1. Introduction
Although seafood has become an important diary protein source, over-capturing has resulted in the rapid decline in the abundance of aquatic species, according to a recent report published by the Food and Agriculture Organization (FAO). In addition, unlike animal husbandry, which often exhibits a low feed conversion ratio, raising aquaculture products by fish farming benefits from a higher feed conversion ratio. Therefore, the growth of the global aquaculture industry has tremendously increased in the past decades. In 2018, the value of aquaculture production reached 82.1 million tons, which is close to the value of 96.4 tons reached by capture production. Because of this increasing trend, aquaculture production is expected to surpass capture production in 2022 [
1].
On the other hand, disease outbreak severely damages regional aquaculture industry from time to time. The loss takes at least a few years from which to recover. To maintain the sustainability of the fish farming industry, the employment of reliable and easy-to-use bioanalytical tools to monitor the pathogen levels in the aquaculture environment is crucial for risk surveillance purposes. There remains a need to develop feasible methods to determine pathogens below trace level at the fish pond side prior to disease outbreak.
Nervous necrosis virus (NNV) is a type of betanodavirus, often infecting more than 50 species of fish, including grouper, causing severe neuron damage [
2]. In the larvae rearing stage, the mortality of infected groupers is nearly 100% [
3]. Typically when infected, grouper larvae present symptoms in 7 days and soon die within 2–4 weeks [
4]. NNV is a positive-sense single-stranded RNA (ss(+)RNA) virus, which contains two nucleic acid molecules: RNA1 and RNA2. RNA1 encodes a RNA-dependent RNA polymerase and RNA2 encodes a coat protein (M.W. ~42 kDa) [
5]. The viral particle of NNV, of which the size is 25–30 nm in diameter, is non-enveloped. It is in the form of an icosahedron shape with T = 3 symmetry, constructed with 180 coat proteins [
6].
While conventional enzyme-linked immunosorbent assay (ELISA) methods provide adequate limit of detection (LOD) for determining NNV coat proteins in brain homogenate samples of infected fish containing more than 3 µg/mL coat proteins [
7], this LOD is not capable of detecting NNV in slightly contaminated aquaculture water at the viral titer level of 10
3.5 TCID
50/mL, which is estimated to contain about 0.1 µg/mL crude proteins [
7] and is equivalent to 10
6 NNV copies/mL [
8]. Besides, performing ELISA on site is difficult, limiting its applications in resource-limited areas. In addition, the tedious ELISA procedures rely on skilled operators to reduce false positive and false negative rates. On the other hand, the testing results of easy-to-use lateral flow assays are only semi-quantitative [
9], although the digital camera-assisted visual densitometric detection method has been employed [
10].
Similarly, fast molecular detection methods based on isothermal nucleic acid amplification, such as loop-mediated isothermal amplification (LAMP) and recombinase polymerase amplification (RPA), do not provide accurate results because they are prone to random polymerizations of non-target genes. Sometimes, adequate primer designs are also challenging [
11,
12,
13].
Recently, nanoplasmonic biosensors have received great attention as they are label-free, and thus, can potentially be developed into compact and highly sensitive devices. In principle, noble metal nanoparticles and the most used gold nanoparticles (AuNPs) have an extinction band known as a particle plasmon resonance (PPR) band or a localized surface plasmon resonance (LSPR) band. The biosensing sensitivity is due to the spectral change of the nanoparticle upon adsorbate-induced refractive index (RI) change of the particle’s surrounding environment. When an analyte binds with a reciprocal recognition molecule on the particle surface, plasmonic absorption will increase. Therefore, absorbance-based PPR biosensors are particularly attractive, because they only need a simple optical setup. To improve the sensitivity of the PPR biosensing approach, we have developed a real-time and label-free fiber optic particle plasmon resonance (FOPPR) sensor, which makes use of repeated total internal reflections through an optical fiber to increase the optical path, and hence, the plasmonic absorption of the AuNP layer on the fiber core surface, thus providing several advantages, including good sensitivity at pM levels, wide linear detection range over five orders, low-cost and miniaturizable instrumentation, and affordable sensor chips. Applications of the label-free FOPPR biosensor to real samples have been demonstrated. These applications include detection of antinuclear antibodies in sera [
14], tumor necrosis factor-α and matrix metalloproteinases-3 in synovial fluids [
15], orchid viruses in plants [
16], HLA-B27 messenger RNA in blood [
17], methamphetamine in urine [
18], and single nucleotide polymorphismin of DNA in blood [
19].
To fulfill the aforementioned unmet need, this study applied the label-free direct method of an FOPPR biosensor to illustrate its applicability in quantifying the amount of NNV in real samples of fish bodies and pond water. Having commercially available anti-NNV antibodies immobilized on the surface of the AuNP-covered optical fiber, we used either coat protein antigens or viral particles of NNV to prepare standards to develop an FOPPR biosensor for NNV. Because of the enhanced plasmon absorbance signals resulting from repeated reflections along the sensing optical fiber, ultrasensitive detection is expected.
Although the direct method in an FOPPR biosensor is highly sensitive, it is still challenging to detect the amount of a virus in concentration levels comparable to the gold standard real-time quantitative polymerase chain reaction (qPCR) method, which amplifies the number of copies of DNA. On the other hand, methods to amplify the number of copies of a protein have not yet been discovered. As a companion to the label-free direct method using FOPPR biosensor, an alternative sandwich method-based FOPPR biosensing strategy called fiber optic nanogold-linked immunosorbent assay (FONLISA), which uses a AuNP-labelled probe as the plasmonic signal enhancer, as well as the second biorecognition element, has been recently developed. By this method, AuNPs are not initially present on the fiber core surface, and thus, the background absorbance value is zero. Then, the interactions among a detection antibody-functionalized AuNP (AuNP@Ab
D), a target analyte molecule (A), and an immobilized capture antibody molecule (Ab
C) form a sandwich-like AuNP@Ab
D–A–Ab
C complex on the unclad surface of a sensing fiber. Therefore, one analyte molecule will lead to the increase of absorbance by one AuNP. In contrast, by the direct method in an FOPPR biosensor, the binding of one analyte molecule on the AuNP surface will result in an increase of absorptivity of the AuNP, which is several orders of magnitude smaller than the absorptivity of the AuNP itself [
20]. As a result, the FONLISA method improves the sensitivity by at least three orders of magnitude [
21,
22,
23,
24,
25]. Using this enhancement method, we have pushed the LOD for NNV similar to the level of the gold standard qPCR method, without carrying out amplification steps. Upon development of AuNP@Ab
D as a biopreserved kit for long term storage, the method has a high potential for application to the point-of-use sites.
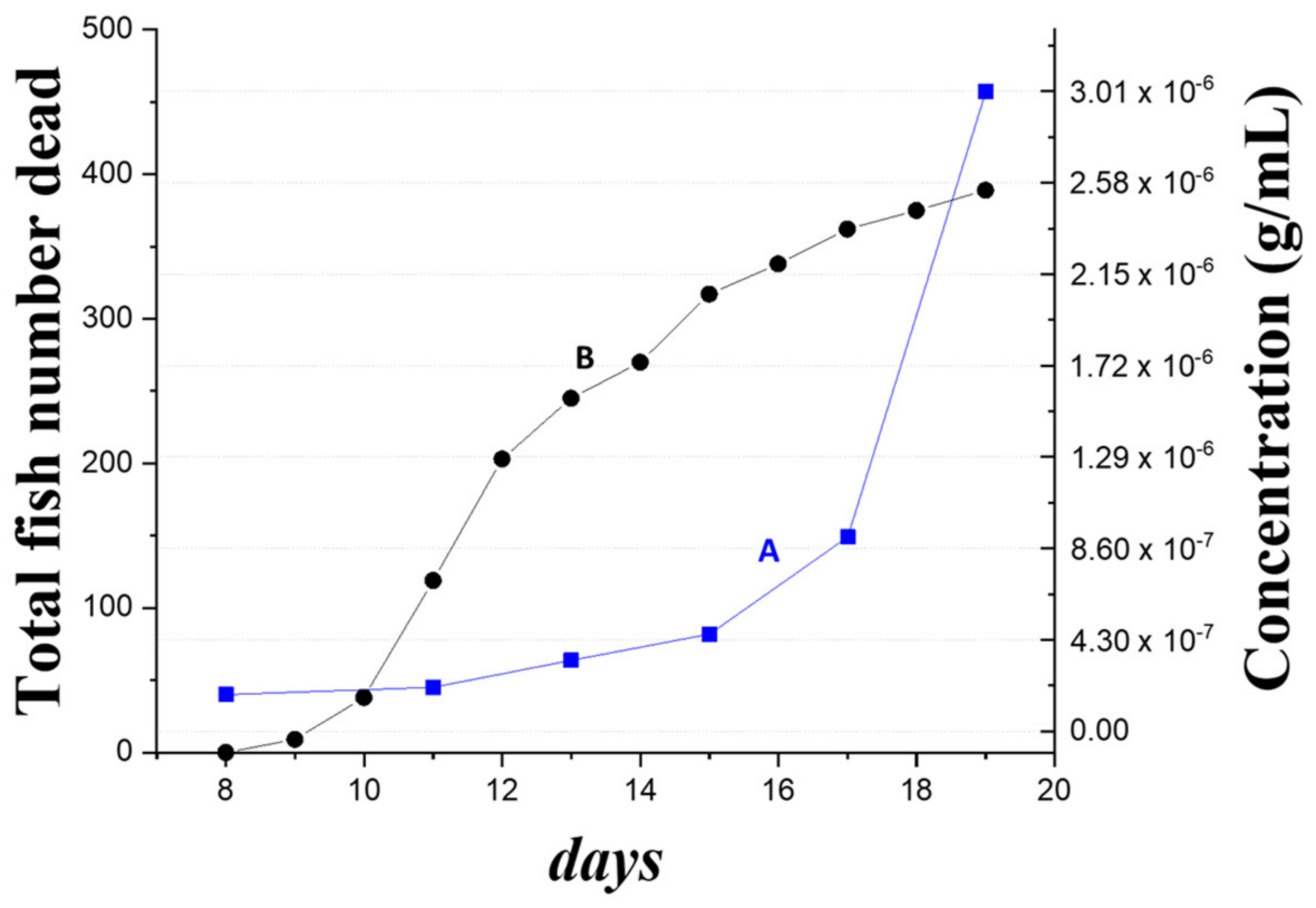
Finally, to demonstrate the feasibility of our method in an open environment without air conditioning control as a surveillance tool for NNV outbreak warning, we analyzed the pond water samples siphoned from an infected grouper container every two or three days for eleven days by FOPPR biosensor. This is our first report on applying an FOPPR biosensor for on-site detection in an aquaculture environment. Our highly sensitive FOPPR biosensor can detect trace levels of a virus prior to the outbreak of an infection that exhibits a rapid increase of death toll.
2. Experimental
2.1. Materials
The following chemicals of reagent grade were obtained from Sigma-Aldrich (St. Louis, MO, USA): 11-mercaptoundecanoic acid (MUA), 6-mercapto-1-hexanol (MCH), N-hydroxysuccinimide (NHS), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC), 16-mercaptohexadecanoic acid (MHA) and ethanolamine. (3-Mercaptopropyl)-trimethyoxysilane (MPTMS, 98%) and cetyltrimethylammonium bromide (CTAB) were purchased from ThermoFisher Scientific Acros Organics (Geel, Belgium). Hydrogen tetrachloroaurate trihydrate (HAuCl
4), sodium borohydride (NaBH4), and Tween 20 were from Showa (Tokyo, Japan). 11-Aminodecyltrithoxysilane (AUTES) was acquired from ThermoFisher Scientific Alfa Aesar (Heysham, UK). Disuccinimidyl suberate (DSS) was purchased from Ark Pharm (Arlington Heights, IL, USA). Triton X-100 was from J. T. Baker. Isopropyl β-D-1-thiogalactopyranoside (IPTG) was from ZymesetBiotek Co. Ltd. (Keelung, Taiwan). Diethyl pyrocarbonate (DEPC) was from P&C Biotech, Inc. (Taichung, Taiwan). Polyclonal rabbit antibody and monoclonal mouse antibody against NNV were from Genesis Biotech Group (Mercer, NJ, USA). Ampicillin, fetal bovine serum, L-15 Medium, and cDNA synthesis reagents were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Yeast extract and tryptone were purchased from Condalab (Torrejon de Ardoz, Spain) to prepare Luria–Bertani (LB) medium. Sulfobetaine thiol (SBSH) [
26] and sulfobetaine silane (SBSi) [
27] were synthesized as described in previous publications. All the aqueous solutions were prepared using deionized water from a Millipore (Billerica, MA, USA) Milli-Q water purification system with a resistance of 18.2 MΩ. Multimode silica optical fibers (model F-MBC) were obtained from Newport (Irvine, CA, USA) with core and cladding diameters of 400 and 430 μm, respectively.
2.2. Preparation of Coat Protein of NNV
NNV coat protein, expressed in bacterium BL21(DE3) strain, was prepared in-house. The preparation procedures are described as follows. The NNV coat protein was expressed in bacterium BL21(DE3) strain, in which, the vector containing 339 aa. (1017 bps) of RNA2 gene was cloned. Single colony was inoculated into LB medium (3 mL) containing ampicillin and cultured overnight at 37 °C. Next, a proper volume of overnight culture was drawn to inoculate into 500 mL LB/ampicillin medium. This culture liquid was incubated at 37 °C for about 2 to 3 h. An aliquot of culture (1 mL) was taken out as the non-induction sample. The remaining culture liquid was used for protein induction by adding the induction reagent isopropyl β-D-1-thiogalactopyranoside (IPTG) up to 1 mM. Incubation was continued at 27 °C for another 2 h. Then, the culture was centrifuged at 6000× g for 15 min at 4 °C. The supernatant in the centrifuge tube was discarded. The collected pellet fraction was re-suspended in pre-chilled phosphate buffered saline-Triton (PBST) solution (25 mL), which contained 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, and 1% Triton X-100. The pellet fraction was lysed with a French Press. The cell lysate was centrifuged at 4 °C, 15,000× g for 30 min. The supernatant (minor fraction for protein expression) in the centrifuge tube was transferred to a new tube. The pellet (major fraction for protein expression) remaining in the tube was re-suspended in 25 mL pre-chilled PBST for further purification. The protein fractions of the supernatant and pellet were analyzed by SDS-PAGE method using Coomassie Blue staining protocol.
2.3. Viral Particle Cultivation and Quantitation
Grouper fin cells (GF-1) were cultured at 28 °C in L-15 medium supplemented with 5% fetal bovine serum (FBS) up to 70–80% confluence. Then, viral particles of NNV isolated from naturally infected groupers (Epinephelus lanceolatus) were diluted with L15 medium lacking 5% FBS to 105 fold and seeded into the cell culture plate to incubate at 28 °C for another 30 min. When the infectious fluid was removed, the culture plate was washed with 1× PBS solution twice, then was allowed to continue incubation at 28 °C for 7 days. Finally, the cells were collected by a cell scraper into a 15 mL tube. The collection tube was centrifuged under 1000× g for 5 min. Then, the supernatant was filtered through a 0.22 μm filter and stored at −80 °C in a frozen tube.
To quantify the viral content level of the cultivated solution, GF-1 cells of 2 × 104 cells/well were seeded into each well of a 48-well plate containing L-15 medium plus 5% FBS and cultured overnight at 28 °C. Next, the cells were visualized under a light microscope to confirm that they were evenly distributed with over 80% confluence. Ten-fold serial dilution of the original viral sample was performed. Viral samples of 103 to 109-fold dilution (100 μL) and mock control were added into each well to infect the cells. After incubation at 28 °C for 7 days, the occurrences of cytopathic effect (CPE) were observed and microscopic images were acquired. The NNV titer of the cultivated solution at 1.4 × 107 TCID50/mL was determined using Reed–Muench method. Without dilution, the crude protein content of the viral sample was determined to be ≈1.6 × 10−4 g/mL, using spectrophotometric method.
Using diethyl pyrocarbonate (DEPC)-treated water, 1 µg NNV RNA extracted from the aforementioned cultivated virus solution was mixed with cDNA synthesis reagents to react at 25 °C for 10 min, 42 °C for 60 min, 70 °C for 10 min, and chilled in an ice bath. The cDNA product of 3 μL was siphoned to perform the real time qPCR protocol. After mixing with a PCR master mix reagent, forward and reverse primer solutions, and fluorescence dyes, the cDNA solution was heated to 95 °C for 5 min, and then the temperature program cycle was repeated 33 times, as follows: 95 °C for 40 s, 56 °C for 40 s, and 72 °C for 40 s. The amplified product solution was determined with fluorimetry [
28]. The synthesized virus gene (RNA2) was used as a standard for the calculation of the viral copy number. Standard solutions of RNA2 (1 ng/µL; M.W. 1.55 × 10
5 Da) were diluted from 10
1 to 10
10 folds prior to amplification via the aforementioned PCR procedures to establish the standard curve indicating the relation between Ct number and dilution factor. Compared to the Ct numbers of each serial dilution solution of virus extract, we estimated the copy number of our cultivated virus solution as 9.6 × 10
8 copies/µL. In other words, 1 TCID
50/mL is equivalent to 6.9 × 10
4 copies/mL or 69 copies/μL.
2.4. Apparatus
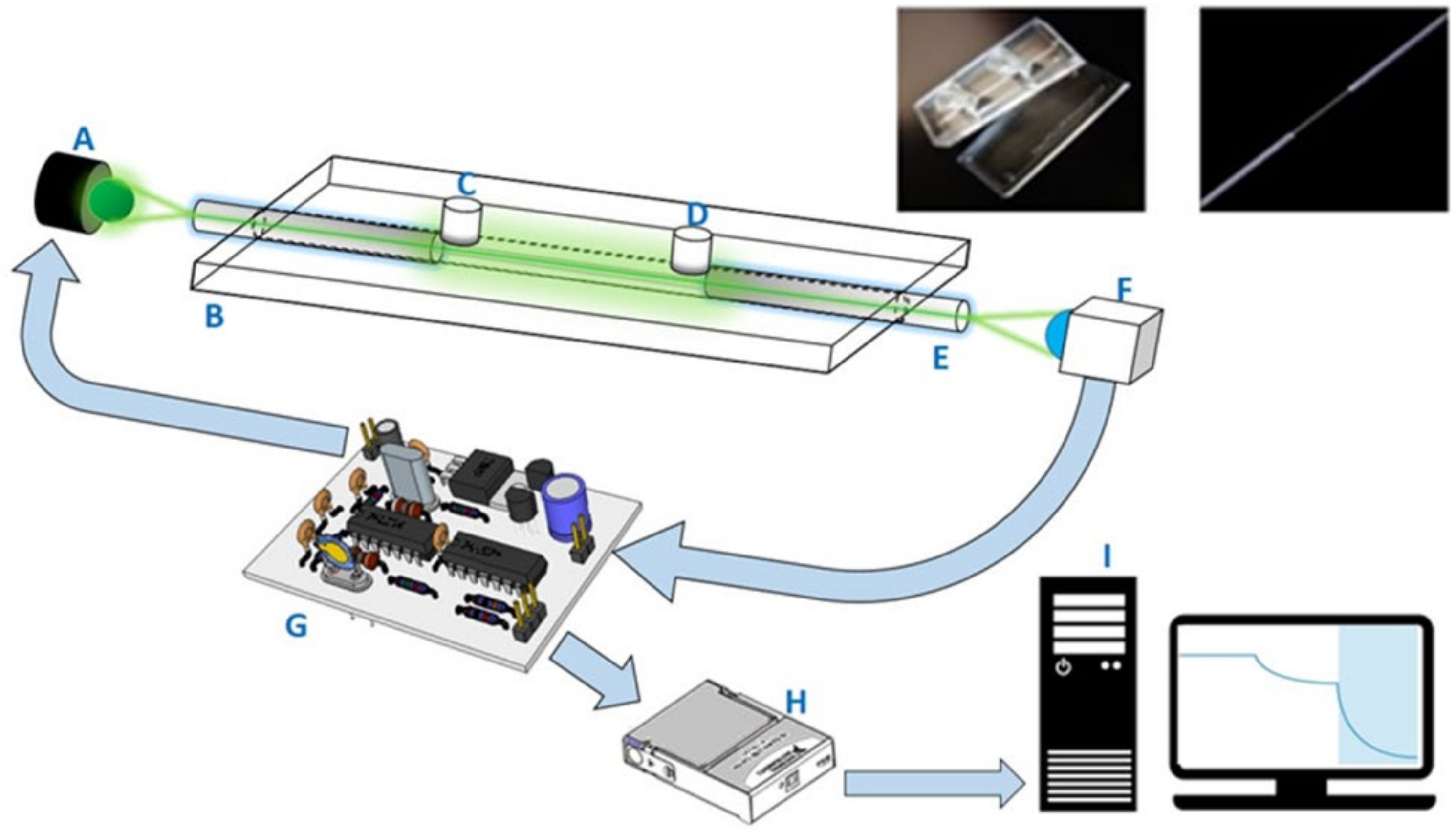
The two-piece components of the sensing chips, made of cyclic olefin copolymer (COC) slides produced via an injection molding process, were similar to those in our previous publications [
21,
29,
30]. There was a microchannel with both a depth and width of 800 μm on the bottom slide. After the bonding of the bottom and top slides, an optical fiber with an unclad segment (2 cm) serving as the sensing region was placed in the microchannel. The empty spaces at both ends were sealed. There were two holes, used as inlet and outlet ports, on the top slides to connect with the sample introduction and purging tubes. The imbedded photographs in
Figure 1 shows the components of the COC sensing chip used in this study.
One desktop FOPPR biosensor (INB Kinetic) was provided by Instant NanoBiosensors Co., Ltd. (Taipei, Taiwan). The layout of the optical and electronic components of this biosensor is similar to that of our previous setup [
21,
29,
30]. This biosensor contained one built-in circuitry to modulate a light-emitting diode (LED) light source at 1 kHz and amplification of signal from a photoreceiver, a data acquisition card, and a software loaded in a personal computer to perform lock-in amplification procedures to improve the signal-to-noise ratio of the monitored signal. A schematic illustration of this biosensor system is shown in
Figure 1.
2.5. Preparation of Sensing Fibers for Direct Method
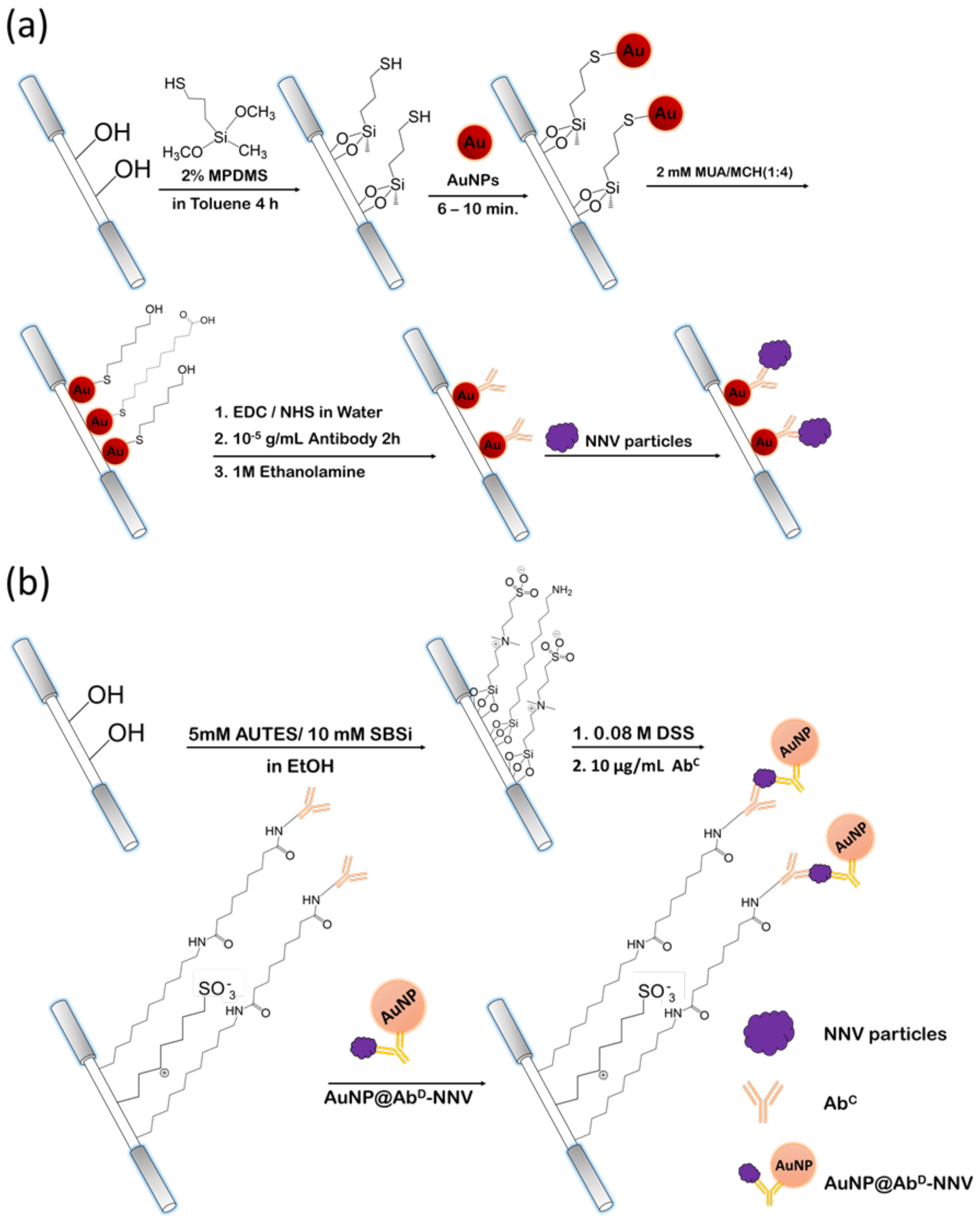
Spherical AuNPs were produced via the procedures described previously [
31]. The mean diameter of the AuNPs was 12.3 ± 0.3 nm was determined by a transmission electron microscope (JEOL 1200 EX, Tokio, Japan). The nanoparticles were immobilized on the unclad segment, i.e., the sensing region, of an optical fiber via self-assembly. The steps to prepare the sensing fiber, including chemical modification of the sensing region surface and conjugation of immune-probe on AuNP surface, are described as follows. First, an unclad segment of an optical fiber was immersed into a toluene solution of 2% MPDMS for 4 h to produce a self-assembled monolayer (SAM) of MPDMS presenting thiol groups to react with AuNPs so that the AuNPs were immobilized on the surface. Next, MUA (2 mM) and MCH (2 mM) were mixed at a volume ratio of 1:4 in an ethanol solution. The optical fiber with AuNP-coated unclad segment was then immersed in the ethanol solution for 16 h under room temperature so that it would be covered with a mixed SAM of MUA and MCH on the AuNP surface. When the full monolayer coverage was accomplished, the fiber was finally rinsed with de-ionized water and dried with nitrogen gas. Then, the carboxylic group of MUA in the mixed SAM was activated when the unclad fiber was immersed into an aqueous solution of EDC (0.1 mM) and NHS (0.025 mM) for 1 h. Upon the activation of the carboxylic group, the segment was immersed into an antibody solution (10
−5 g/mL) in PBS (pH 7.2) for 2 h. When antibody-AuNP conjugation was accomplished, the nonreacted carboxylic group of MUA was capped by injecting an ethanolamine solution (1 M) into the microchannel for 30 min.
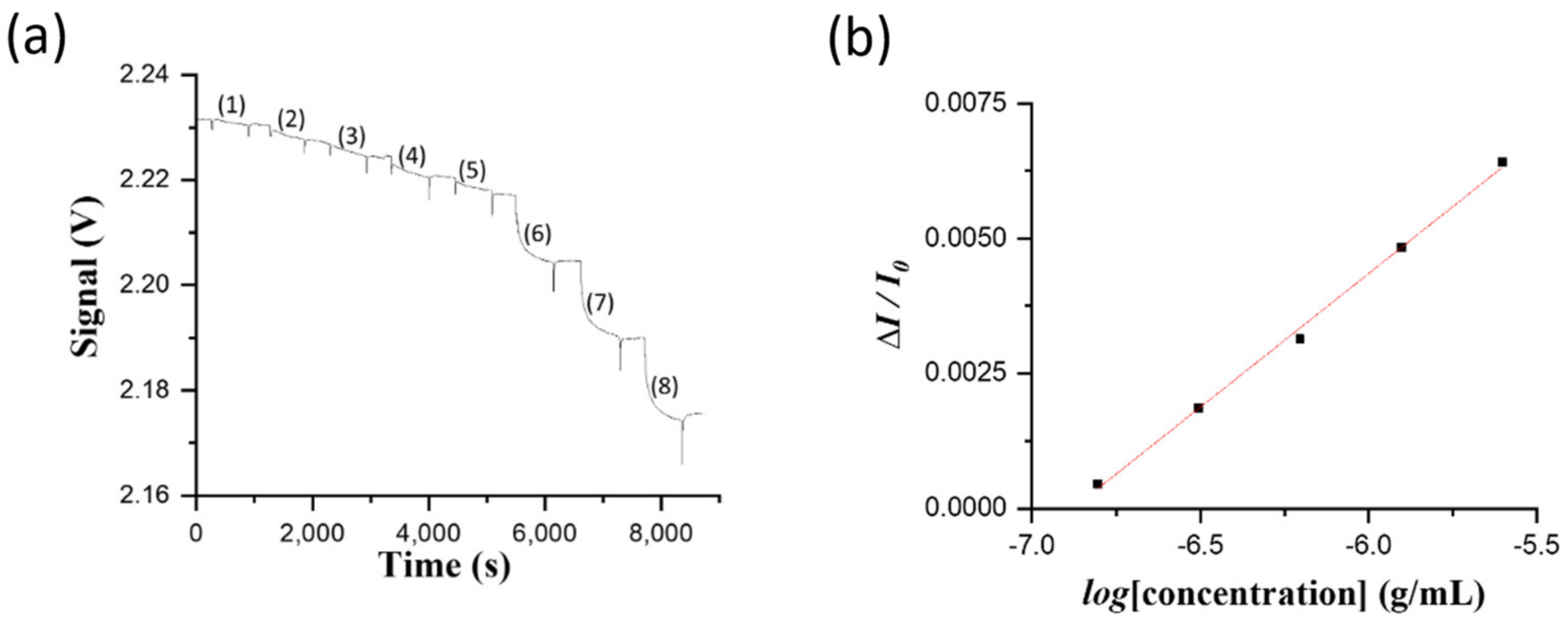
2.6. Analysis of Samples by Direct Method
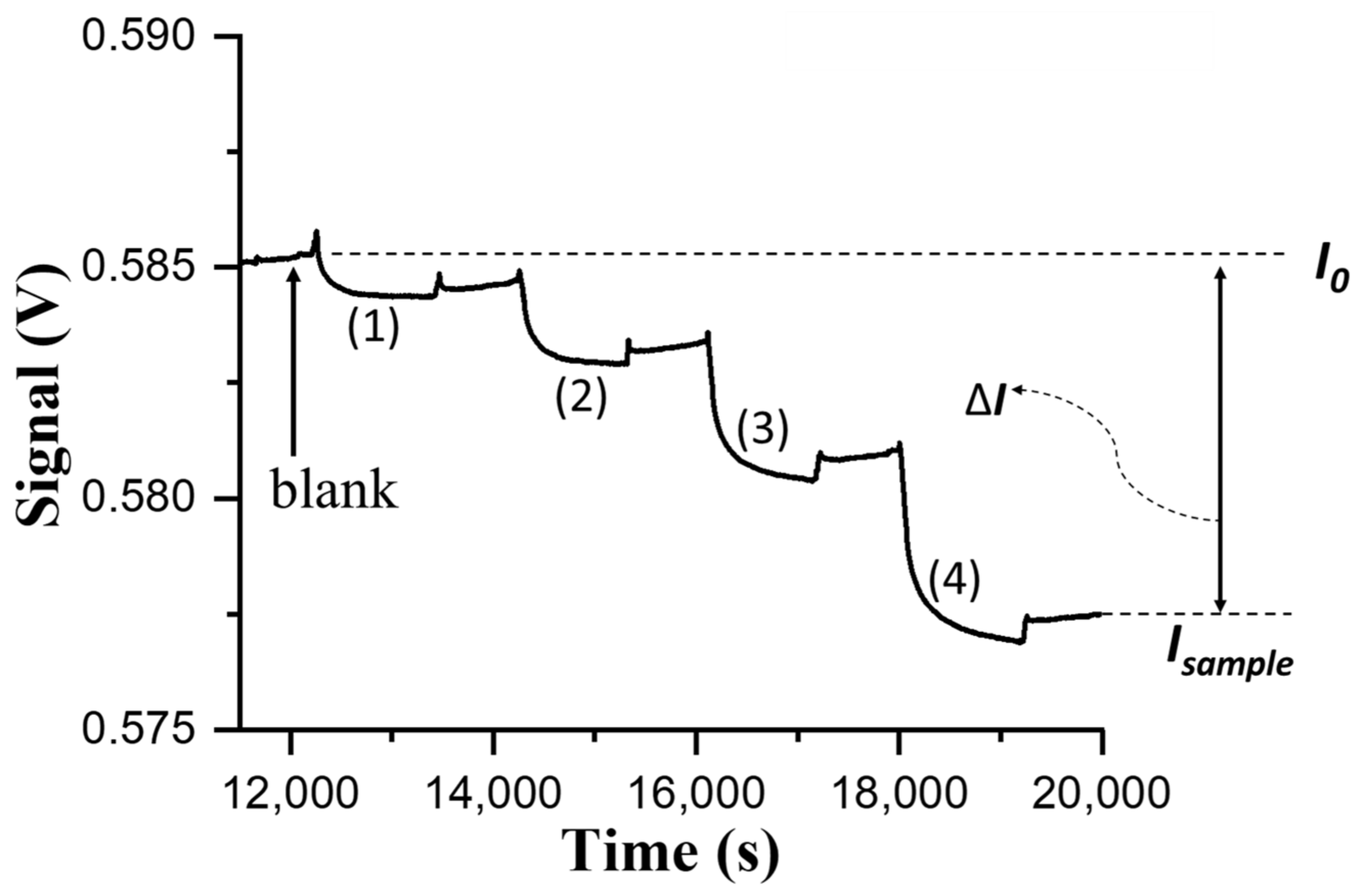
For biosensing experiments based on the direct method in the FOPPR biosensor, a phosphate-buffered saline (PBS) solution (pH 7.4) was injected into a sensing chip to thoroughly rinse the sensing region surface and to establish a baseline that shows the transmitted light intensity through the sensing fiber (I
0). Subsequently, when a sample solution of NNV coat protein or NNV particles was filled into the microchannel of the sensing chip, the analyte (NNV coat protein or NNV particles) would dock on the sensing region surface through binding with the immune probe on AuNPs, resulting in the decrease of transmitted light intensity through the sensing fiber (I
S). For quantitative analysis, the intensity I
S is normalized with respect to I
0, and the normalized response is defined as ΔI/I
0, where ΔI = I
0 − I
S, which is related to the amount of analyte docked on the sensing region surface [
20]. At least three chips were prepared to replicate each measurement in determining the virus level of NNV particle sample for method validation.
2.7. Preparation of AuNP@AbD Conjugate and Sensing Fibers for FONLISA Detection
The procedures to prepare sensing fibers and AuNP@AbD conjugate for FONLISA are described as follows. AuNPs were first dispersed in 0.001 % (v/v) Tween 20 solution. Then a methanol solution of MHA and SBSH, of which the final concentrations were 0.02 mM and 0.08 mM, respectively, was added and incubated for at least 16 h. MHA and SBSH were chemisorbed on AuNP via thiol groups. The aggregation of AuNPs was avoided because of negative charged terminal group on the zwitterionic SBSH. Upon activation of the carboxylic group on MHA using EDC/NHS procedures, a PBS solution (pH 7.2) containing the detection antibody (~10−5 g/mL) was added and allowed to react for 16 h to conjugate the detection antibody on AuNP surface. The non-activated MHA sites were deactivated by reacting with a solution of 1 M ethanolamine in water at a pH of 8.5 for 30 min.
The sensing fiber for FONLISA was prepared stepwise, as follows. First, the unclad segment of an optical fiber was immersed into a solution of 5 mM AUTES and 10 mM SBSi in absolute alcohol for 16 h to assemble a mixed SAM on the fiber core surface. Next, the AUTES/SBSi-modified unclad segment was then immersed in an 80 mM DSS solution in ethanol for 16 h under room temperature to activate the amine group on AUTES. When the activation of amine group was accomplished, the fiber was rinsed with deionized water and dried with nitrogen gas. Finally, the segment was immersed in a PBS solution (pH 7.2) containing the capture antibody (~10−5 g/mL) for 2 h to conjugate the detection antibody on the sensing region surface. The non-activated AUTES sites were deactivated by reacting with a solution of 1 M ethanolamine in water at a pH of 8.5 for 30 min.
In this case, the sensing region surface was covered with a mixed self-assembled monolayer (SAM) containing a zwitterion component to avoid non-specific binding of AuNP@AbD conjugate on sensing region surface. Then the capture antibody was conjugated on the sensing region surface. For biosensing experiments based on FONLISA, similar procedures as the direct method were used except that the sample solution was first mixed with a solution of AuNP@AbD conjugate at a fixed concentration in a 1:1 volume ratio and incubated for 15 min prior to filling into the microchannel to obtain enhanced signals.
2.8. Determination of Real Samples
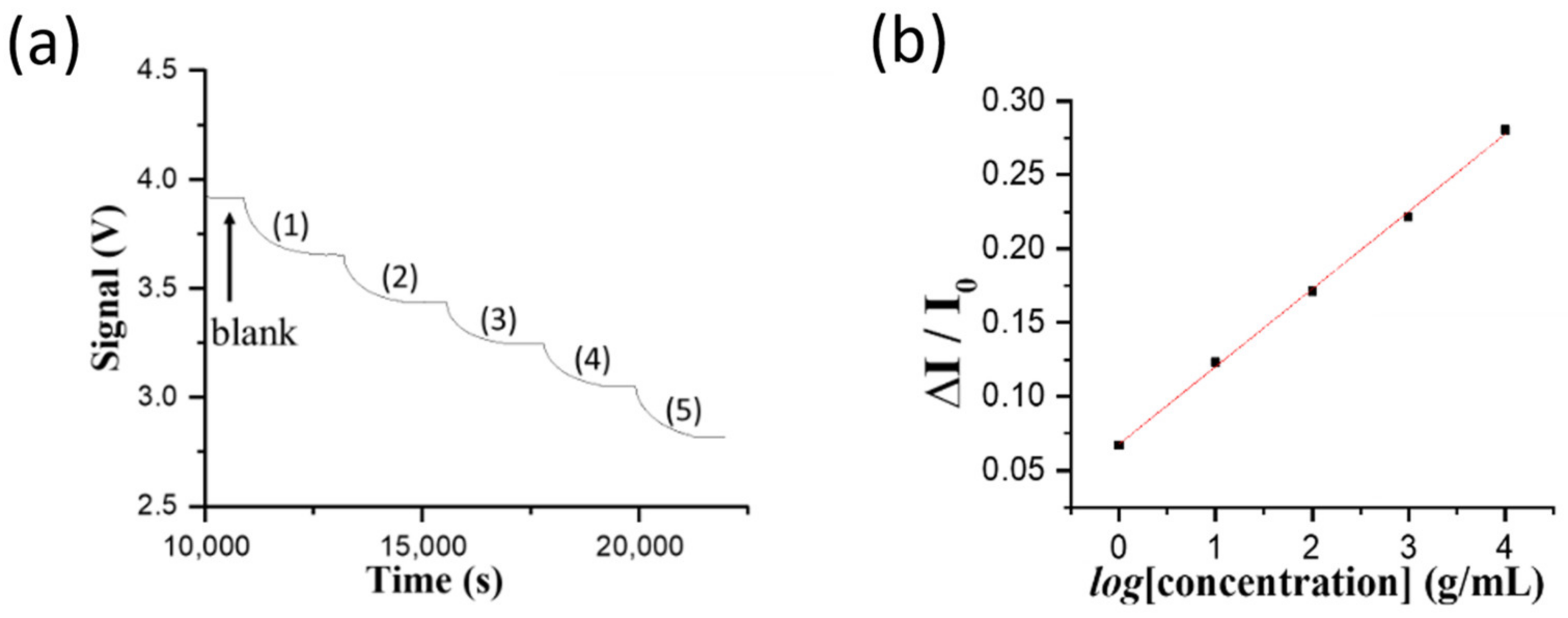
One larvae of infected grouper of 15 g was blended in PBS buffer and diluted to 150 g. One aliquot of 1 mL homogenate solution was siphoned to determine the NNV content. First, the homogenate solution was diluted using PBS buffer to ten folds prior to running through a filtration membrane of which the pore size is 0.2 μm. The filtrate solution was then diluted serially to 2, 4, 8, 16, and 32 folds prior to loading into a sensor chip to ensure the detected signal intensities of the diluted filtrate samples were within the dynamic range of the standard curve using the virus particle solutions. In the direct FOPPR biosensor method, the following procedure was used to correct the refractive index (RI) difference between the sample and the blank (i.e., PBS solution). When a sample sensorgram reached steady state, a PBS solution was injected to flush the previously loaded solution and then this transmitted light intensity through the sensor fiber was taken as IS in order to correct the over-estimated absorbance caused by the matrix effect.
One aliquot (800 µL) of infected grouper homogenate in PBS was added with lysis reagent to dissolve tissue. Repeating washing and centrifugation steps using chloroform, isopropanol, and ethanol, were performed. Collected pellets were dissolved in DEPC treated water to extract RNA, determined via the procedures to quantify the cultivated NNV solution.
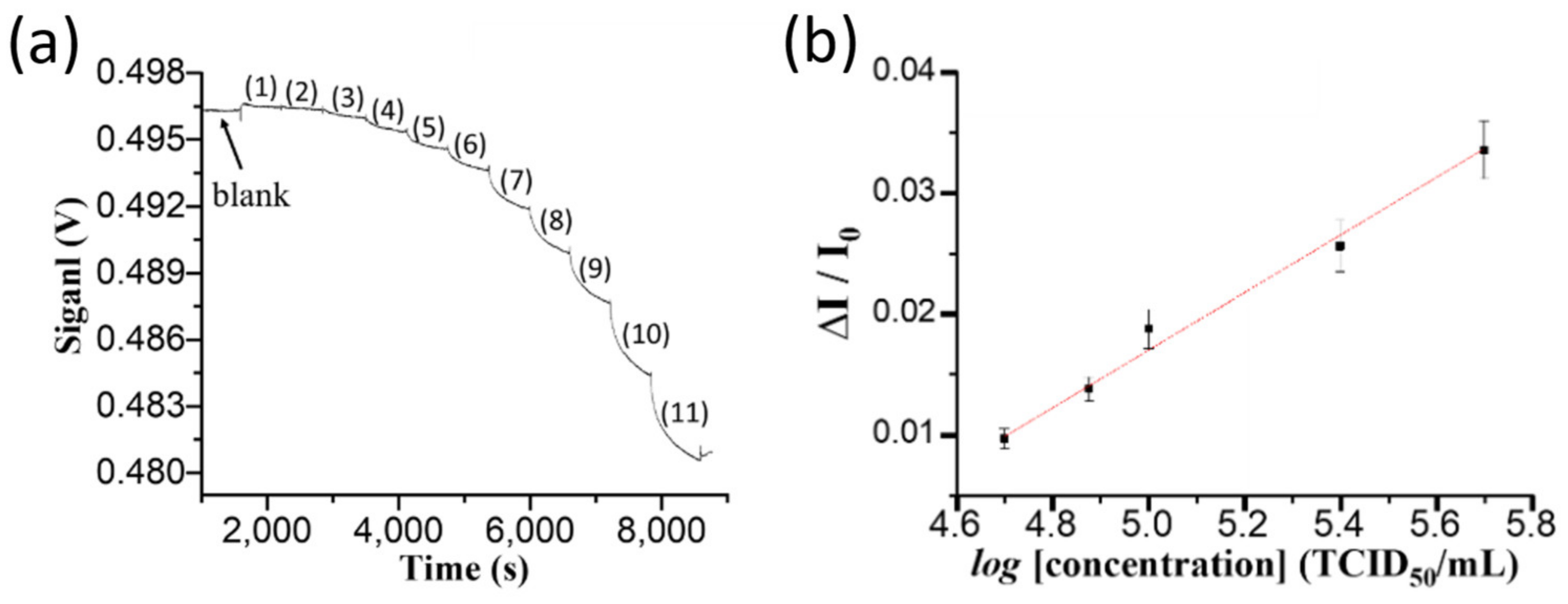
Pond water samples of 250 mL were collected every two or three days from an aquaculture container of about 1000 pieces of grouper larvae at the beginning of the culturing process. The container was an in indoor facility belonging to a local fish farmer. Whenever suspected infected groupers by NNV arose because of contaminated feeds, an aliquot of 2.0 mL was withdrawn from each sample to run through a filter paper by gravity for clean-up. A filtrate solution of 100 μL was loaded into a sensor chip to obtain a sensorgram.
Because of high salinity (~10‰), the RI of clean pond water is higher than that of PBS solution. To correct the RI difference between the sample and PBS solution, when a sample sensorgram reached steady state, a PBS solution was injected to flush the previously loaded solution, and then this transmitted light intensity through the sensor fiber was taken as IS in order to correct the over-estimated absorbance caused by the matrix effect.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






