Single Plane Illumination Microscopy for Microfluidic Device Imaging

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
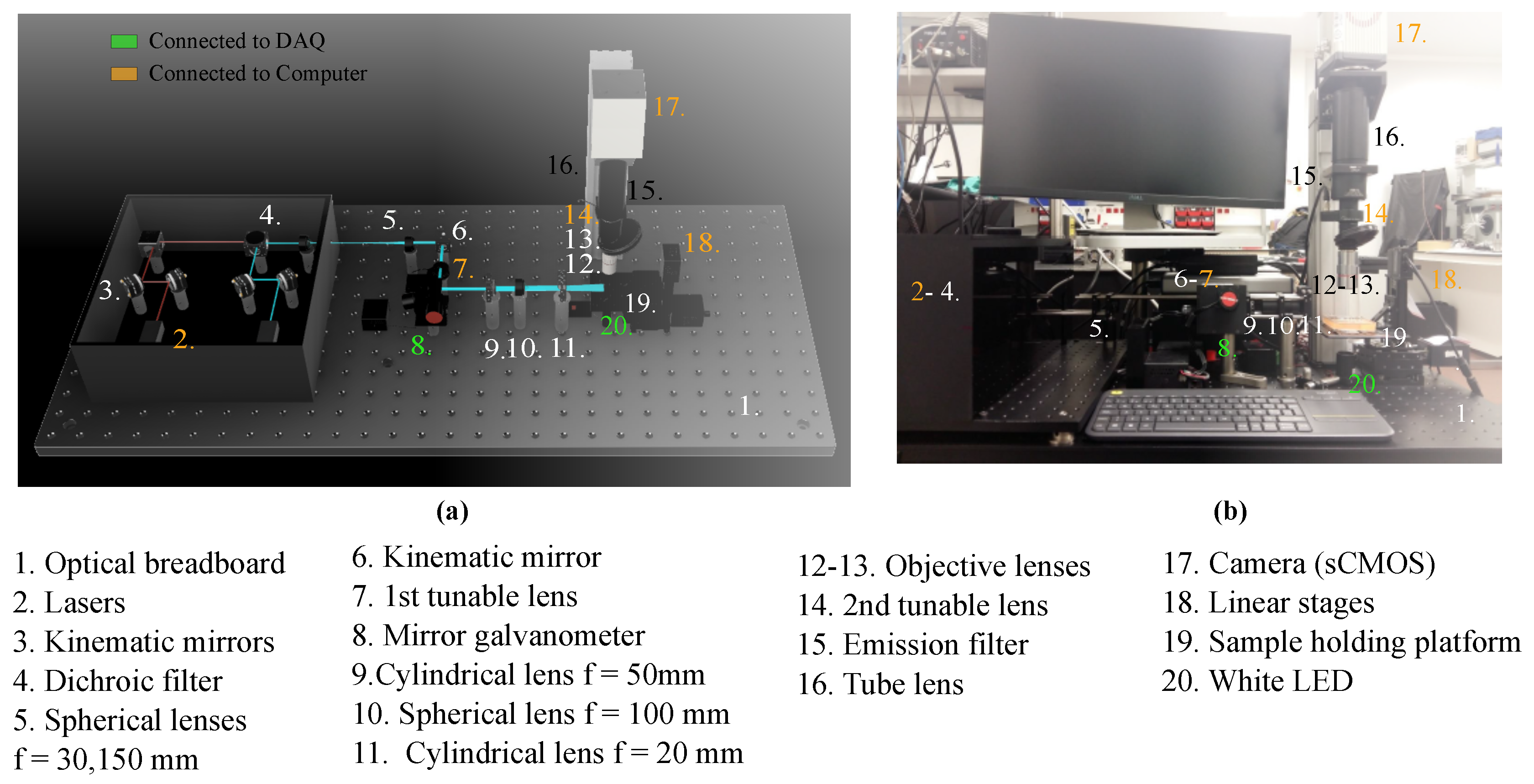
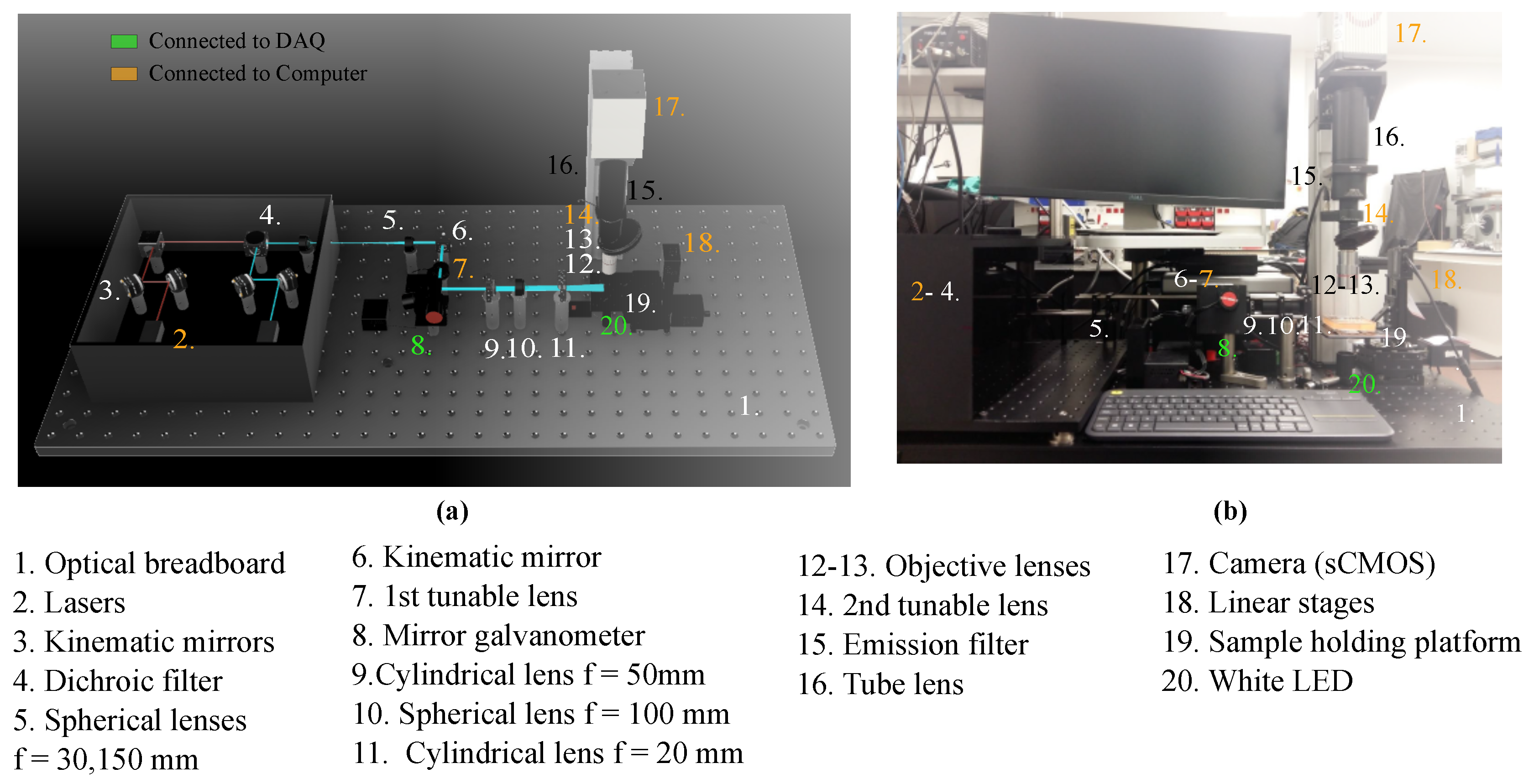
2.1. Imaging System Components and Architecture
2.1.1. General Setup
2.1.2. Excitation Path
2.1.3. Emission Path
2.1.4. Software
2.2. Sample Preparation
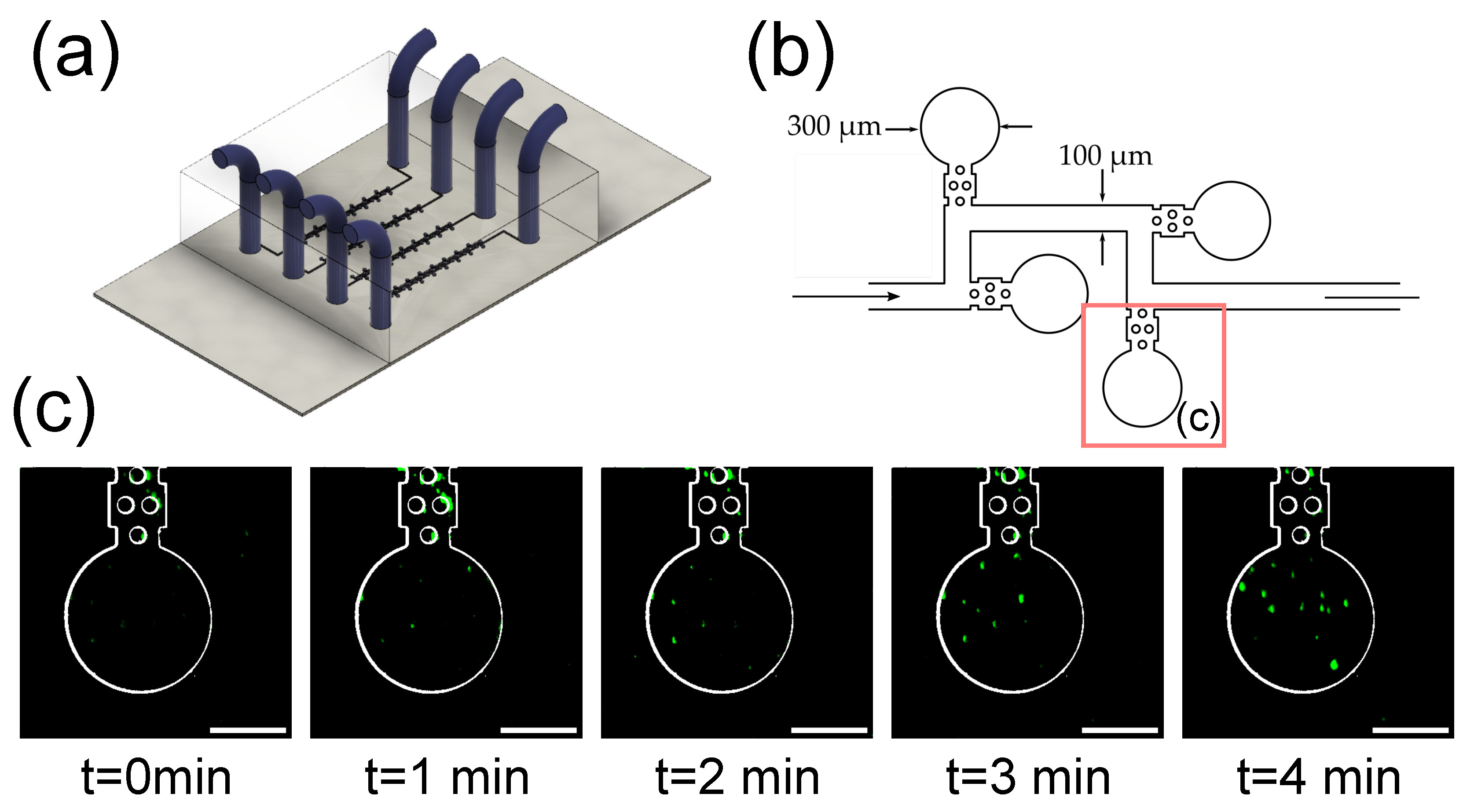
2.2.1. Microfluidic Device Fabrication
2.2.2. Spheroid Preparation
2.2.3. Skin Dermal Compartment Preparation
3. Results
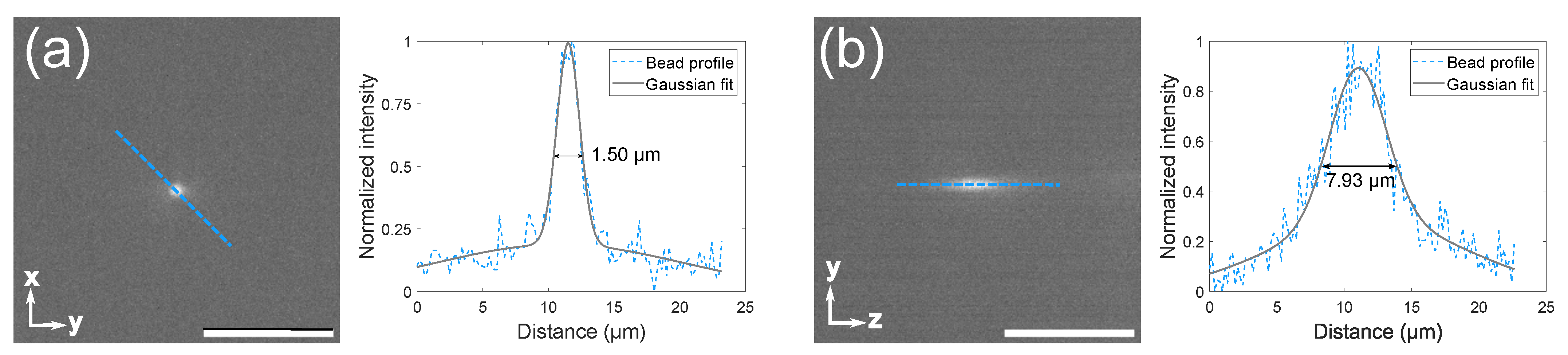
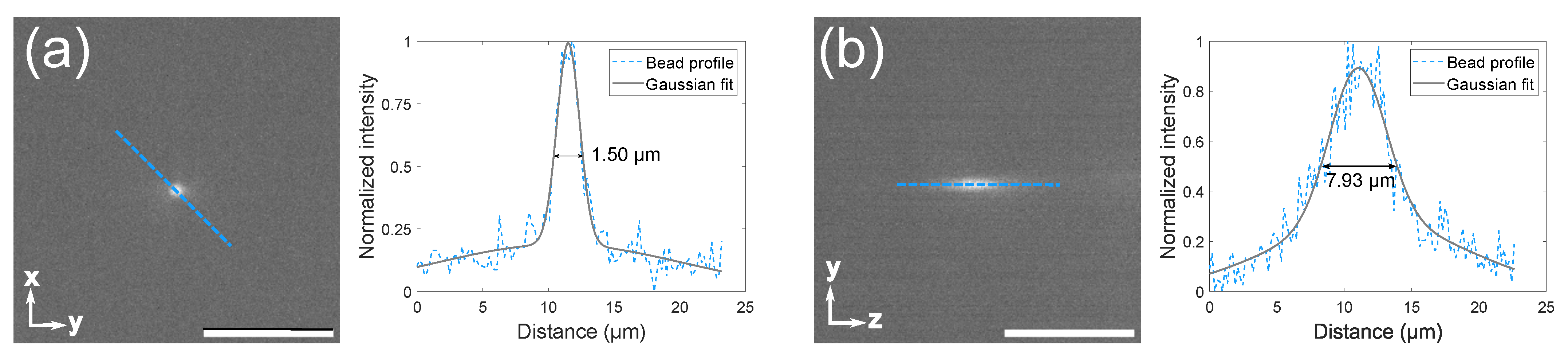
3.1. Microscope Setup and Characterization
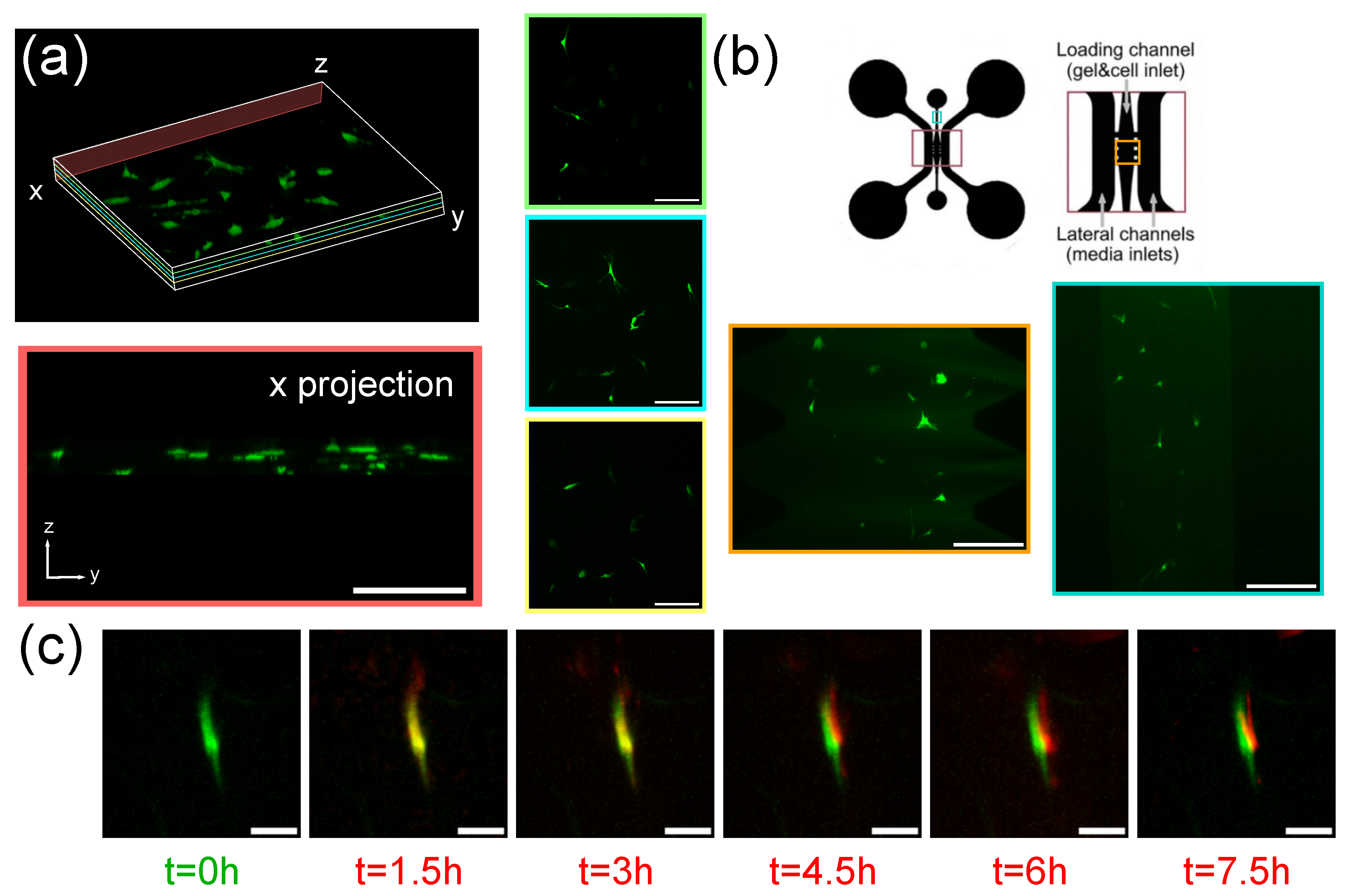
3.2. Imaging of Spheroids
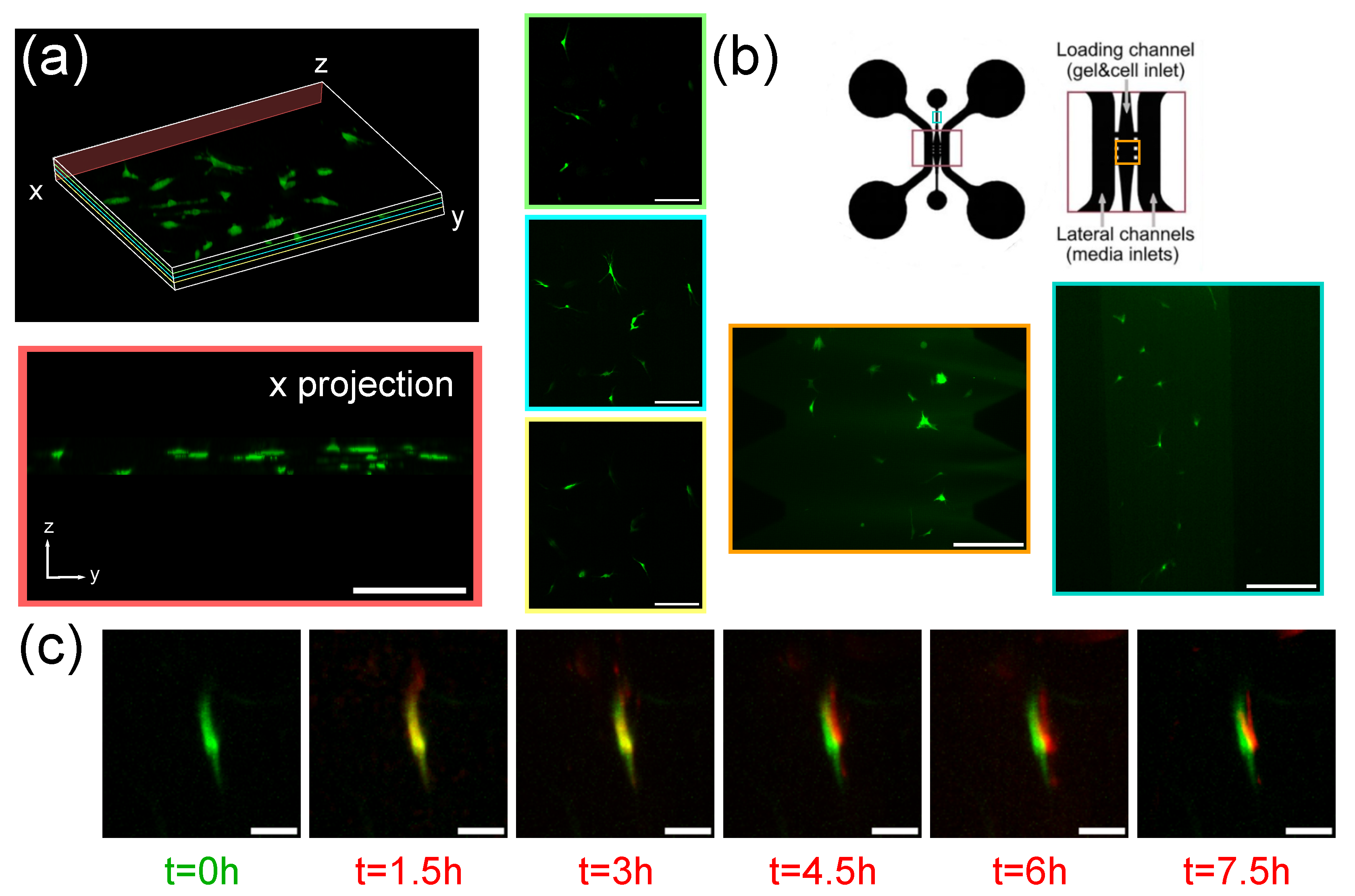
3.3. Imaging of Fibroblasts-on-a-Chip
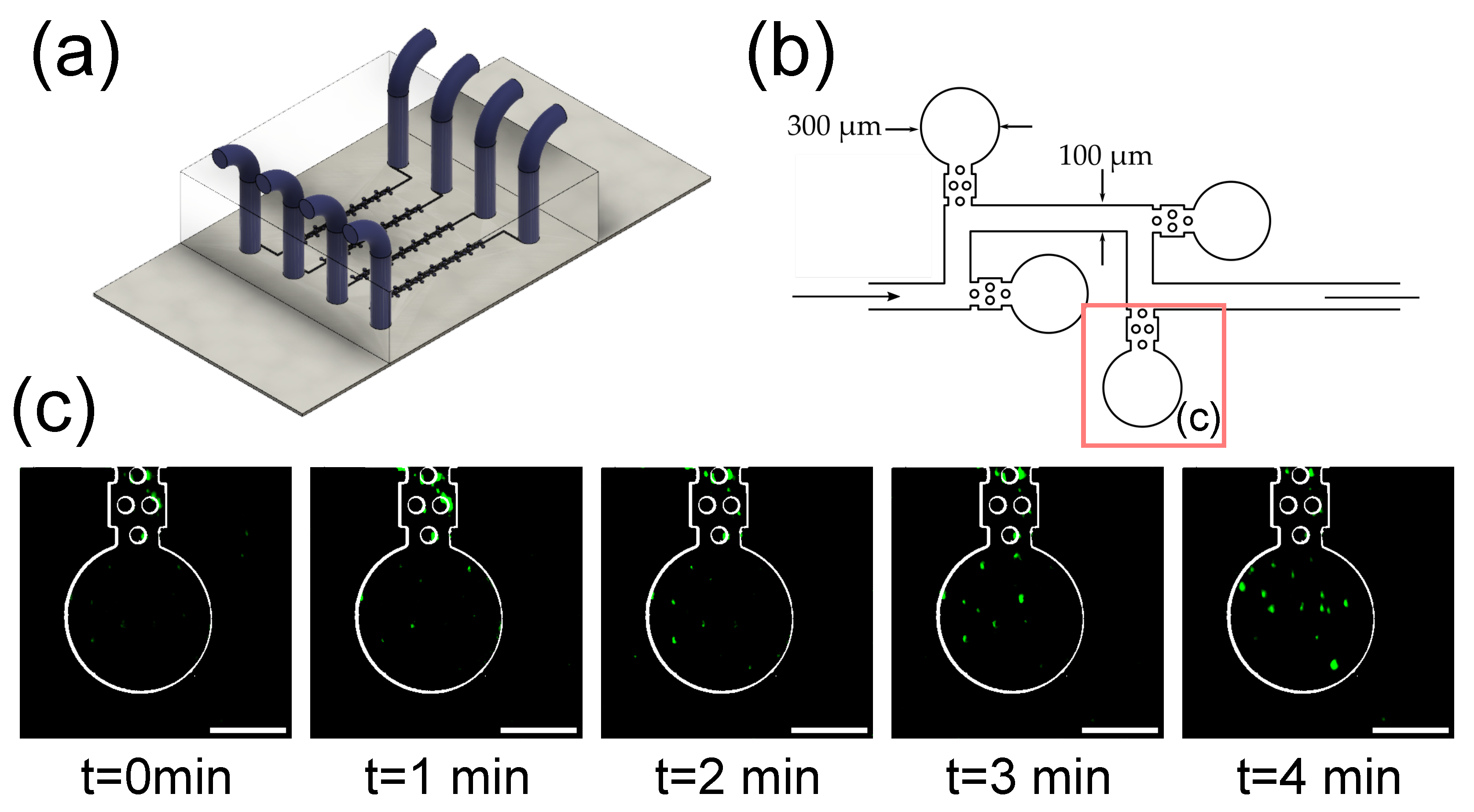
3.4. Imaging of Fluorescent Microbeads in Zero-Flow Chambers
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| LSFM | Light sheet fluorescence microscopy |
| SPIM | Single-plane illumination microscopy |
| PDMS | Polydimethylsiloxane |
| OPD | Optical path differences |
| ETL | Electrically tunable lens |
| DAQ | Data acquisition device |
| fps | frames per second |
| FWHM | Full width at half maximum |
| PSF | Point spread function |
| LUT | Look up table |
References
- Whitesides, G.M. The origins and the future of microfluidics. Nature 2006, 442, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Lin, S.C.S.; Dong, C.; Huang, T.J. Single-layer planar on-chip flow cytometer using microfluidic drifting based three-dimensional (3D) hydrodynamic focusing. Lab Chip 2009, 9, 1583–1589. [Google Scholar] [CrossRef] [PubMed]
- Inatomi, K.I.; Izuo, S.I.; Lee, S.S. Application of a microfluidic device for counting of bacteria. Lett. Appl. Microbiol. 2006, 43, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Korolj, A.; Lai, B.F.L.; Radisic, M. Advances in organ-on-a-chip engineering. Nat. Rev. Mater. 2018, 3, 257–278. [Google Scholar] [CrossRef]
- Raj M, K.; Chakraborty, S. PDMS microfluidics: A mini review. J. Appl. Polym. Sci. 2020, 137, 48958. [Google Scholar] [CrossRef]
- Fujii, T. PDMS-based microfluidic devices for biomedical applications. Microelectron. Eng. 2002, 61, 907–914. [Google Scholar] [CrossRef]
- Regehr, K.J.; Domenech, M.; Koepsel, J.T.; Carver, K.C.; Ellison-Zelski, S.J.; Murphy, W.L.; Schuler, L.A.; Alarid, E.T.; Beebe, D.J. Biological implications of polydimethylsiloxane-based microfluidic cell culture. Lab Chip 2009, 9, 2132–2139. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Mateos, M.A.; Gonzalez-Rico, J.; Nunez-Sardinha, E.; Gomez-Cruz, C.; Lopez-Donaire, M.L.; Lucarini, S.; Arias, A.; Muñoz-Barrutia, A.; Velasco, D.; Garcia-Gonzalez, D. Magneto-mechanical system to reproduce and quantify complex strain patterns in biological materials. Appl. Mater. Today 2022, 27, 101437. [Google Scholar] [CrossRef]
- Schneckenburger, H.; Weber, P.; Wagner, M.; Schickinger, S.; Richter, V.; Bruns, T.; Strauss, W.; Wittig, R. Light exposure and cell viability in fluorescence microscopy. J. Microsc. 2012, 245, 311–318. [Google Scholar] [CrossRef]
- Lazzari, G.; Vinciguerra, D.; Balasso, A.; Nicolas, V.; Goudin, N.; Garfa-Traore, M.; Fehér, A.; Dinnyés, A.; Nicolas, J.; Couvreur, P.; et al. Light sheet fluorescence microscopy versus confocal microscopy: In quest of a suitable tool to assess drug and nanomedicine penetration into multicellular tumor spheroids. Eur. J. Pharm. Biopharm. 2019, 142, 195–203. [Google Scholar] [CrossRef]
- Vinci, M.; Gowan, S.; Boxall, F.; Patterson, L.; Zimmermann, M.; Court, W.; Lomas, C.; Mendiola, M.; Hardisson, D.; Eccles, S.A. Advances in establishment and analysis of three-dimensional tumor spheroid-based functional assays for target validation and drug evaluation. BMC Biol. 2012, 10, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, J.; Bertrand, L. THUNDER Imagers: How Do They Really Work? THUNDER Imager Technical Note 2019. Available online: https://www.google.com.hk/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&ved=2ahUKEwjVx5CCv9f7AhUcsFYBHRyXBiEQFnoECAkQAQ&url=https%3A%2F%2F7157e75ac0509b6a8f5c-5b19c577d01b9ccfe75d2f9e4b17ab55.ssl.cf1.rackcdn.com%2FEQZZKYFC-PDF-1-450123-4426040265.pdf&usg=AOvVaw3eWTRefw9YSIPAjKaQzyoO (accessed on 23 October 2022).
- Schueder, F.; Lara-Gutiérrez, J.; Beliveau, B.J.; Saka, S.K.; Sasaki, H.M.; Woehrstein, J.B.; Strauss, M.T.; Grabmayr, H.; Yin, P.; Jungmann, R. Multiplexed 3D super-resolution imaging of whole cells using spinning disk confocal microscopy and DNA-PAINT. Nat. Commun. 2017, 8, 2090. [Google Scholar] [CrossRef]
- Oreopoulos, J.; Berman, R.; Browne, M. Spinning-disk confocal microscopy: Present technology and future trends. Methods Cell Biol. 2014, 123, 153–175. [Google Scholar]
- Pearre, B.W.; Michas, C.; Tsang, J.M.; Gardner, T.J.; Otchy, T.M. Fast micron-scale 3D printing with a resonant-scanning two-photon microscope. Addit. Manuf. 2019, 30, 100887. [Google Scholar] [CrossRef]
- Lorenzo, C.; Frongia, C.; Jorand, R.; Fehrenbach, J.; Weiss, P.; Maandhui, A.; Gay, G.; Ducommun, B.; Lobjois, V. Live cell division dynamics monitoring in 3D large spheroid tumor models using light sheet microscopy. Cell Div. 2011, 6, 22. [Google Scholar] [CrossRef] [Green Version]
- Bruns, T.; Schickinger, S.; Wittig, R.; Schneckenburger, H. Preparation strategy and illumination of three-dimensional cell cultures in light sheet-based fluorescence microscopy. J. Biomed. Opt. 2012, 17, 101518. [Google Scholar] [CrossRef] [Green Version]
- Maioli, V.; Chennell, G.; Sparks, H.; Lana, T.; Kumar, S.; Carling, D.; Sardini, A.; Dunsby, C. Time-lapse 3-D measurements of a glucose biosensor in multicellular spheroids by light sheet fluorescence microscopy in commercial 96-well plates. Sci. Rep. 2016, 6, 37777. [Google Scholar] [CrossRef] [Green Version]
- Pampaloni, F.; Richa, R.; Ansari, N.; Stelzer, E.H. Live spheroid formation recorded with light sheet-based fluorescence microscopy. In Advanced Fluorescence Microscopy; Springer: New York, NY, USA, 2015; pp. 43–57. [Google Scholar]
- Osaki, T.; Uzel, S.G.; Kamm, R.D. On-chip 3D neuromuscular model for drug screening and precision medicine in neuromuscular disease. Nat. Protoc. 2020, 15, 421–449. [Google Scholar] [CrossRef]
- Albert-Smet, I.; Marcos-Vidal, A.; Vaquero, J.J.; Desco, M.; Muñoz-Barrutia, A.; Ripoll, J. Applications of light-sheet microscopy in microdevices. Front. Neuroanat. 2019, 13, 1. [Google Scholar] [CrossRef] [Green Version]
- Kácik, D.; Tatar, P.; Martinček, I. Measurement of PDMS refractive index by low-coherence interferometry. In Proceedings of the 2014 ELEKTRO, Rajecke Teplice, Slovakia, 19–20 May 2014; pp. 662–665. [Google Scholar]
- Xu, T.; Lim, Y.J.; Zheng, Y.; Jung, M.; Gaus, K.; Gardiner, E.E.; Lee, W.M. Modified inverted selective plane illumination microscopy for sub-micrometer imaging resolution in polydimethylsiloxane soft lithography devices. Lab Chip 2020, 20, 3960–3969. [Google Scholar] [CrossRef]
- Stelzer, E.H. Light-sheet fluorescence microscopy for quantitative biology. Nat. Methods 2015, 12, 23–26. [Google Scholar] [CrossRef]
- Bissardon, C.; Mermet, X.; Morales, S.; Bottausci, F.; Carriere, M.; Rivera, F.; Blandin, P. Light sheet fluorescence microscope for microfluidic chip. In Proceedings of the EPJ Web of Conferences, EOS Annual Meeting (EOSAM 2020), Online, 7–11 September 2020; EDP Sciences: Julius, France, 2020; Volume 238, p. 04005. [Google Scholar]
- Mickoleit, M.; Schmid, B.; Weber, M.; Fahrbach, F.O.; Hombach, S.; Reischauer, S.; Huisken, J. High-resolution reconstruction of the beating zebrafish heart. Nat. Methods 2014, 11, 919–922. [Google Scholar] [CrossRef]
- Chang, B.J.; Manton, J.D.; Sapoznik, E.; Pohlkamp, T.; Terrones, T.S.; Welf, E.S.; Murali, V.S.; Roudot, P.; Kayley, H.; Whitehead, L.; et al. Real-time multi-angle projection imaging of biological dynamics. Nat. Methods 2021, 18, 829–834. [Google Scholar] [CrossRef]
- Aakhate, M.; Müller, H.A.J. Multiview tiling light sheet microscopy for 3D high-resolution live imaging. Development 2021, 148, dev199725. [Google Scholar] [CrossRef]
- Huisken, J.; Stainier, D.Y. Even fluorescence excitation by multidirectional selective plane illumination microscopy (mSPIM). Opt. Lett. 2007, 32, 2608–2610. [Google Scholar] [CrossRef]
- Liu, L.; Yu, L.; Li, Z.; Li, W.; Huang, W. Patient-derived organoid (PDO) platforms to facilitate clinical decision making. J. Transl. Med. 2021, 19, 40. [Google Scholar] [CrossRef]
- Calandrini, C.; Drost, J. Normal and tumor-derived organoids as a drug screening platform for tumor-specific drug vulnerabilities. STAR Protoc. 2022, 3, 101079. [Google Scholar] [CrossRef]
- Filaire, E.; Nachat-Kappes, R.; Laporte, C.; Harmand, M.F.; Simon, M.; Poinsot, C. Alternative in vitro models used in the main safety tests of cosmetic products and new challenges. Int. J. Cosmet. Sci. 2022, 44, 604–613. [Google Scholar] [CrossRef]
- Topouzi, H.; Logan, N.J.; Williams, G.; Higgins, C.A. Methods for the isolation and 3D culture of dermal papilla cells from human hair follicles. Exp. Dermatol. 2017, 26, 491–496. [Google Scholar] [CrossRef] [Green Version]
- Montero, A.; Acosta, S.; Hernández, R.; Elvira, C.; Jorcano, J.L.; Velasco, D. Contraction of fibrin-derived matrices and its implications for in vitro human skin bioengineering. J. Biomed. Mater. Res. 2020, 109, 500–514. [Google Scholar] [CrossRef]
- Desmaison, A.; Guillaume, L.; Triclin, S.; Weiss, P.; Ducommun, B.; Lobjois, V. Impact of physical confinement on nuclei geometry and cell division dynamics in 3D spheroids. Sci. Rep. 2018, 8, 8785. [Google Scholar] [CrossRef] [PubMed]
- Pampaloni, F.; Ansari, N.; Stelzer, E.H. High-resolution deep imaging of live cellular spheroids with light-sheet-based fluorescence microscopy. Cell Tissue Res. 2013, 352, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Anguiano, M.; Castilla, C.; Maška, M.; Ederra, C.; Peláez, R.; Morales, X.; Munoz-Arrieta, G.; Mujika, M.; Kozubek, M.; Munoz-Barrutia, A.; et al. Characterization of three-dimensional cancer cell migration in mixed collagen-Matrigel scaffolds using microfluidics and image analysis. PLoS ONE 2017, 12, e0171417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abercrombie, M. Fibroblasts. J. Clin. Pathol. Suppl. (R. Coll. Pathol.) 1978, 12, 1. [Google Scholar]
- Sun, P.; Liu, Y.; Sha, J.; Zhang, Z.; Tu, Q.; Chen, P.; Wang, J. High-throughput microfluidic system for long-term bacterial colony monitoring and antibiotic testing in zero-flow environments. Biosens. Bioelectron. 2011, 26, 1993–1999. [Google Scholar] [CrossRef]
- Stelzer, E.H. Single Plane Illumination Microscope. US Patent 9,823,455, 21 November 2017. [Google Scholar]
- Dyer, L.; Parker, A.; Paphiti, K.; Sanderson, J. Lightsheet microscopy. Curr. Protoc. 2022, 2, e488. [Google Scholar] [CrossRef]
- Gualda, E.J.; Vale, T.; Almada, P.; Feijó, J.A.; Martins, G.G.; Moreno, N. OpenSpinMicroscopy: An open-source integrated microscopy platform. Nat. Methods 2013, 10, 599–600. [Google Scholar] [CrossRef]
- Gualda, E.J.; Pereira, H.; Vale, T.; Estrada, M.F.; Brito, C.; Moreno, N. SPIM-fluid: Open source light-sheet based platform for high-throughput imaging. Biomed. Opt. Express 2015, 6, 4447–4456. [Google Scholar] [CrossRef] [Green Version]
- Pitrone, P.G.; Schindelin, J.; Stuyvenberg, L.; Preibisch, S.; Weber, M.; Eliceiri, K.W.; Huisken, J.; Tomancak, P. OpenSPIM: An open-access light-sheet microscopy platform. Nat. Methods 2013, 10, 598–599. [Google Scholar] [CrossRef]
- Hoang, V.T.; Stępniewski, G.; Czarnecka, K.H.; Kasztelanic, R.; Long, V.C.; Xuan, K.D.; Shao, L.; Śmietana, M.; Buczyński, R. Optical properties of buffers and cell culture media for optofluidic and sensing applications. Appl. Sci. 2019, 9, 1145. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Eglen, R. Three-Dimensional Cell Cultures in Drug Discovery and Development. SLAS Discov. Adv. Life Sci. R D 2011, 22, 456–472. [Google Scholar] [CrossRef] [Green Version]
- Ryu, N.E.; Lee, S.H.; Park, H. Spheroid Culture System Methods and Applications for Mesenchymal Stem Cells. Cells 2019, 8, 1620. [Google Scholar] [CrossRef]
- Mó, I.M.; Sabino, I.; de Melo-Diogo, D.; Lima-Sousa, R.; Alves, C.; Correia, I. The importance of spheroids in analyzing nanomedicine efficacy. Nanomedicine 2020, 15, 1513–1525. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Component | Model |
|---|---|---|
| 1 | Optical breadboard | Thorlabs PBG51506 |
| 2 | Lasers | iBeam smart Ultra Compact |
| 3 | Kinematic mirrors | Thorlabs KCB1/M |
| 4 | Dichroic filter | Edmund optics 25mm Diam. |
| 5 | Spherical lenses, | Thorlabs AC254-030-A-ML |
| Thorlabs AC254-150-A-ML | ||
| 6 | Kinematic mirror | Thorlabs KMSS/M |
| 7 | 1st tunable lens | Ed. EL-16-40-TC-VIS-5D-C |
| 8 | Mirror galvanometer | Thorlabs GVS012/M |
| 9 | Cylindrical lens, | Thorlabs LJ1695RM-A |
| 10 | Spherical lens, f = 100 | Thorlabs AC254-100-A-ML |
| 11 | Cylindrical lens, f = 20 | Thorlabs LJ1328L2-A |
| 12 | Objective lens 5× | Mitutoyo 378-802-6 |
| 13 | Objective lens 20× | Edmund Optics 59-878 |
| 14 | 2nd tunable lens | Ed. EL-16-40-TC-VIS-5D-C |
| 15 | Emission filter | Edmund optics Dual-Band |
| 16 | Tube lens | Thorlabs TL200-CLS2 |
| 17 | Camera (sCMOS) | Hamamatsu C13440-20CU |
| 18 | Linear stages | Zaber T-LSM050A |
| 19 | Sample holding platform | ZABER AP102B |
| 20 | White LED |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomez-Cruz, C.; Laguna, S.; Bachiller-Pulido, A.; Quilez, C.; Cañadas-Ortega, M.; Albert-Smet, I.; Ripoll, J.; Muñoz-Barrutia, A. Single Plane Illumination Microscopy for Microfluidic Device Imaging. Biosensors 2022, 12, 1110. https://doi.org/10.3390/bios12121110
Gomez-Cruz C, Laguna S, Bachiller-Pulido A, Quilez C, Cañadas-Ortega M, Albert-Smet I, Ripoll J, Muñoz-Barrutia A. Single Plane Illumination Microscopy for Microfluidic Device Imaging. Biosensors. 2022; 12(12):1110. https://doi.org/10.3390/bios12121110
Chicago/Turabian StyleGomez-Cruz, Clara, Sonia Laguna, Ariadna Bachiller-Pulido, Cristina Quilez, Marina Cañadas-Ortega, Ignacio Albert-Smet, Jorge Ripoll, and Arrate Muñoz-Barrutia. 2022. "Single Plane Illumination Microscopy for Microfluidic Device Imaging" Biosensors 12, no. 12: 1110. https://doi.org/10.3390/bios12121110
APA StyleGomez-Cruz, C., Laguna, S., Bachiller-Pulido, A., Quilez, C., Cañadas-Ortega, M., Albert-Smet, I., Ripoll, J., & Muñoz-Barrutia, A. (2022). Single Plane Illumination Microscopy for Microfluidic Device Imaging. Biosensors, 12(12), 1110. https://doi.org/10.3390/bios12121110