Aggregation-Induced Ignition of Near-Infrared Phosphorescence of Non-Symmetric [Pt(C^N*N’^C’)] Complex in Poly(caprolactone)-based Block Copolymer Micelles: Evaluating the Alternative Design of Near-Infrared Oxygen Biosensors

 ,
,  ,
, 
 , and
, and
Abstract
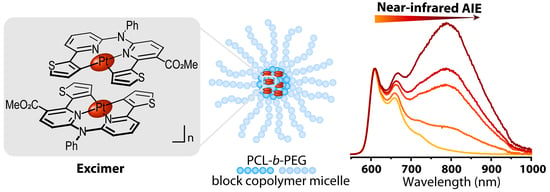
:
1. Introduction
2. Materials and Methods
2.1. General Comments
2.2. Handling of PCL-b-PEG Block Copolymers and Preparation of Block Copolymer Micelles
2.3. Measurerents of Two-Photon Properties
2.4. Confocal Microscopy and PLIM
3. Results
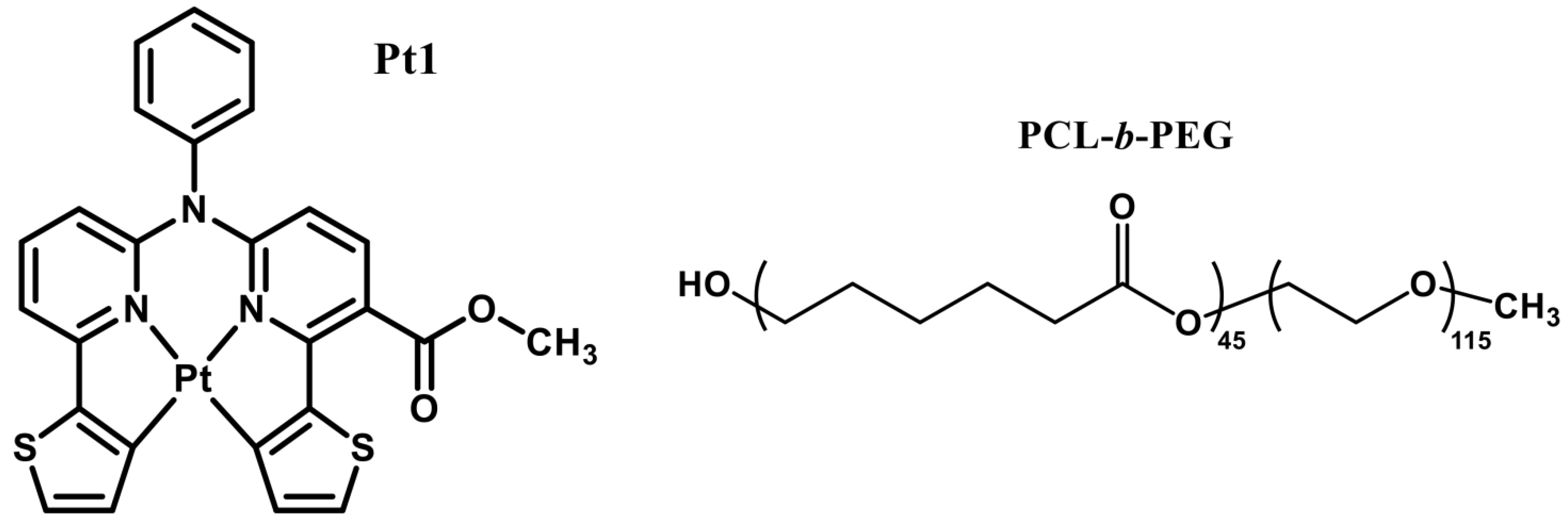
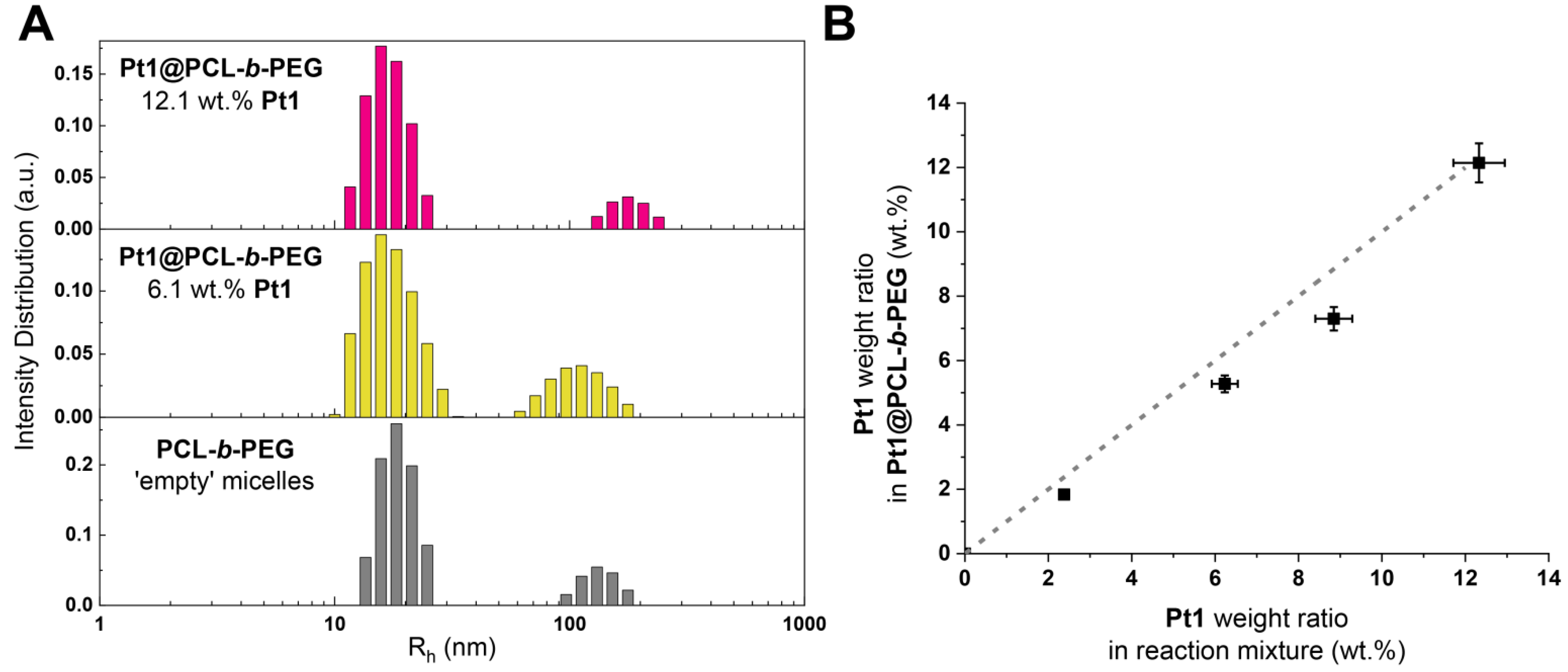
3.1. Preparation and Structural Characterization of Phosphorescent Micelles
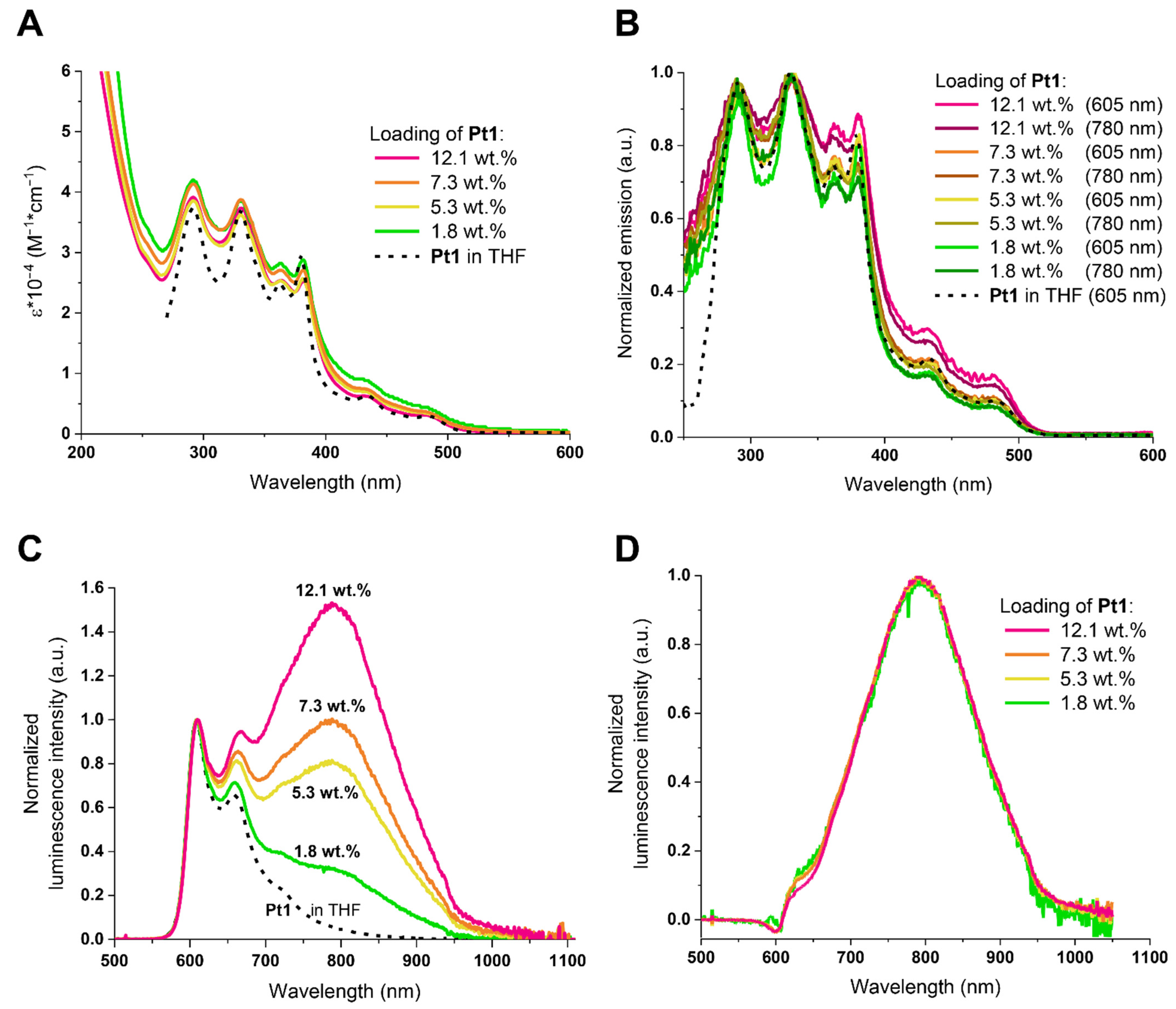
3.2. Photophysical Characterization of Phosphorescent Micelles
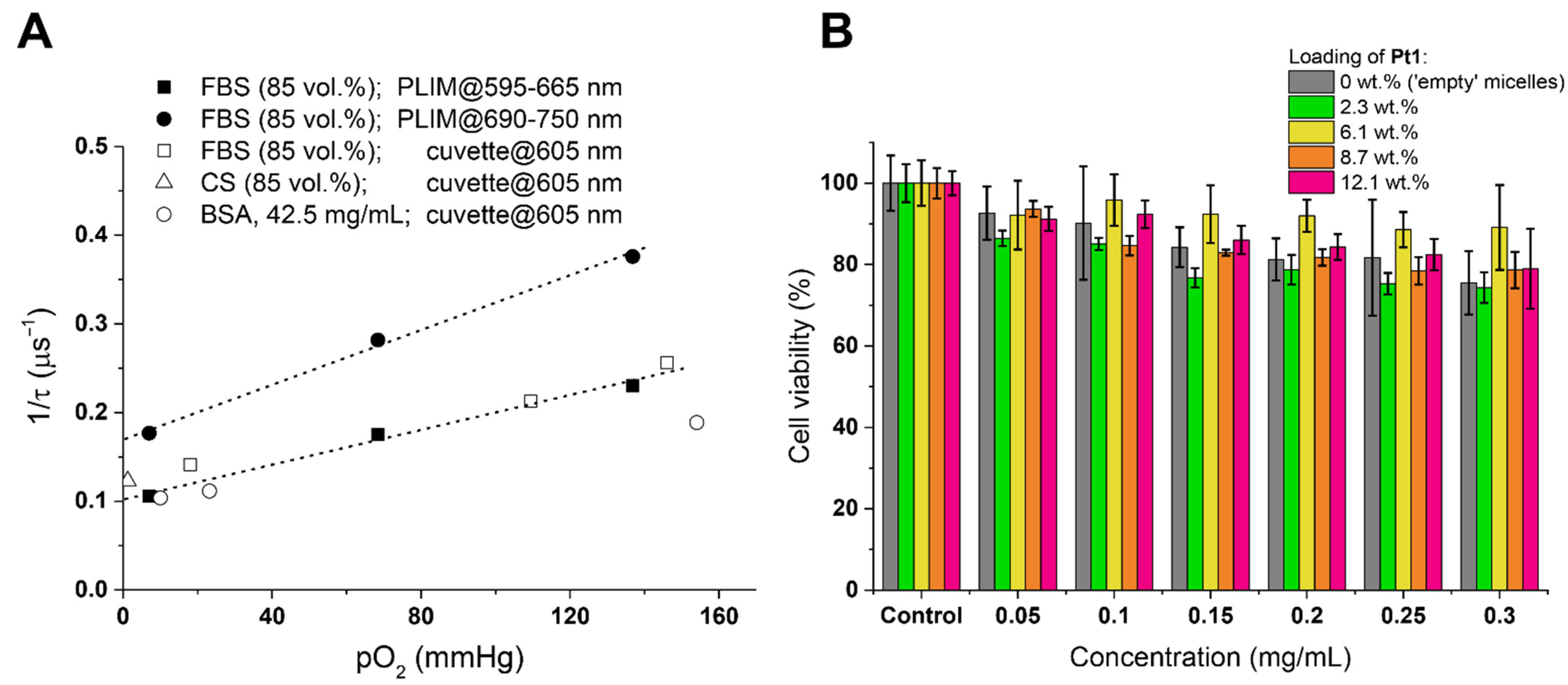
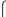 s for Pt1@PCL-b-PEG loaded by 12.1 wt.% of Pt1 in aerated water dispersion at 37.6 °C), and its contribution in the decay varies from 92 to 95% (Table S2). These observations indicate that for the Pt1@PCL-b-PEG samples containing 12.1 wt.% of the complex (1) the emission decay curves in water and PBS (Figures S7 and S8) may be successfully fitted with the monoexponential function (Table 1 and Table S2) and (2) lifetime measurements may be carried out using any of these detection wavelengths due to the major contribution of the NIR emission band to the decay independent of the wavelength choice. Lifetimes data at different O2 partial pressures were used to construct Stern–Volmer plots for Pt1@PCL-b-PEG dispersions (with Pt1 loading ranging from 2.3 to 12.1 wt.%) in water and PBS at 37.6 °C (Figure 3A). Lifetime sensitivity to oxygen estimated as the τ0/τ160 ratio (where τ0 refers to 0 mmHg O2, τ160 to 160 mmHg O2) varied from 3.0 for the dispersions with 2.3 wt.% of Pt1 (τ160 = 1.30 ± 0.07 s; τ0 = 3.9 ± 0. 2 μs) to approx. 1.5 for that with 12.1 wt.% of Pt1 (τ160 = 0.60 ± 0.03 μs; τ0 = 0.92 ± 0.05 μs), see Figure 3B and Table 1. The corresponding Stern–Volmer plots were linear for all micellar compositions (Figure 3A). Stern–Volmer constants (KSV) display values from 0.0033 ± 0.0002 mmHg−1 (Pt1 loading: 12.1 wt.%) to 0.0124 ± 0.0001 mmHg−1 (Pt1 loading: 2.3 wt.%) in Table 1. KSV values strongly depend on Pt1 loading and level off above the compositions of 9 wt.% Pt1, whereas quenching constants, KQ do not depend on the Pt1 content and equal to 0.0034 ± 0.0002 μs−1mmHg−1 for all studied micelles (Table 1).
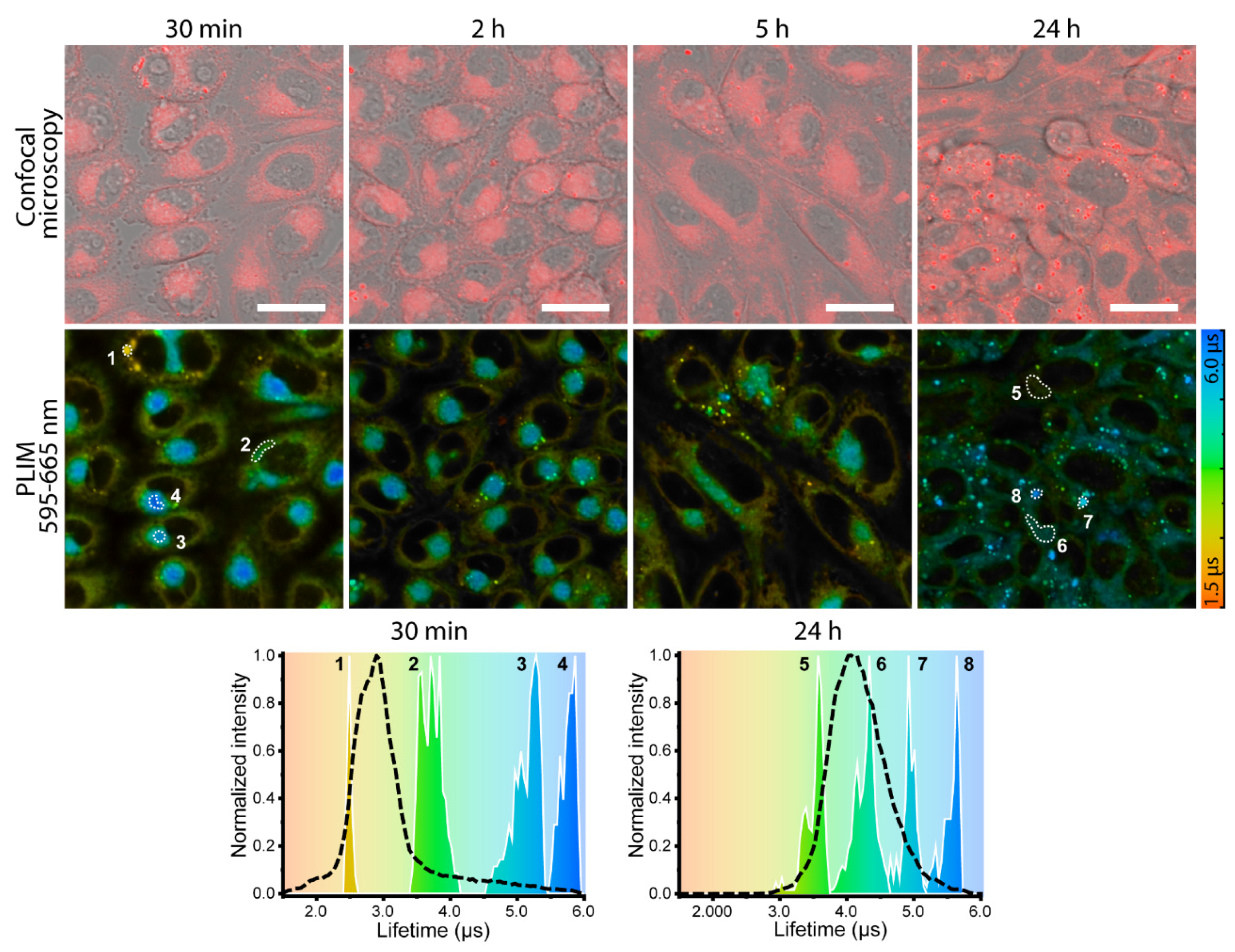
s for Pt1@PCL-b-PEG loaded by 12.1 wt.% of Pt1 in aerated water dispersion at 37.6 °C), and its contribution in the decay varies from 92 to 95% (Table S2). These observations indicate that for the Pt1@PCL-b-PEG samples containing 12.1 wt.% of the complex (1) the emission decay curves in water and PBS (Figures S7 and S8) may be successfully fitted with the monoexponential function (Table 1 and Table S2) and (2) lifetime measurements may be carried out using any of these detection wavelengths due to the major contribution of the NIR emission band to the decay independent of the wavelength choice. Lifetimes data at different O2 partial pressures were used to construct Stern–Volmer plots for Pt1@PCL-b-PEG dispersions (with Pt1 loading ranging from 2.3 to 12.1 wt.%) in water and PBS at 37.6 °C (Figure 3A). Lifetime sensitivity to oxygen estimated as the τ0/τ160 ratio (where τ0 refers to 0 mmHg O2, τ160 to 160 mmHg O2) varied from 3.0 for the dispersions with 2.3 wt.% of Pt1 (τ160 = 1.30 ± 0.07 s; τ0 = 3.9 ± 0. 2 μs) to approx. 1.5 for that with 12.1 wt.% of Pt1 (τ160 = 0.60 ± 0.03 μs; τ0 = 0.92 ± 0.05 μs), see Figure 3B and Table 1. The corresponding Stern–Volmer plots were linear for all micellar compositions (Figure 3A). Stern–Volmer constants (KSV) display values from 0.0033 ± 0.0002 mmHg−1 (Pt1 loading: 12.1 wt.%) to 0.0124 ± 0.0001 mmHg−1 (Pt1 loading: 2.3 wt.%) in Table 1. KSV values strongly depend on Pt1 loading and level off above the compositions of 9 wt.% Pt1, whereas quenching constants, KQ do not depend on the Pt1 content and equal to 0.0034 ± 0.0002 μs−1mmHg−1 for all studied micelles (Table 1).3.3. In Vitro Investigation of Pt1@PCL-b-PEG Oxygen Probes Inside CHO-K1 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Luo, J.; Xie, Z.; Lam, J.W.Y.; Cheng, L.; Tang, B.Z.; Chen, H.; Qiu, C.; Kwok, H.S.; Zhan, X.; Liu, Y.; et al. Aggregation-Induced Emission of 1-Methyl-1,2,3,4,5-Pentaphenylsilole. Chem. Commun. 2001, 18, 1740–1741. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Lam, J.W.Y.; Tang, B.Z. Aggregation-Induced Emission. Chem. Soc. Rev. 2011, 40, 5361–5388. [Google Scholar] [CrossRef]
- Mei, J.; Hong, Y.; Lam, J.W.Y.; Qin, A.; Tang, Y.; Tang, B.Z. Aggregation-Induced Emission: The Whole Is More Brilliant than the Parts. Adv. Mater. 2014, 26, 5429–5479. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.; Leung, N.L.C.; Kwok, R.T.K.; Lam, J.W.Y.; Tang, B.Z. Aggregation-Induced Emission: Together We Shine, United We Soar! Chem. Rev. 2015, 115, 11718–11940. [Google Scholar] [CrossRef] [PubMed]
- Maisuls, I.; Wang, C.; Gutierrez Suburu, M.E.; Wilde, S.; Daniliuc, C.-G.; Brünink, D.; Doltsinis, N.L.; Ostendorp, S.; Wilde, G.; Kösters, J.; et al. Ligand-Controlled and Nanoconfinement-Boosted Luminescence Employing Pt (ii) and Pd (ii) Complexes: From Color-Tunable Aggregation-Enhanced Dual Emitters towards Self-Referenced Oxygen Reporters. Chem. Sci. 2021, 12, 3270–3281. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Sun, J.Z.; Tang, B.Z. Reaction-Based AIE-Active Fluorescent Probes for Selective Detection and Imaging. Isr. J. Chem. 2018, 58, 845–859. [Google Scholar] [CrossRef]
- Yu, H.; Chen, B.; Huang, H.; He, Z.; Sun, J.; Wang, G.; Gu, X.; Tang, B.Z. AIE-Active Photosensitizers: Manipulation of Reactive Oxygen Species Generation and Applications in Photodynamic Therapy. Biosensors 2022, 12, 348. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Wang, X.; Chen, Z.; Xiang, S.; Wang, T.; Feng, H.T.; Tang, B.Z. Organometallic AIEgens for Biological Theranostics. Mater. Chem. Front. 2021, 5, 3281–3297. [Google Scholar] [CrossRef]
- Liu, J.; Jin, C.; Yuan, B.; Liu, X.; Chen, Y.; Ji, L.; Chao, H. Selectively Lighting up Two-Photon Photodynamic Activity in Mitochondria with AIE-Active Iridium (iii) Complexes. Chem. Commun. 2017, 53, 2052–2055. [Google Scholar] [CrossRef]
- Qiu, K.; Ouyang, M.; Liu, Y.; Huang, H.; Liu, C.; Chen, Y.; Ji, L.; Chao, H. Two-Photon Photodynamic Ablation of Tumor Cells by Mitochondria-Targeted Iridium (iii) Complexes in Aggregate States. J. Mater. Chem. B 2017, 5, 5488–5498. [Google Scholar] [CrossRef]
- Sheet, S.K.; Sen, B.; Patra, S.K.; Rabha, M.; Aguan, K.; Khatua, S. Aggregation-Induced Emission-Active Ruthenium(II) Complex of 4,7-Dichloro Phenanthroline for Selective Luminescent Detection and Ribosomal RNA Imaging. ACS Appl. Mater. Interfaces 2018, 10, 14356–14366. [Google Scholar] [CrossRef] [PubMed]
- Sen, B.; Patra, S.K.; Khatua, S. Ruthenium (II) Polypyridine Complex-Based Aggregation-Induced Emission Luminogen for Rapid and Selective Detection of Phosgene in Solution and in the Gas Phase. Inorg. Chem. 2021, 60, 19175–19188. [Google Scholar] [CrossRef] [PubMed]
- Sathish, V.; Ramdass, A.; Lu, Z.-Z.; Velayudham, M.; Thanasekaran, P.; Lu, K.-L.; Rajagopal, S. Aggregation-Induced Emission Enhancement in Alkoxy-Bridged Binuclear Rhenium (I) Complexes: Application as Sensor for Explosives and Interaction with Microheterogeneous Media. J. Phys. Chem. B 2013, 117, 14358–14366. [Google Scholar] [CrossRef]
- Liu, H.-Q.; Peng, Y.-X.; Zhang, Y.; Yang, X.-Q.; Feng, F.-D.; Luo, X.-B.; Yan, L.-S.; Hu, B.; Huang, W. Aggregation-Induced Emission Generation via Simultaneous N-Alkylation and rhenium (I) Tricarbonyl Complexation for 2-(2-thienyl)imidazo[4,5-f][1,10]-Phenanthroline. Dye. Pigment. 2020, 174, 108074. [Google Scholar] [CrossRef]
- Gabr, M.T.; Pigge, F.C. Rhenium Tricarbonyl Complexes of AIE Active Tetraarylethylene Ligands: Tuning Luminescence Properties and HSA-Specific Binding. Dalt. Trans. 2017, 46, 15040–15047. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zou, H.; Wang, X.; He, B.; Liu, S.H.; Lam, J.W.Y.; Tang, B.Z. New AIE-Active Copolymers with Au(I) Isocyanide Acrylate Units. J. Inorg. Organomet. Polym. Mater. 2020, 30, 1490–1496. [Google Scholar] [CrossRef]
- Cuerva, C.; Campo, J.A.; Cano, M.; Lodeiro, C. Multi-Stimuli-Responsive Properties of Aggregation-Enhanced Emission-Active Unsymmetrical Pt II Metallomesogens through Self-Assembly. Chem.–A Eur. J. 2019, 25, 12046–12051. [Google Scholar] [CrossRef]
- Martínez-Junquera, M.; Lalinde, E.; Moreno, M.T.; Alfaro-Arnedo, E.; López, I.P.; Larráyoz, I.M.; Pichel, J.G. Luminescent Cyclometalated Platinum (ii) Complexes with Acyclic Diaminocarbene Ligands: Structural, Photophysical and Biological Properties. Dalt. Trans. 2021, 50, 4539–4554. [Google Scholar] [CrossRef]
- Gong, Z.-L.; Tang, K.; Zhong, Y.-W. A Carbazole-Bridged Biscyclometalated Diplatinum Complex: Synthesis, Characterization, and Dual-Mode Aggregation-Enhanced Phosphorescence. Inorg. Chem. 2021, 60, 6607–6615. [Google Scholar] [CrossRef]
- Yang, J.; Sun, L.; Hao, L.; Yang, G.-G.; Zou, Z.-C.; Cao, Q.; Ji, L.-N.; Mao, Z.-W. A Halogen Ion-Selective Phosphorescence Turn-on Probe Based on Induction of Pt–Pt Interactions. Chem. Commun. 2019, 55, 11191–11194. [Google Scholar] [CrossRef]
- Kritchenkov, I.S.; Elistratova, A.A.; Sokolov, V.V.; Chelushkin, P.S.; Shirmanova, M.V.; Lukina, M.M.; Dudenkova, V.V.; Shcheslavskiy, V.I.; Kalinina, S.; Reeß, K.; et al. A Biocompatible Phosphorescent Ir (iii) Oxygen Sensor Functionalized with Oligo (ethylene Glycol) Groups: Synthesis, Photophysics and Application in PLIM Experiments. New J. Chem. 2020, 44, 10459–10471. [Google Scholar] [CrossRef]
- Kritchenkov, I.S.; Solomatina, A.I.; Kozina, D.O.; Porsev, V.V.; Sokolov, V.V.; Shirmanova, M.V.; Lukina, M.M.; Komarova, A.D.; Shcheslavskiy, V.I.; Belyaeva, T.N.; et al. Biocompatible Ir (III) Complexes as Oxygen Sensors for Phosphorescence Lifetime Imaging. Molecules 2021, 26, 2898. [Google Scholar] [CrossRef] [PubMed]
- Esipova, T.V.; Barrett, M.J.P.; Erlebach, E.; Masunov, A.E.; Weber, B.; Vinogradov, S.A. Oxyphor 2P: A High-Performance Probe for Deep-Tissue Longitudinal Oxygen Imaging. Cell Metab. 2019, 29, 736–744. [Google Scholar] [CrossRef]
- Tsytsarev, V.; Arakawa, H.; Borisov, S.; Pumbo, E.; Erzurumlu, R.S.; Papkovsky, D.B. In Vivo Imaging of Brain Metabolism Activity Using a Phosphorescent Oxygen-Sensitive Probe. J. Neurosci. Methods 2013, 216, 146–151. [Google Scholar] [CrossRef]
- Chien, J.S.; Mohammed, M.; Eldik, H.; Ibrahim, M.M.; Martinez, J.; Nichols, S.P.; Wisniewski, N.; Klitzman, B. Injectable Phosphorescence-Based Oxygen Biosensors Identify Post Ischemic Reactive Hyperoxia. Sci. Rep. 2017, 7, 8255. [Google Scholar] [CrossRef]
- Rivera, K.R.; Pozdin, V.A.; Young, A.T.; Erb, P.D.; Wisniewski, N.A.; Magness, S.T.; Daniele, M. Integrated Phosphorescence-Based Photonic Biosensor (iPOB) for Monitoring Oxygen Levels in 3D Cell Culture Systems. Biosens. Bioelectron. 2019, 123, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Chelushkin, P.S.; Shakirova, J.R.; Kritchenkov, I.S.; Baigildin, V.A.; Tunik, S.P. Phosphorescent NIR Emitters for Biomedicine: Applications, Advances and Challenges. Dalt. Trans. 2022, 51, 1257–1280. [Google Scholar] [CrossRef]
- Borisov, S.M.; Mayr, T.; Mistlberger, G.; Waich, K.; Koren, K.; Chojnacki, P.; Klimant, I. Precipitation as a Simple and Versatile Method for Preparation of Optical Nanochemosensors. Talanta 2009, 79, 1322–1330. [Google Scholar] [CrossRef] [PubMed]
- Solomatina, A.I.; Galenko, E.E.; Kozina, D.O.; Kalinichev, A.A.; Baigildin, V.A.; Prudovskaya, N.A.; Shakirova, J.R.; Khlebnikov, A.F.; Porsev, V.V.; Evarestov, R.A.; et al. Non-Symmetric [Pt(C^N*N’^C’)] Complexes: Aggregation Induced Emission in Solid State and in Nanoparticles Tuned by Ligand Structure. Chem. Eur. J. 2022; submitted.
- Weissleder, R.A. Clearer Vision for in vivo Imaging. Nat. Biotechnol. 2001, 19, 316–317. [Google Scholar] [CrossRef]
- Elistratova, A.A.; Kritchenkov, I.S.; Lezov, A.A.; Gubarev, A.S.; Solomatina, A.I.; Kachkin, D.V.; Shcherbina, N.A.; Liao, Y.-C.; Liu, Y.-C.; Yang, Y.-Y.; et al. Lifetime Oxygen Sensors Based on Block Copolymer Micelles and Non-Covalent Human Serum Albumin Adducts Bearing Phosphorescent Near-Infrared Iridium (III) Complex. Eur. Polym. J. 2021, 159, 110761. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Conway, J.R.W.; Warren, S.C.; Herrmann, D.; Murphy, K.J.; Cazet, A.S.; Vennin, C.; Shearer, R.F.; Killen, M.J.; Magenau, A.; Mélénec, P.; et al. Intravital Imaging to Monitor Therapeutic Response in Moving Hypoxic Regions Resistant to PI3K Pathway Targeting in Pancreatic Cancer. Cell Rep. 2018, 23, 3312–3326. [Google Scholar] [CrossRef] [PubMed]
- Vezzu, D.A.K.; Deaton, J.C.; Jones, J.S.; Bartolotti, L.; Harris, C.F.; Marchetti, A.P.; Kondakova, M.; Pike, R.D.; Huo, S. Highly Luminescent Tetradentate Bis-Cyclometalated Platinum Complexes: Design, Synthesis, Structure, Photophysics, and Electroluminescence Application. Inorg. Chem. 2010, 49, 5107–5119. [Google Scholar] [CrossRef]
- Wilde, S.; González-Abradelo, D.; Daniliuc, C.-G.; Böckmann, M.; Doltsinis, N.L.; Strassert, C.A. Fluorination-Controlled Aggregation and Intermolecular Interactions in Pt (II) Complexes with Tetradentate Luminophores. Isr. J. Chem. 2018, 58, 932–943. [Google Scholar] [CrossRef]
- Fasano, M.; Curry, S.; Terreno, E.; Galliano, M.; Fanali, G.; Narciso, P.; Notari, S.; Ascenzi, P. The Extraordinary Ligand Binding Properties of Human Serum Albumin. IUBMB Life (Int. Union Biochem. Mol. Biol. Life) 2005, 57, 787–796. [Google Scholar] [CrossRef]
- Chelushkin, P.S.; Krupenya, D.V.; Tseng, Y.-J.; Kuo, T.-Y.; Chou, P.-T.; Koshevoy, I.O.; Burov, S.V.; Tunik, S.P. Water-Soluble Noncovalent Adducts of the Heterometallic Copper Subgroup Complexes and Human Serum Albumin with Remarkable Luminescent Properties. Chem. Commun. 2014, 50, 849–851. [Google Scholar] [CrossRef]
- Solomatina, A.I.; Baigildin, V.A.; Zhukovsky, D.D.; Krupenya, D.V.; Koshel, E.I.; Shcheslavskiy, V.I.; Tunik, S.P.; Chelushkin, P.S. How to Avoid Protein Aggregation to Improve Cellular Uptake of Albumin-Based Conjugates: Towards the Rational Design of Cell-Penetrable Phosphorescent Probes. Colloid Polym. Sci. 2019, 297, 325–337. [Google Scholar] [CrossRef]
- Kritchenkov, I.S.; Chelushkin, P.S.; Sokolov, V.V.; Pavlovskiy, V.V.; Porsev, V.V.; Evarestov, R.A.; Tunik, S.P. Near-Infrared [Ir(N∧C)2(N∧N)] + Emitters and Their Noncovalent Adducts with Human Serum Albumin: Synthesis and Photophysical and Computational Study. Organometallics 2019, 38, 3740–3751. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Loading of Pt1 | DispersionMedium | τ0, μs | τ160, μs | KQ, μs−1 mmHg−1 | KSV, mmHg−1 |
|---|---|---|---|---|---|
| 2.3 wt.% | H2O | 3.9 ± 0.2 | 1.30 ± 0.07 | 0.00320 ± 0.00002 | 0.0124 ± 0.0001 |
| PBS | 4.1 ± 0.2 | 1.30 ± 0.07 | 0.00325 ± 0.00004 | 0.0132 ± 0.0003 | |
| 6.1 wt.% | H2O | 1.38 ± 0.07 | 0.80 ± 0.04 | 0.0033 ± 0.0001 | 0.0046 ± 0.0002 |
| PBS | 1.47 ± 0.07 | 0.78 ± 0.04 | 0.0038 ± 0.0003 | 0.0056 ± 0.0005 | |
| 8.7 wt.% | H2O | 1.04 ± 0.05 | 0.67 ± 0.03 | 0.0032 ± 0.0001 | 0.0034 ± 0.0001 |
| 12.1 wt.% | H2O | 0.92 ± 0.05 | 0.60 ± 0.03 | 0.0036 ± 0.0002 | 0.0033 ± 0.0002 |
| PBS | 0.93 ± 0.05 | 0.59 ± 0.03 | 0.0039 ± 0.0002 | 0.0036 ± 0.0002 |
| Dispersion | C, mg/mL | pH | T, °C | 2.3 wt.% Pt1 | 12.1 wt.% Pt1 | ||
|---|---|---|---|---|---|---|---|
| pO2, mmHg | τ, μs | pO2, mmHg | τ, μs | ||||
| H2O | 0.2 | n.d. | 37.6 | 160.2 ± 0.4 | 1.29 ± 0.06 | 157.9 ± 0.5 | 0.60 ± 0.03 |
| PBS | 0.2 | 7.2 | 25.6 | 161 ± 2 | 1.82 ± 0.09 | 149.5 ± 0.7 | 0.85 ± 0.04 |
| PBS | 0.2 | 7.2 | 37.6 | 163.0 ± 0.6 | 1.29 ± 0.06 | 152 ± 1 | 0.60 ± 0.03 |
| PBS | 0.4 | 7.2 | 37.6 | 155.5 ± 0.6 | 1.34 ± 0.06 | 151.6 ± 0.8 | 0.60 ± 0.03 |
| PBS | 0.2 | 8.1 | 37.6 | 162.1 ± 0.2 | 1.26 ± 0.06 | 153.9 ± 0.4 | 0.63 ± 0.05 |
| PBS | 0.2 | 6.6 | 37.6 | 166 ± 2 | 1.28 ± 0.06 | 157.7 ± 0.8 | 0.60 ± 0.04 |
| PBS | 0.2 | 5.8 | 37.6 | 170 ± 2 | 1.26 ± 0.06 | 155.0± 0.7 | 0.61 ± 0.05 |
| PBS + H2O2 (100 nM) | 0.2 | 7.2 | 37.6 | 158 ± 1 | 1.31 ± 0.06 | not determined | |
| PBS + BSA | 0.2 | 7.2 | 37.6 | 160 ± 2 | 1.88 ± 0.09 a | 154.2 ± 0.4 | 1.14 ± 0.06 b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zharskaia, N.A.; Solomatina, A.I.; Liao, Y.-C.; Galenko, E.E.; Khlebnikov, A.F.; Chou, P.-T.; Chelushkin, P.S.; Tunik, S.P. Aggregation-Induced Ignition of Near-Infrared Phosphorescence of Non-Symmetric [Pt(C^N*N’^C’)] Complex in Poly(caprolactone)-based Block Copolymer Micelles: Evaluating the Alternative Design of Near-Infrared Oxygen Biosensors. Biosensors 2022, 12, 695. https://doi.org/10.3390/bios12090695
Zharskaia NA, Solomatina AI, Liao Y-C, Galenko EE, Khlebnikov AF, Chou P-T, Chelushkin PS, Tunik SP. Aggregation-Induced Ignition of Near-Infrared Phosphorescence of Non-Symmetric [Pt(C^N*N’^C’)] Complex in Poly(caprolactone)-based Block Copolymer Micelles: Evaluating the Alternative Design of Near-Infrared Oxygen Biosensors. Biosensors. 2022; 12(9):695. https://doi.org/10.3390/bios12090695
Chicago/Turabian StyleZharskaia, Nina A., Anastasia I. Solomatina, Yu-Chan Liao, Ekaterina E. Galenko, Alexander F. Khlebnikov, Pi-Tai Chou, Pavel S. Chelushkin, and Sergey P. Tunik. 2022. "Aggregation-Induced Ignition of Near-Infrared Phosphorescence of Non-Symmetric [Pt(C^N*N’^C’)] Complex in Poly(caprolactone)-based Block Copolymer Micelles: Evaluating the Alternative Design of Near-Infrared Oxygen Biosensors" Biosensors 12, no. 9: 695. https://doi.org/10.3390/bios12090695
APA StyleZharskaia, N. A., Solomatina, A. I., Liao, Y.-C., Galenko, E. E., Khlebnikov, A. F., Chou, P.-T., Chelushkin, P. S., & Tunik, S. P. (2022). Aggregation-Induced Ignition of Near-Infrared Phosphorescence of Non-Symmetric [Pt(C^N*N’^C’)] Complex in Poly(caprolactone)-based Block Copolymer Micelles: Evaluating the Alternative Design of Near-Infrared Oxygen Biosensors. Biosensors, 12(9), 695. https://doi.org/10.3390/bios12090695