment | APTES silanization | BS3 | Porous Si microcavity | [239] | |
| Covalent | PCC | HF treatment | Thermal hydrolyzation with 1,8-nonadiyne | PCC attachment via click chemistry with copper(I)-catalyzed azide alkyne cycloaddition | Porous Si microcavity sensor | [91,240] | |
| Glycan and Lectin Immobilization | ||||||
|---|---|---|---|---|---|---|
| Immobilization Strategy | Bioreceptor Subtype | Surface Pre-Treatment | Chemical Surface Modification | Linking Strategy | Sensor Type | Refs. |
| Covalent | Glycan | Piranha treatment | UDPA organophosphonate surface modification | DVS activation | Si MRR | [126] |
| Covalent | Glycan | Piranha and UV/ozone plasma treatment | MPTMS silanization | SM(PEG)12 linker | Si3N4 MRR | [133] |
| Covalent | Glycan | Piranha treatment | APTES silanization | BS(PEG)9 linker | Si3N4 MRR | [132] |
| Covalent | Lectin | Hydrogen peroxide and thermal treatment | APTES silanization | Glutaraldehyde linker | Porous Si sensor | [131] |
| Non-covalent | Glycoproteins and neoglycoconjugates | - | - | Passive adsorption | Si MRR | [246] |
| Covalent + bioaffinity | Lectin (biotinylated) | UV/ozone clean | APTMS silanization | NHS-PEG4-biotin linker + avidin | Si3N4 reflectometric interference spectroscopy sensor | [285] |
| HCCD Reporter Immobilization | ||||||
|---|---|---|---|---|---|---|
| Immobilization Strategy | Reporter Type | Surface Pre-Treatment | Chemical Surface Modification | Linking Strategy | Sensor Type | Refs. |
| Covalent + bioaffinity | Biotinylated dsDNA-quantum dot reporters | Thermal oxidation | APTES silanization | Glutaraldehyde linker + streptavidin used as a bioaffinity linker | Porous Si sensor | [137] |
| Covalent + bioaffinity | Biotinylated ssDNA-gold nanoparticle reporters | Plasma treatment | Silane-PEG-biotin surface modification | Streptavidin used as a bioaffinity linker | Si MRR | [139] |
| CRISPR-Ca9-Mediated Sensing | |||||
|---|---|---|---|---|---|
| Immobilization Strategy | Surface Pre-Treatment | Chemical Surface Modification | Nucleic Acid Probe Linking Strategy | Sensor Type | Refs. |
| Covalent | Oxygen plasma | APTES silanization | Glutaraldehyde linker | Si MRR | [143] |
| Lipid Nanodisc Immobilization | |||||
|---|---|---|---|---|---|
| Immobilization Strategy | Surface Pre-Treatment | Chemical Surface Modification | Linking Strategy | Sensor Type | Refs. |
| Non-covalent | Piranha treatment | - | Passive adsorption | Si MRR | [149] |
| Non-covalent | Piranha treatment | - | Passive adsorption | Si MRR | [150] |
| Non-covalent | Acetone and isopropanol wash | - | Passive adsorption | Si MRR | [148] |
| Patterning Technique | Achievable Resolution | Ease of Multiplexing | Spot quality/ Uniformity | Reproducibility | Throughput | Reagent Consumption | Simplicity | Compatibility with System-Level Integration | Cost/ Availability |
|---|---|---|---|---|---|---|---|---|---|
| Microcontact printing (µCP) | 0.1–0.5 µm [32,77] | Poor | Poor | Moderate | High throughput for patterning with 1 bioreceptor; low throughput for multiplexing | Low | Good | Poor; risk of surface and system damage due to stamp contact | Low cost, widely available (e.g., stamp preparation via standard soft lithography techniques; commercial µCP services available via companies including ThunderNIL and BALTFAB) * |
| Pin printing | ~1–100 µm [313,314,315] | Moderate | Poor | Moderate | Low-high, depending on pin design | Low | Poor | Poor; risk of surface and system damage due to pin contact | Expensive for commercial pin printers (e.g., Bioforce Nano eNabler, BioOdyssey Calligrapher miniarrayer, Arrayit microarrayers) * |
| Microfluidic patterning in channels | ~1–10 µm [316,317] | Moderate | Good | Very good | Moderate; simultaneous bioreceptor deposition possible, but limited number of uniquely addressable locations | High | Good | Moderate; µFN design and placement require careful design to avoid system damage; microfluidics often required for assays too | Low cost, widely available (e.g., µFN preparation via standard soft lithography techniques; commercial microfluidics fabrication services available via companies including ThunderNIL and MicruX Technologies) * |
| Inkjet printing | ~30–150 µm [318,319] | Good | Poor | Moderate | Very high | Low | Poor | Good | Expensive, commercial options available (e.g., Scienion sciFLEXARRAYER, Fujifilm Dimatix DMP-2831) * |
| Microfluidic probe (µFP) | 10 µm [83] | Good | Good | Very good | High | Low-moderate | Poor | Moderate; non-contact, but small risk of system damage due to probe motion | No commercial products available |
| Patterning Technique | Sensor Type | Printed Bioreceptors | Multiplexed Bioreceptor Patterning (i.e., 4-Plex, 8-Plex…) | Ref. |
|---|---|---|---|---|
| Tip-mold reactive microcontact printing | Si3N4 MRR | ssDNA | - | [202] |
| Patterning Technique | Sensor Type | Printed Bioreceptors | Multiplexed Bioreceptor Patterning (i.e., 4-Plex, 8-Plex…) | Ref. |
|---|---|---|---|---|
| Hand spotting with micropipette (0.2 µL/drop) | Si MRR | Antibodies | 2-plex, including control | [17] |
| Hand spotting with micropipette (1 µL/drop) | Si MRR | Antibodies | Up to 4-plex, including controls | [168] |
| Hand-spotting with micropipette | Si MRR | ssDNA probes | 4-plex | [109] |
| Hand-spotting with micropipette | Si MRR | ssDNA probes | 4-plex | [303] |
| Hand spotting with micropipette (1 µL/drop) | Si MRR | ssDNA probes | 2-plex | [198] |
| Hand spotting with micropipette (0.1–0.2 µL/spot) | Si MRR | Lipid nanodiscs | 9-plex | [148] |
| Hand spotting with micropipette | Si MRR | ssDNA probes | 3-plex | [196] |
| Hand-spotting with micropipette | Si MRR | ssDNA probes | 4-plex | [194] |
| BioForce Nano eNabler | Si3N4 MRR | Glycans | 2-plex | [133] |
| BioForce Nano eNabler | Si3N4 MRR | Glycan | - | [132] |
| BioForce Nano eNabler | Si3N4 bimodal waveguide interferometric biosensor | BSA | - | [313] |
| BioForce Nano eNabler | Si MRR | ssDNA (subsequently used to immobilize DNA-conjugated antibodies) | 8-plex | [163] |
| BioForce Nano eNabler | Si MRR | Lipid nanodiscs | 7-plex | [149] |
| BioOdyssey Calligrapher miniarrayer | Si3N4 MZI | SARS-CoV-2 peptide | 2-plex, 20–46 overlapping spots | [238] |
| Patterning Technique | Sensor Type | Printed Bioreceptors | Multiplexed Bioreceptor Patterning (i.e., 4-Plex, 8-Plex…) | Ref. |
|---|---|---|---|---|
| Microfluidic patterning in channels using 4-channel Mylar gasket | Si MRR | Antibody, DNA aptamer, ssDNA (control sequence), and BSA (control) | 4-plex, including 2 controls | [174] |
| Microfluidic patterning in channels using 6-channel PDMS gasket | Si MRR | Antibodies | 6-plex, including one control | [22] |
| Microfluidic patterning in channels using 2-channel Mylar gasket | Si MRR | Antibody | Patterning used to functionalize half of the rings with antibody and leave the other half bare for temperature corrections | [161] |
| Microfluidic patterning in channels using 2-channel Mylar gasket | Si MRR | Antibody | Patterning used to functionalize some rings with antibody and leave the rest bare to control for nonspecific binding, temperature, and instrumental drift | [18] |
| Microfluidic patterning in channels using 2-channel Mylar gasket | Si MRR | Antibody | µFN used to perform functionalization in the presence of catalyst on some rings and without catalyst on others | [295] |
| Microfluidic patterning in channels using 1-, 2-, and 4-channel Mylar gaskets | Si MRR | Lipid nanodiscs | Up to 4-plex | [150] |
| Microfluidic patterning in channels using 2- and 4-channel gaskets | Si MRR | Antibodies | Up to 4-plex | [169] |
| Microfluidic patterning in channels using Mylar gasket | Si MRR | Antibodies | 4-plex | [162] |
| Microfluidic patterning in channels using 4-channel PDMS gasket | Si3N4 bimodal waveguide interferometric biosensor | BSA | 2-plex to compare adsorption- and covalent-based BSA immobilization | [313] |
| Patterning Technique | Sensor Type | Printed Bioreceptors | Multiplexed Bioreceptor Patterning (i.e., 4-Plex, 8-Plex…) | Ref. |
|---|---|---|---|---|
| Piezoelectric non-contact printing (Scienion sciFLEXARRAYER S5) | Si MRR | SS-A antigen | - | [293] |
| Piezoelectric non-contact printing (Scienion sciFLEXARRAYER S3) | Si MRR | Glycoconjugates and fluorescently labeled streptavidin | Up to 4-plex | [246] |
| Piezoelectric non-contact printing (Scienion SciFLEXARRAYER SX) | Si3N4 MRR | Antibody, antigen, BSA | 2-plex (performed on 2-ring chips; one ring spotted with antibody or antigen as the receptor and another with BSA as a control) | [167] |
| Piezoelectric non-contact printing (Scienion sciFLEXARRAYER S12) | Si3N4 MZI | Peptides for volatile chemicals | 64 MZI sensor array | [319] |
| Piezoelectric non-contact printing (FUJIFILM Dimatix DMP-2831) | SiP microcantilevers | Biotin | - | [318] |
| Piezoelectric non-contact printing (FUJIFILM Dimatix DMP-2831) | Silicon nitride MZI | Biotin-modified polyethyleneimine functional polymer and benzophenone dextran hydrogel | - | [347] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puumala, L.S.; Grist, S.M.; Morales, J.M.; Bickford, J.R.; Chrostowski, L.; Shekhar, S.; Cheung, K.C. Biofunctionalization of Multiplexed Silicon Photonic Biosensors. Biosensors 2023, 13, 53. https://doi.org/10.3390/bios13010053
Puumala LS, Grist SM, Morales JM, Bickford JR, Chrostowski L, Shekhar S, Cheung KC. Biofunctionalization of Multiplexed Silicon Photonic Biosensors. Biosensors. 2023; 13(1):53. https://doi.org/10.3390/bios13010053
Chicago/Turabian StylePuumala, Lauren S., Samantha M. Grist, Jennifer M. Morales, Justin R. Bickford, Lukas Chrostowski, Sudip Shekhar, and Karen C. Cheung. 2023. "Biofunctionalization of Multiplexed Silicon Photonic Biosensors" Biosensors 13, no. 1: 53. https://doi.org/10.3390/bios13010053
APA StylePuumala, L. S., Grist, S. M., Morales, J. M., Bickford, J. R., Chrostowski, L., Shekhar, S., & Cheung, K. C. (2023). Biofunctionalization of Multiplexed Silicon Photonic Biosensors. Biosensors, 13(1), 53. https://doi.org/10.3390/bios13010053