SPR Analysis of SUMO-Murine Rap1-Interacting Factor 1 C-Terminal Domain Interaction with G4

Abstract
:1. Introduction
2. Materials and Methods
2.1. MuRif1-CTD Expression, Solubility Testing
- Different buffers and additives were explored to obtain the best condition for cell lysis;
- Diverse additives were incorporated during the expression of muRif1-CTD to the culture medium after induction by IPTG;
- Various induction methods.
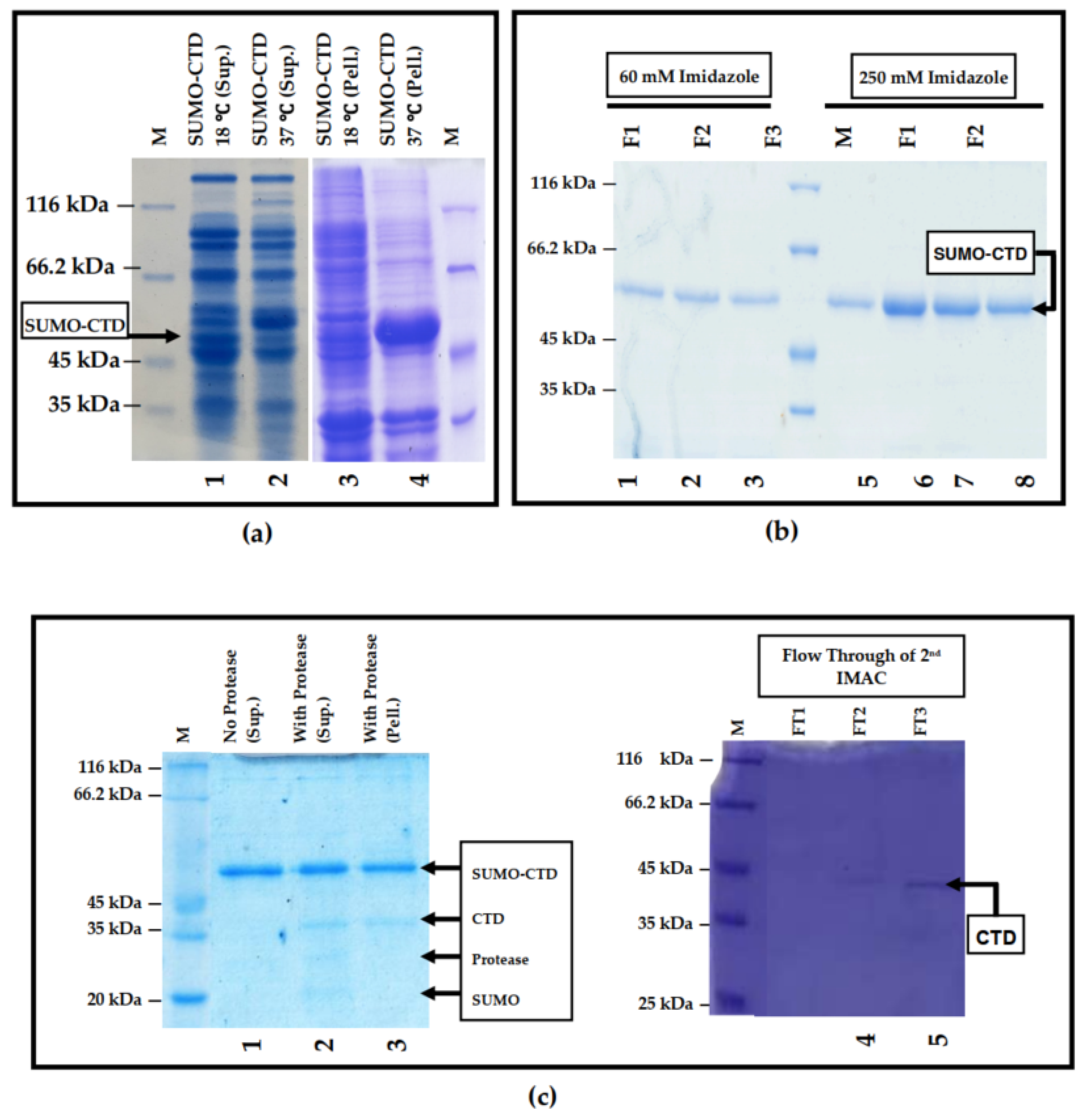
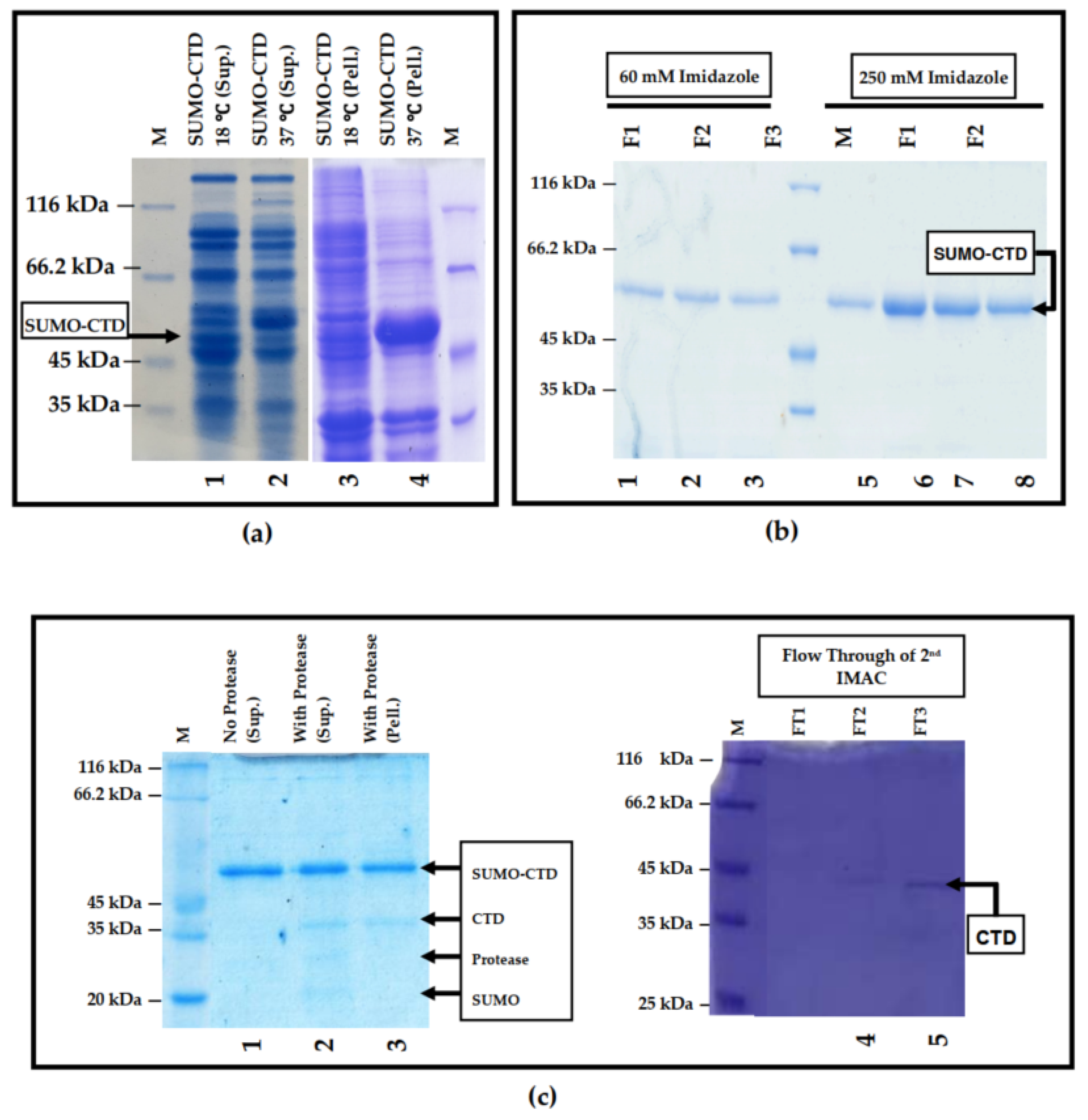
2.2. Protein Purification and Tag Removal Analyses
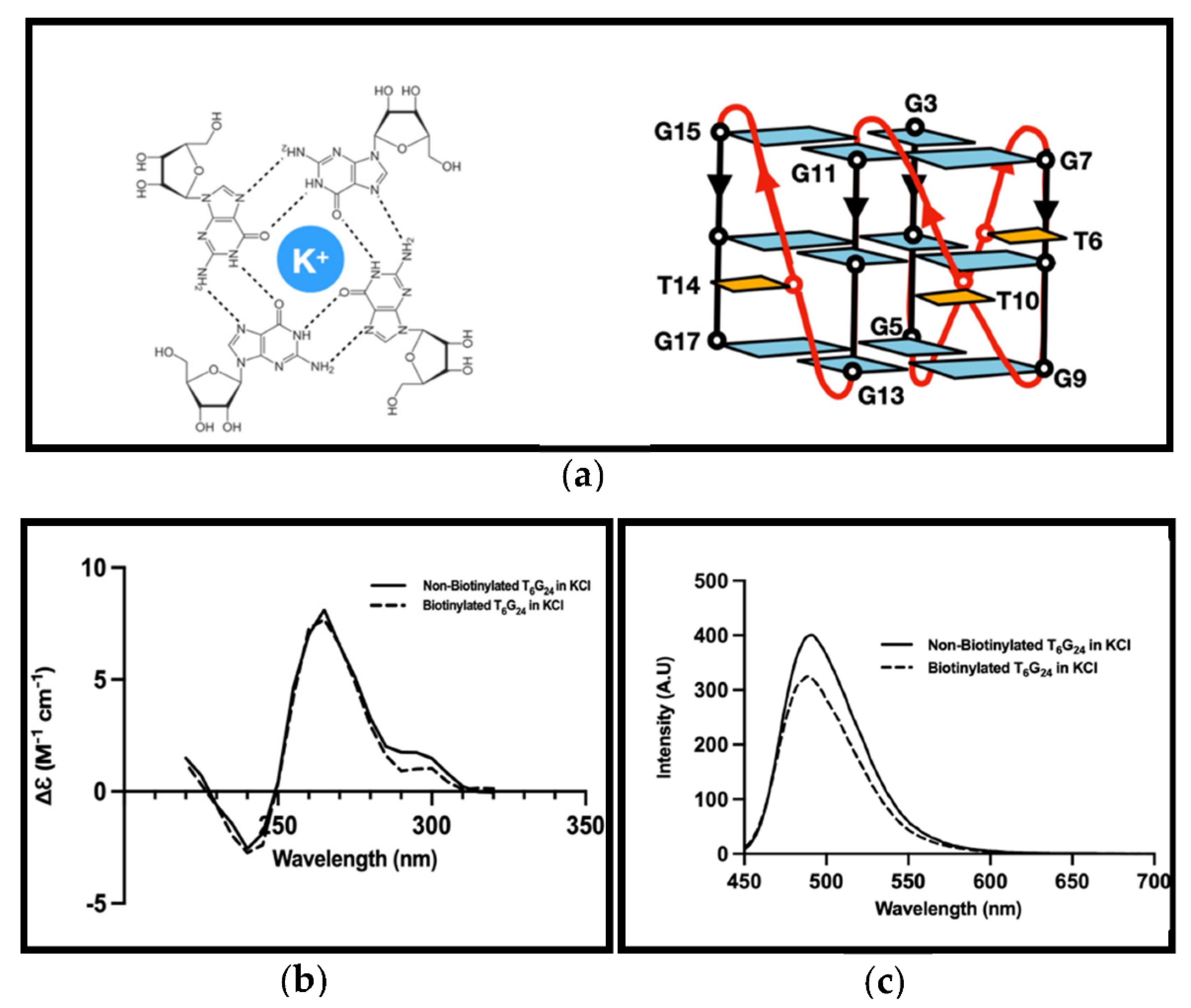
2.3. G4 Formation Investigations
2.4. Electrophoretic Mobility Shift Assay (EMSA) Set Up for G4/muRif1-CTD Interaction Detection
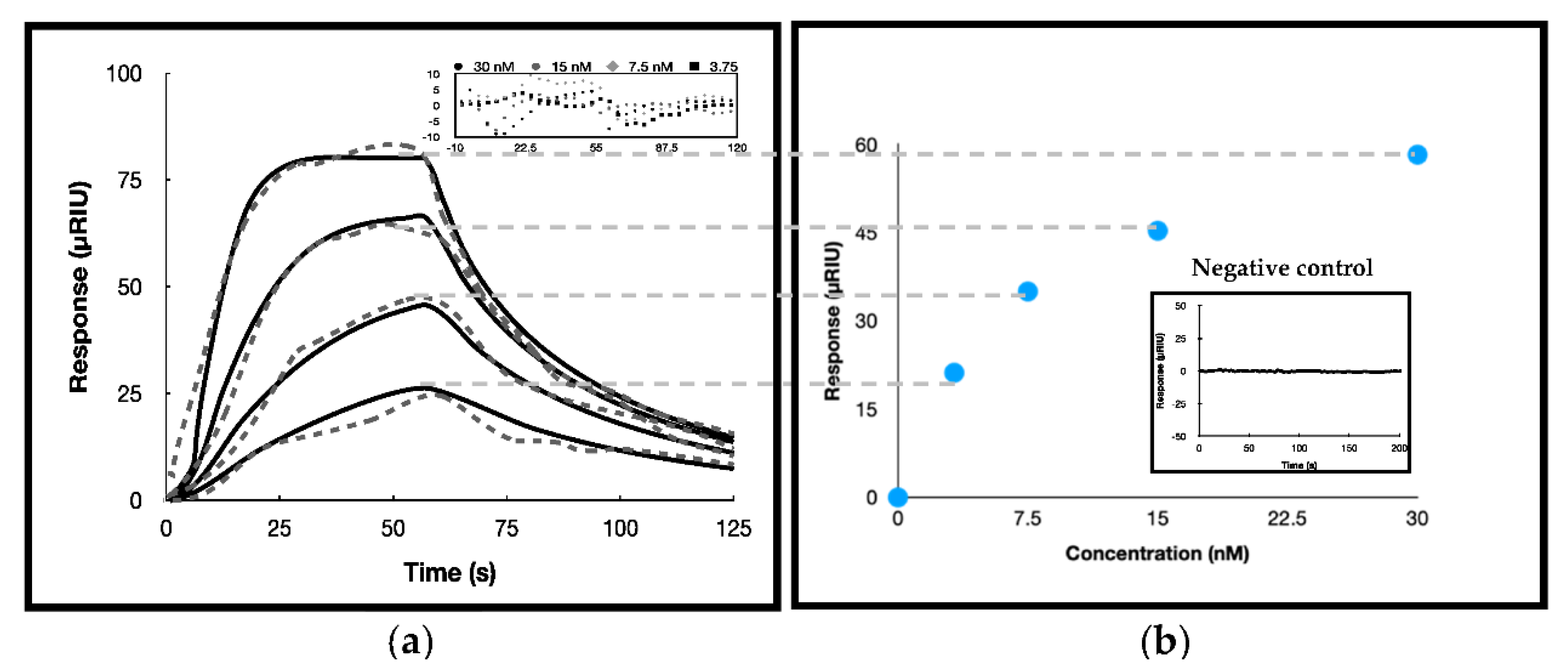
2.5. Interaction Analyses by SPR
3. Results
3.1. Prokaryotic Expression and Purification of SUMO-muRif1-CTD
3.2. G4 Formation Investigation
3.3. Surface Plasmon Resonance Investigations
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mariani, S.; Minunni, M. Surface plasmon resonance applications in clinical analysis. Anal. Bioanal. Chem. 2014, 406, 2303–2323. [Google Scholar] [CrossRef]
- Nguyen, H.H.; Park, J.; Kang, S.; Kim, M. Surface Plasmon Resonance: A Versatile Technique for Biosensor Applications. Sensors 2015, 15, 10481–10510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Zhao, A.; Chen, L.; Zhong, X.; Liao, J.; Gao, M.; Cai, M.; Lee, D.-H.; Li, J.; Chowdhury, D.; et al. Human RIF1 encodes an anti-apoptotic factor required for DNA repair. Carcinogenesis 2009, 30, 1314–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, Y.; Peng, C.; Liu, Y.-B.; Wang, J.; Zhou, H.-H. Silencing RIF1 decreases cell growth, migration and increases cisplatin sensitivity of human cervical cancer cells. Oncotarget 2017, 8, 107044–107051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-B.; Mei, Y.; Long, J.; Zhang, Y.; Hu, D.-L.; Zhou, H. RIF1 promotes human epithelial ovarian cancer growth and progression via activating human telomerase reverse transcriptase expression. J. Exp. Clin. Cancer Res. 2018, 37, 182. [Google Scholar] [CrossRef] [PubMed]
- Hardy, C.F.; Sussel, L.; Shore, D. A RAP1-interacting protein involved in transcriptional silencing and telomere length regulation. Genes Dev. 1992, 6, 801–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Muniandy, P.; Leo, E.; Yin, J.; Thangavel, S.; Shen, X.; Ii, M.; Agama, K.; Guo, R.; Fox, D.; et al. Rif1 provides a new DNA-binding interface for the Bloom syndrome complex to maintain normal replication. EMBO J. 2010, 29, 3140–3155. [Google Scholar] [CrossRef] [Green Version]
- Sreesankar, E.; Senthilkumar, R.; Bharathi, V.; Mishra, R.K.; Mishra, K. Functional diversification of yeast telomere associated protein, Rif1, in higher eukaryotes. BMC Genom. 2012, 13, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverman, J.; Takai, H.; Buonomo, S.B.; Eisenhaber, F.; de Lange, T. Human Rif1, ortholog of a yeast telomeric protein, is regulated by ATM and 53BP1 and functions in the S-phase checkpoint. Genes Dev. 2004, 18, 2108–2119. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Blackburn, E.H. Human Rif1 protein binds aberrant telomeres and aligns along anaphase midzone microtubules. J. Cell Biol. 2004, 167, 819–830. [Google Scholar] [CrossRef] [Green Version]
- Buonomo, S.B.; Wu, Y.; Ferguson, D.; de Lange, T. Mammalian Rif1 contributes to replication stress survival and homology-directed repair. J. Cell Biol. 2009, 187, 385–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornacchia, D.; Dileep, V.; Quivy, J.-P.; Foti, R.; Tili, F.; Santarella-Mellwig, R.; Antony, C.; Almouzni, G.; Gilbert, D.M.; Buonomo, S. Mouse Rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO J. 2012, 31, 3678–3690. [Google Scholar] [CrossRef]
- Hayano, M.; Kanoh, Y.; Matsumoto, S.; Renard-Guillet, C.; Shirahige, K.; Masai, H. Rif1 is a global regulator of timing of replication origin firing in fission yeast. Genes Dev. 2012, 26, 137–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, S.; Ishii, A.; Kanoh, Y.; Oda, M.; Nishito, Y.; Masai, H. Rif1 regulates the replication timing domains on the human genome. EMBO J. 2012, 31, 3667–3677. [Google Scholar] [CrossRef] [Green Version]
- Kanoh, Y.; Matsumoto, S.; Fukatsu, R.; Kakusho, N.; Kono, N.; Renard-Guillet, C.; Masuda, K.; Iida, K.; Nagasawa, K.; Shirahige, K.; et al. Rif1 binds to G quadruplexes and suppresses replication over long distances. Nat. Struct. Mol. Biol. 2015, 22, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Díaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.; Tkáč, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef] [Green Version]
- Hendrickx, A.; Beullens, M.; Ceulemans, H.; Abt, T.D.; Van Eynde, A.; Nicolaescu, E.; Lesage, B.; Bollen, M. Docking Motif-Guided Mapping of the Interactome of Protein Phosphatase-1. Chem. Biol. 2009, 16, 365–371. [Google Scholar] [CrossRef] [Green Version]
- Bollen, M.; Peti, W.; Ragusa, M.J.; Beullens, M. The extended PP1 toolkit: Designed to create specificity. Trends Biochem. Sci. 2010, 35, 450–458. [Google Scholar] [CrossRef] [Green Version]
- Moriyama, K.; Yoshizawa-Sugata, N.; Masai, H. Oligomer formation and G-quadruplex binding by purified murine Rif1 protein, a key organizer of higher-order chromatin architecture. J. Biol. Chem. 2018, 293, 3607–3624. [Google Scholar] [CrossRef] [Green Version]
- Sukackaite, R.; Jensen, M.R.; Mas, P.J.; Blackledge, M.; Buonomo, S.; Hart, D.J. Structural and Biophysical Characterization of Murine Rif1 C Terminus Reveals High Specificity for DNA Cruciform Structures. J. Biol. Chem. 2014, 289, 13903–13911. [Google Scholar] [CrossRef] [Green Version]
- Shi, T.; Bunker, R.; Mattarocci, S.; Ribeyre, C.; Faty, M.; Gut, H.; Scrima, A.; Rass, U.; Rubin, S.; Shore, D.; et al. Rif1 and Rif2 Shape Telomere Function and Architecture through Multivalent Rap1 Interactions. Cell 2013, 153, 1340–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valton, A.; Hassan-Zadeh, V.; Lema, I.; Boggetto, N.; Alberti, P.; Saintomé, C.; Riou, J.-F.; Prioleau, M. G4 motifs affect origin positioning and efficiency in two vertebrate replicators. EMBO J. 2014, 33, 732–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masai, H.; Fukatsu, R.; Kakusho, N.; Kanoh, Y.; Moriyama, K.; Ma, Y.; Iida, K.; Nagasawa, K. Rif1 promotes association of G-quadruplex (G4) by its specific G4 binding and oligomerization activities. Sci. Rep. 2019, 9, 8618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, S.; Fukatsu, R.; Kanoh, Y.; Kakusho, N.; Matsumoto, S.; Chaen, S.; Masai, H. Both a Unique Motif at the C Terminus and an N-Terminal HEAT Repeat Contribute to G-Quadruplex Binding and Origin Regulation by the Rif1 Protein. Mol. Cell. Biol. 2019, 39, e00364-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Cheok, C.F. Dynamics of RIF1 SUMOylation is regulated by PIAS4 in the maintenance of Genomic Stability. Sci. Rep. 2017, 7, 17367. [Google Scholar] [CrossRef] [Green Version]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Gabelica, V.; Maeda, R.; Fujimoto, T.; Yaku, H.; Murashima, T.; Sugimoto, N.; Miyoshi, D. Multiple and Cooperative Binding of Fluorescence Light-up Probe Thioflavin T with Human Telomere DNA G-Quadruplex. Biochemistry 2013, 52, 5620–5628. [Google Scholar] [CrossRef]
- De La Faverie, A.R.; Guédin, A.; Bedrat, A.; Yatsunyk, L.A.; Mergny, J.-L. Thioflavin T as a fluorescence light-up probe for G4 formation. Nucleic Acids Res. 2014, 42, e65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, J.; Queiroz, J.A.; Cruz, C. Circular Dichroism of G-Quadruplex: A Laboratory Experiment for the Study of Topology and Ligand Binding. J. Chem. Educ. 2017, 94, 1547–1551. [Google Scholar] [CrossRef]
- Zuo, X.; Mattern, M.R.; Tan, R.; Li, S.; Hall, J.; Sterner, D.E.; Shoo, J.; Tran, H.; Lim, P.; Sarafianos, S.G.; et al. Expression and purification of SARS coronavirus proteins using SUMO-fusions. Protein Expr. Purif. 2005, 42, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa-Sugata, N.; Yamazaki, S.; Masai, H. Rif1, a Conserved Chromatin Factor Regulating DNA Replication, DNA Repair, and Transcription. In The Initiation of DNA Replication in Eukaryotes; Kaplan, D.L., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 143–158. [Google Scholar] [CrossRef]
- Butt, T.R.; Jonnalagadda, S.; Monia, B.P.; Sternberg, E.J.; Marsh, J.A.; Stadel, J.M.; Ecker, D.J.; Crooke, S.T. Ubiquitin fusion augments the yield of cloned gene products in Escherichia coli. Proc. Natl. Acad. Sci. USA 1989, 86, 2540–2544. [Google Scholar] [CrossRef] [Green Version]
- Malakhov, M.P.; Mattern, M.R.; Malakhova, O.A.; Drinker, M.; Weeks, S.; Butt, T.R. SUMO fusions and SUMO-specific protease for efficient expression and purification of proteins. J. Struct. Funct. Genom. 2004, 5, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Butt, T.R.; Edavettal, S.C.; Hall, J.P.; Mattern, M.R. SUMO fusion technology for difficult-to-express proteins. Protein Expr. Purif. 2005, 43, 1–9. [Google Scholar] [CrossRef]
- Creton, S.; Jentsch, S. SnapShot: The SUMO System. Cell 2010, 143, 848.e1. [Google Scholar] [CrossRef]
- Kolesar, P.; Sarangi, P.; Altmannova, V.; Zhao, X.; Krejci, L. Dual roles of the SUMO-interacting motif in the regulation of Srs2 sumoylation. Nucleic Acids Res. 2012, 40, 7831–7843. [Google Scholar] [CrossRef] [Green Version]
- Altmannova, V.; Eckert-Boulet, N.; Arneric, M.; Kolesar, P.; Chaloupkova, R.; Damborsky, J.; Sung, P.; Zhao, X.; Lisby, M.; Krejci, L. Rad52 SUMOylation affects the efficiency of the DNA repair. Nucleic Acids Res. 2010, 38, 4708–4721. [Google Scholar] [CrossRef] [Green Version]
- Wei, F.; Schoöler, H.R.; Atchison, M.L. Sumoylation of Oct4 Enhances Its Stability, DNA Binding, and Transactivation. J. Biol. Chem. 2007, 282, 21551–21560. [Google Scholar] [CrossRef] [Green Version]
- Hang, L.E.; Lopez, C.R.; Liu, X.; Williams, J.M.; Chung, I.; Wei, L.; Bertuch, A.A.; Zhao, X. Regulation of Ku-DNA Association by Yku70 C-terminal Tail and SUMO Modification. J. Biol. Chem. 2014, 289, 10308–10317. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Strategy | Agent Used | Result |
|---|---|---|
| Buffer type and pH | Tris and Phosphate 25 mM pH range: 6–8. | Insoluble |
| Lysis buffer Additives | Salts: NaCl (100 mM), KCl (100 mM), ammonium sulfate (100 mM and 500 mM), sodium acetate (100 mM). | Insoluble |
| Urea (0.1, 0.5 M) 1 | ||
| Amino acids: Arg (375 mM), Arg (1 M), Glu (5 mM), Asp (50 mM), Glycine-betaine (10, 100 and 1000 mM), Arg+Gly (each 50 mM). | Insoluble | |
| Reducing Agents: 2-ME (0.05%), DTT (5 mM). | Insoluble | |
| Detergents: Triton X-100 (0.2% and 1% (v/v)), SDS (0.1% (w/v)), Sarcosyl (0.1–0.3% (w/v)) 2 | Insoluble | |
| Fatty Acid: Glycerol (5 and 10% (v/v)). | Insoluble | |
| Sugars: Trehalose (0.5 M), sorbitol (0.5 M), sorbitol + trehalose+Arg (each 50 mM). | Insoluble | |
| Induction condition | Culture media (TB, LB). | Insoluble |
| Time of induction (3, 6, and 18 h) for MuRif1-CTD 270 aa and 221 aa. | Insoluble | |
| Temperatures (18 °C, 25 °C, 37 °C), and heat shock. | Insoluble | |
| Effect of temperature and time of induction combination. Effect of medium, temperature and time of expression combination. | Insoluble | |
| Different IPTG concentrations (0.1, 0.25, 0.5, 1, and 2 mM). Different IPTG concentrations (0.01, 0.1, 0.5 mM) and time of induction (12, 20, and 24 h). | Insoluble | |
| Induction methods (Lactose in place of IPTG). | Insoluble | |
| OD for induction (0.8, 1, and 1.2) and glucose addition. | Insoluble | |
| Hosts (BL21, BL21 (DE3), Rosetta gami, BL21 (DE3) pLyse) with different time of induction. | Insoluble | |
| Induction additives | Amino acid: Arg (0.5 M). | Insoluble |
| Alcohol: Ethanol (3% v/v). | Insoluble | |
| Sugar: Trehalose (0.5 M), sorbitol (0.5 M) and a mixture of trehalose, sorbitol and Arginine, Betain (100 mM) | Insoluble | |
| Solubilization tag | SUMO | Soluble |
| ka(M−1 s−1) | kd (s−1) | KD (nM) | Rmax 1 | Response Standard Deviation | ΔG°bind 2(kcal/mol) * | |
|---|---|---|---|---|---|---|
| Kinetic Fit | Steady-State Fit | |||||
| (4.2 ± 0.2) 106 | 0.030 ± 0.001 | 7.1 ± 0.4 | 6 ± 1 | 99 ± 2 | 4.856 | −11.123 ± 0.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alavi, S.; Ghadiri, H.; Dabirmanesh, B.; Khajeh, K. SPR Analysis of SUMO-Murine Rap1-Interacting Factor 1 C-Terminal Domain Interaction with G4. Biosensors 2022, 12, 37. https://doi.org/10.3390/bios12010037
Alavi S, Ghadiri H, Dabirmanesh B, Khajeh K. SPR Analysis of SUMO-Murine Rap1-Interacting Factor 1 C-Terminal Domain Interaction with G4. Biosensors. 2022; 12(1):37. https://doi.org/10.3390/bios12010037
Chicago/Turabian StyleAlavi, Sana, Hamed Ghadiri, Bahareh Dabirmanesh, and Khosro Khajeh. 2022. "SPR Analysis of SUMO-Murine Rap1-Interacting Factor 1 C-Terminal Domain Interaction with G4" Biosensors 12, no. 1: 37. https://doi.org/10.3390/bios12010037
APA StyleAlavi, S., Ghadiri, H., Dabirmanesh, B., & Khajeh, K. (2022). SPR Analysis of SUMO-Murine Rap1-Interacting Factor 1 C-Terminal Domain Interaction with G4. Biosensors, 12(1), 37. https://doi.org/10.3390/bios12010037