Developing Activated Carbon Veil Electrode for Sensing Salivary Uric Acid

 ,
,  ,
,  and
and
Abstract
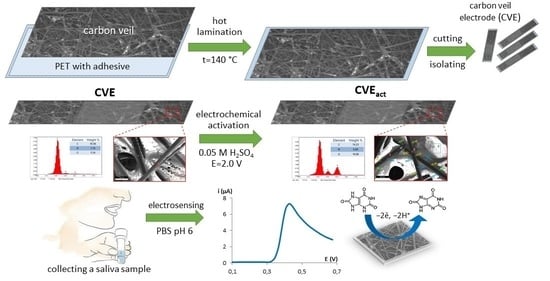
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Apparatus
2.3. Procedures
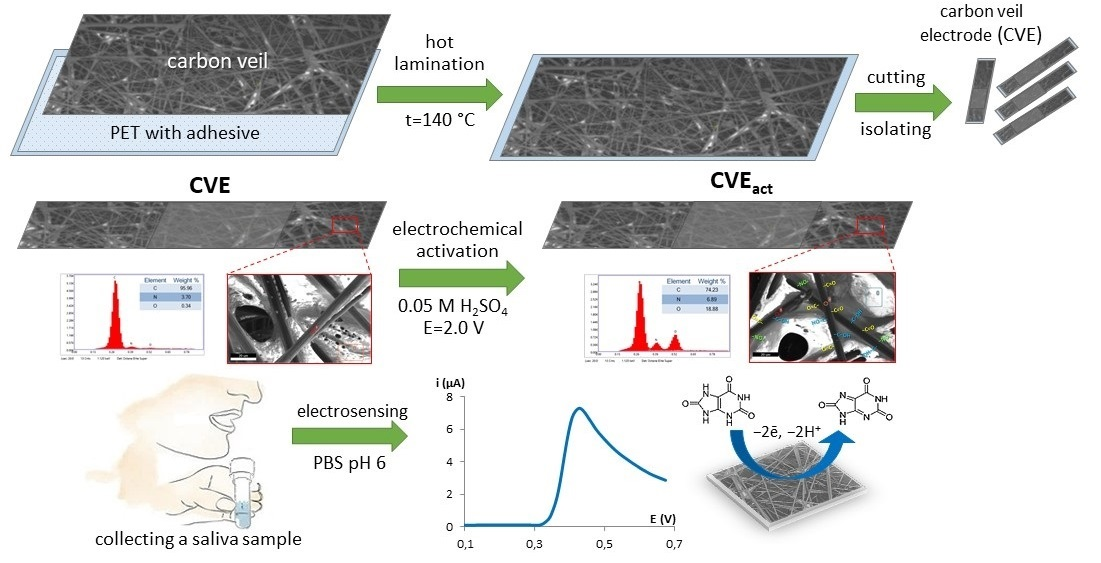
2.3.1. Electrode Fabrication and Sensor Preparation
2.3.2. Sampling and Sample Preparation
2.3.3. Electrochemical Measurements
2.4. Statistical Analysis and Data Treatment
3. Results
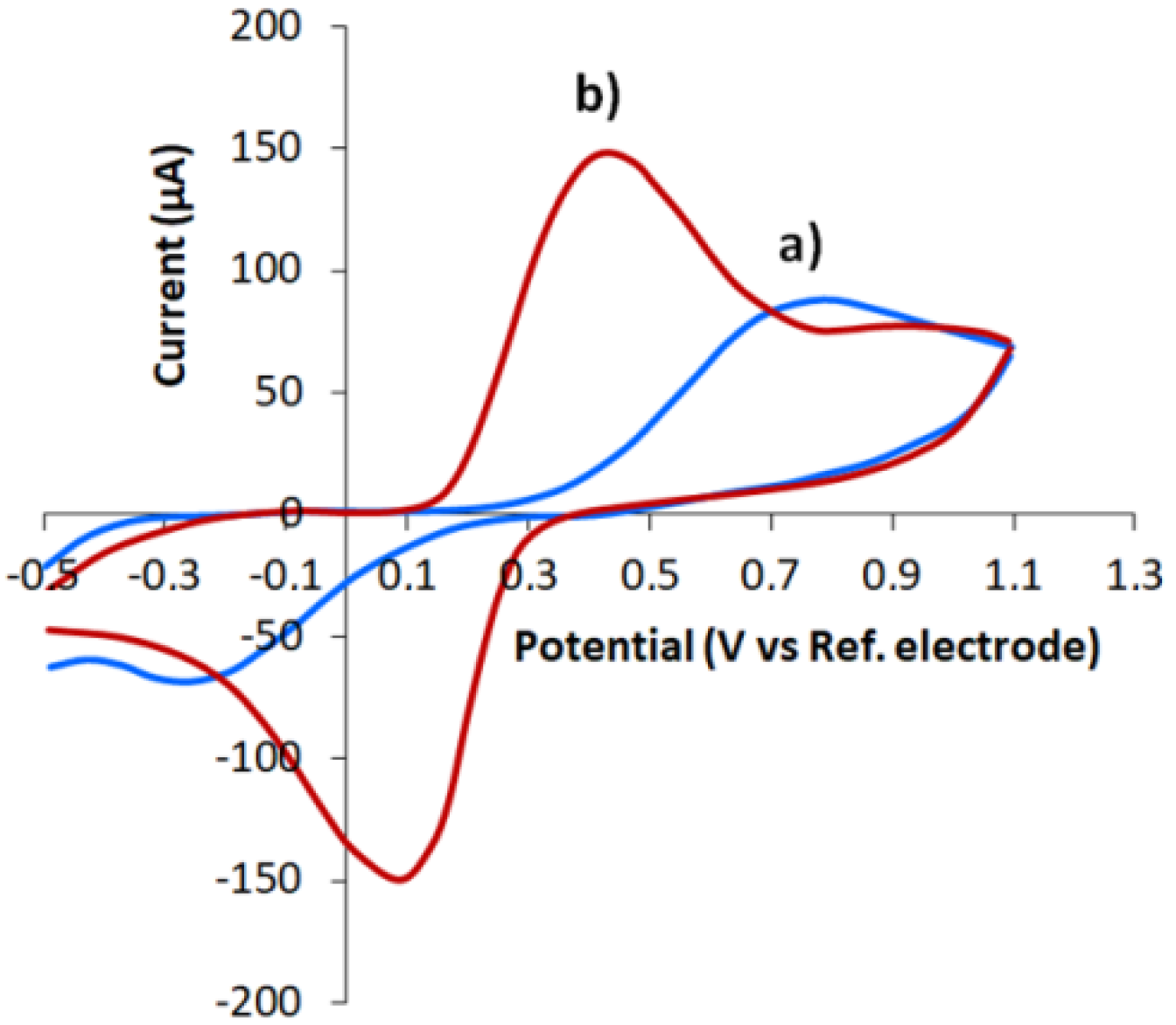
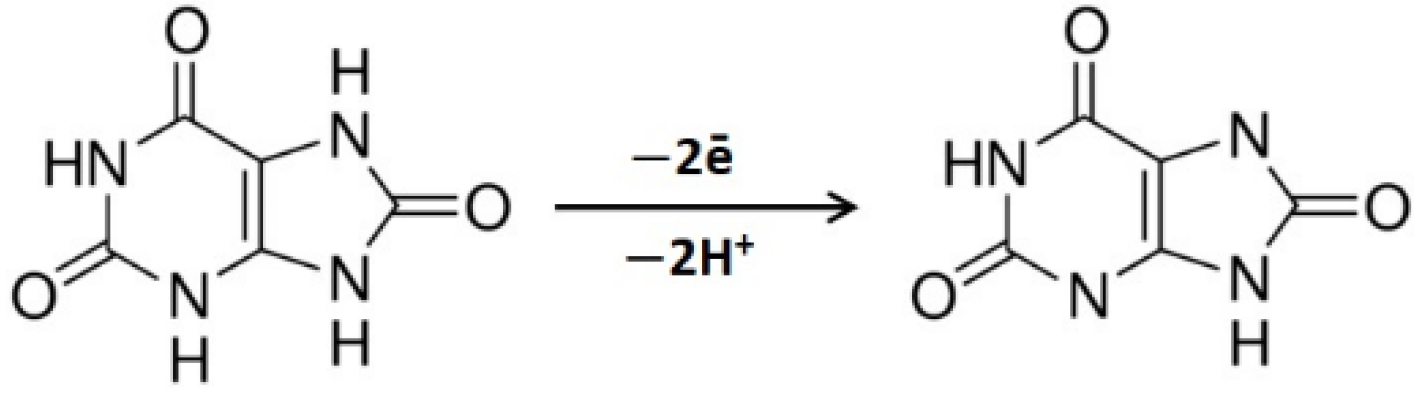
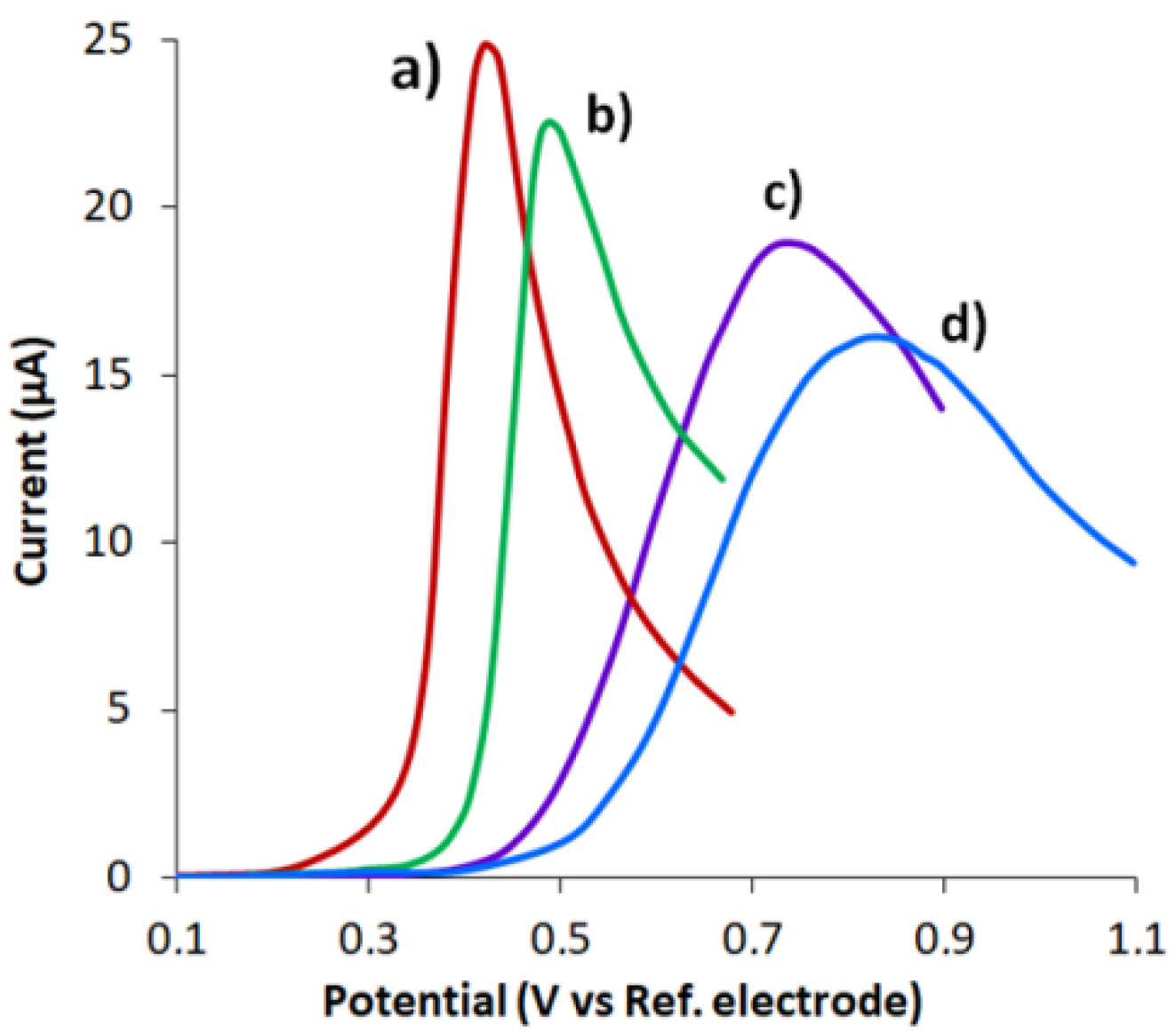
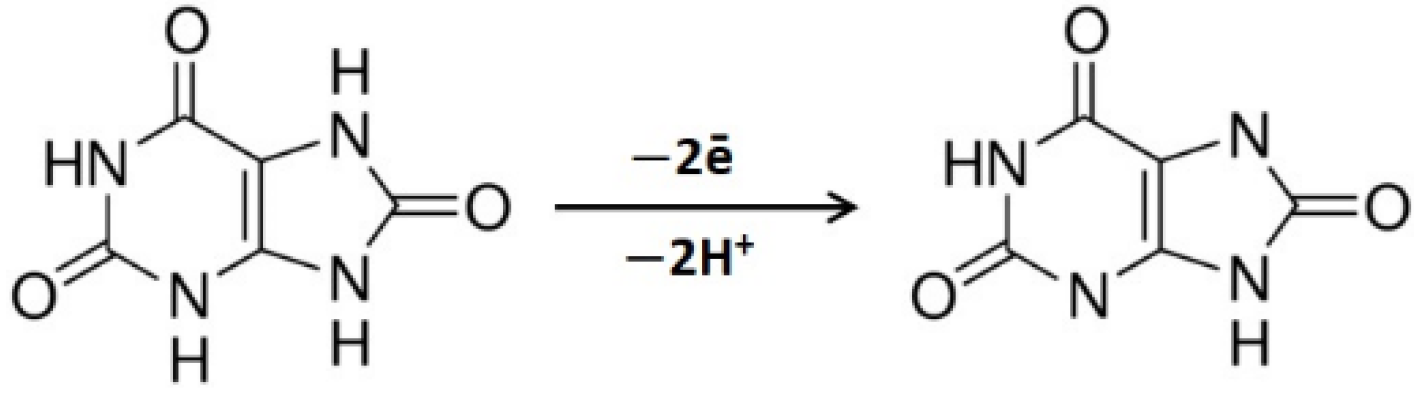
3.1. CVE Activation
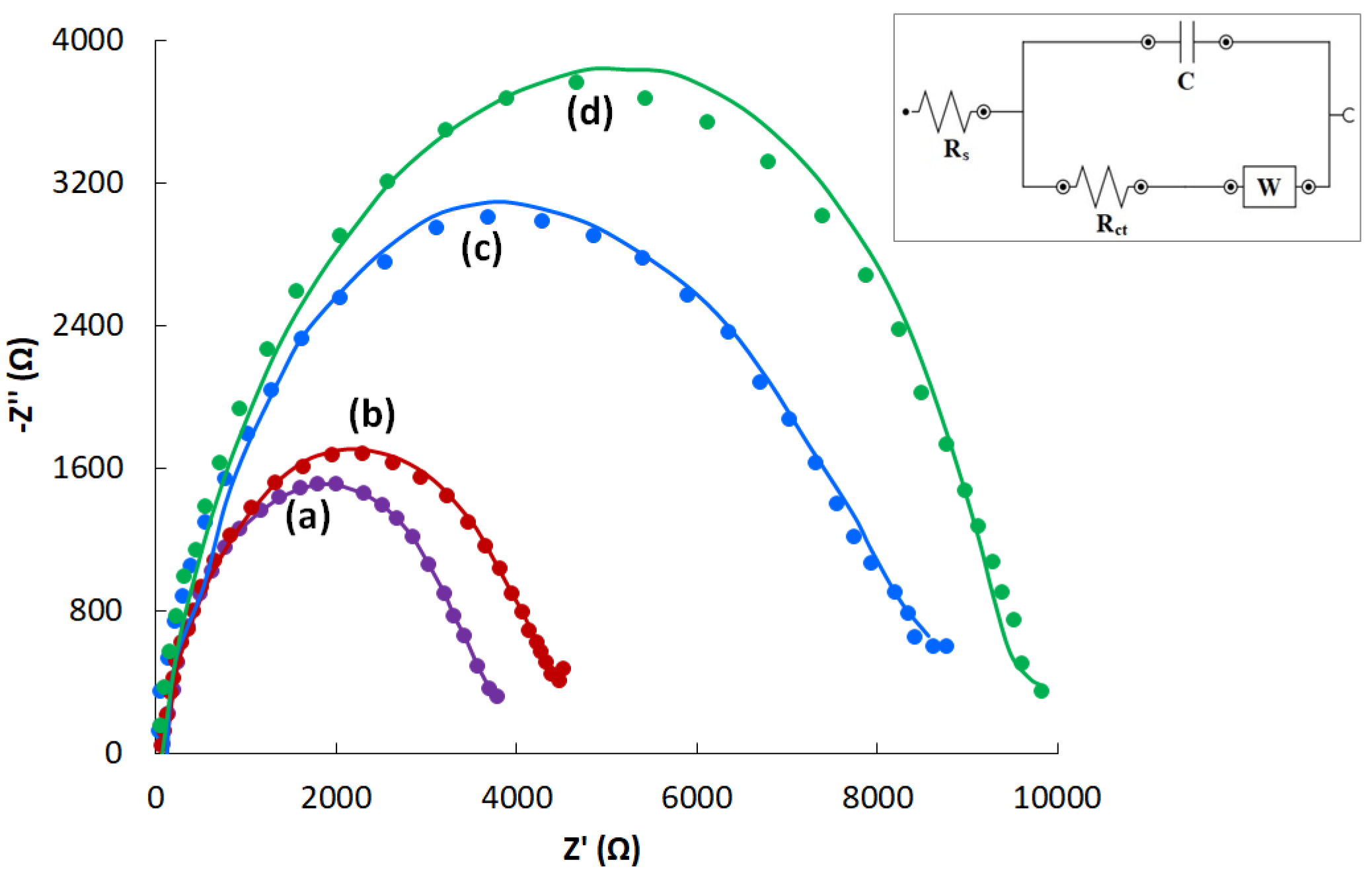
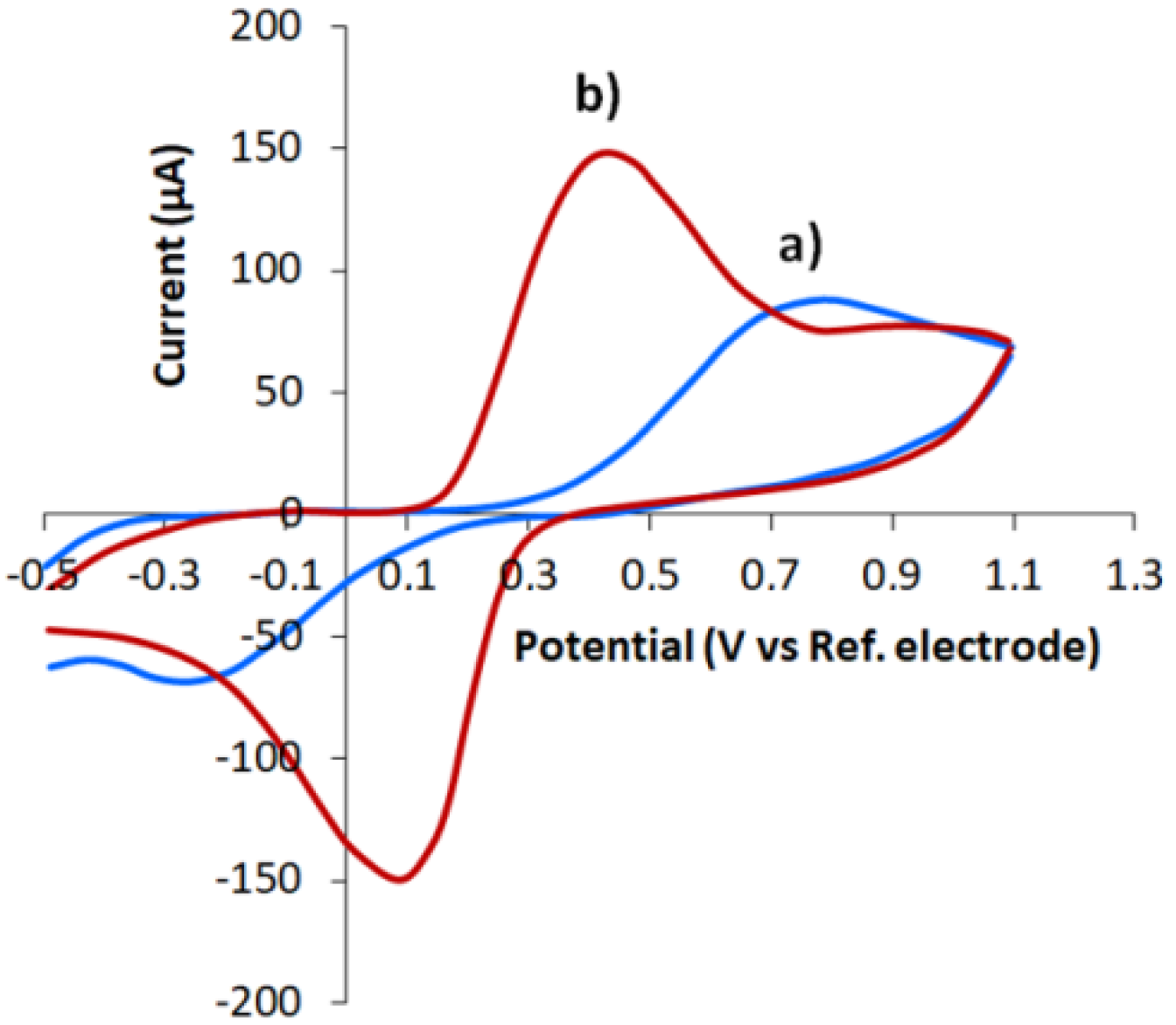
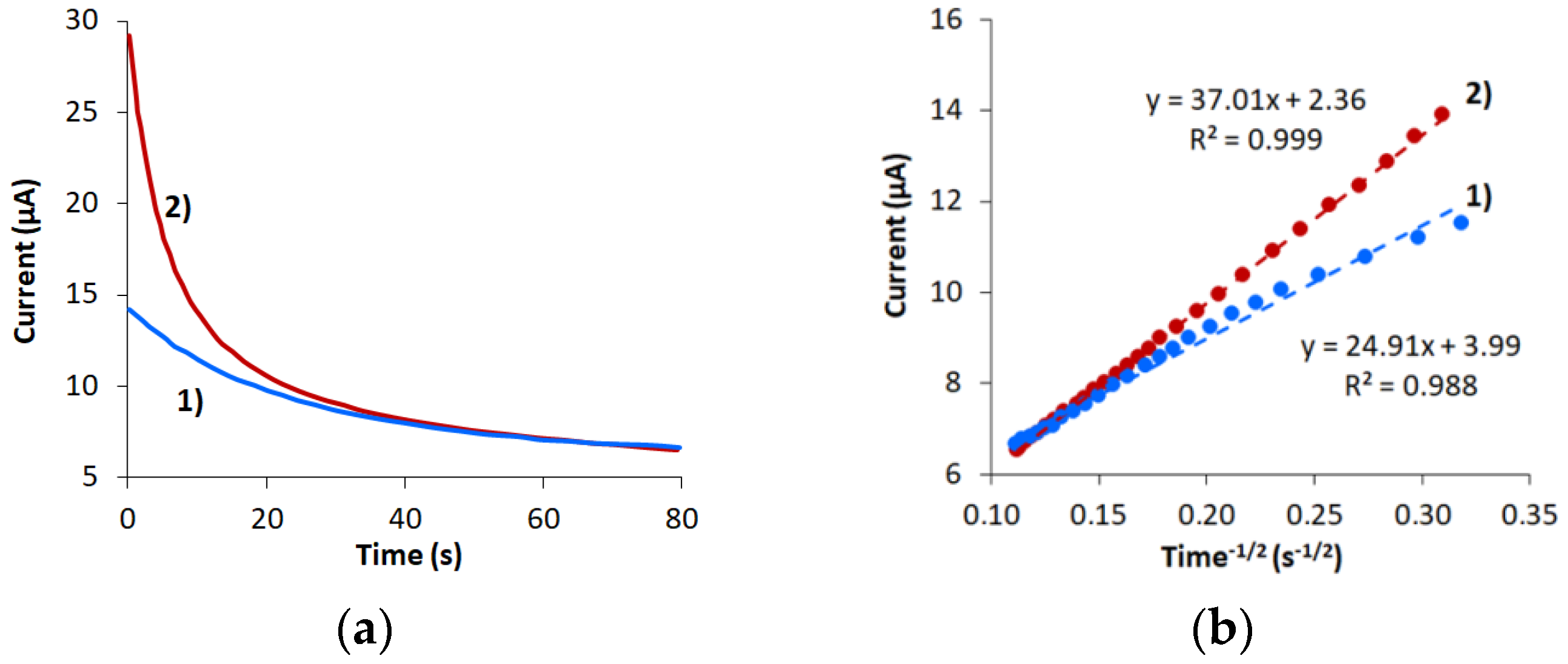
3.2. Characterization of CVE and CVEact
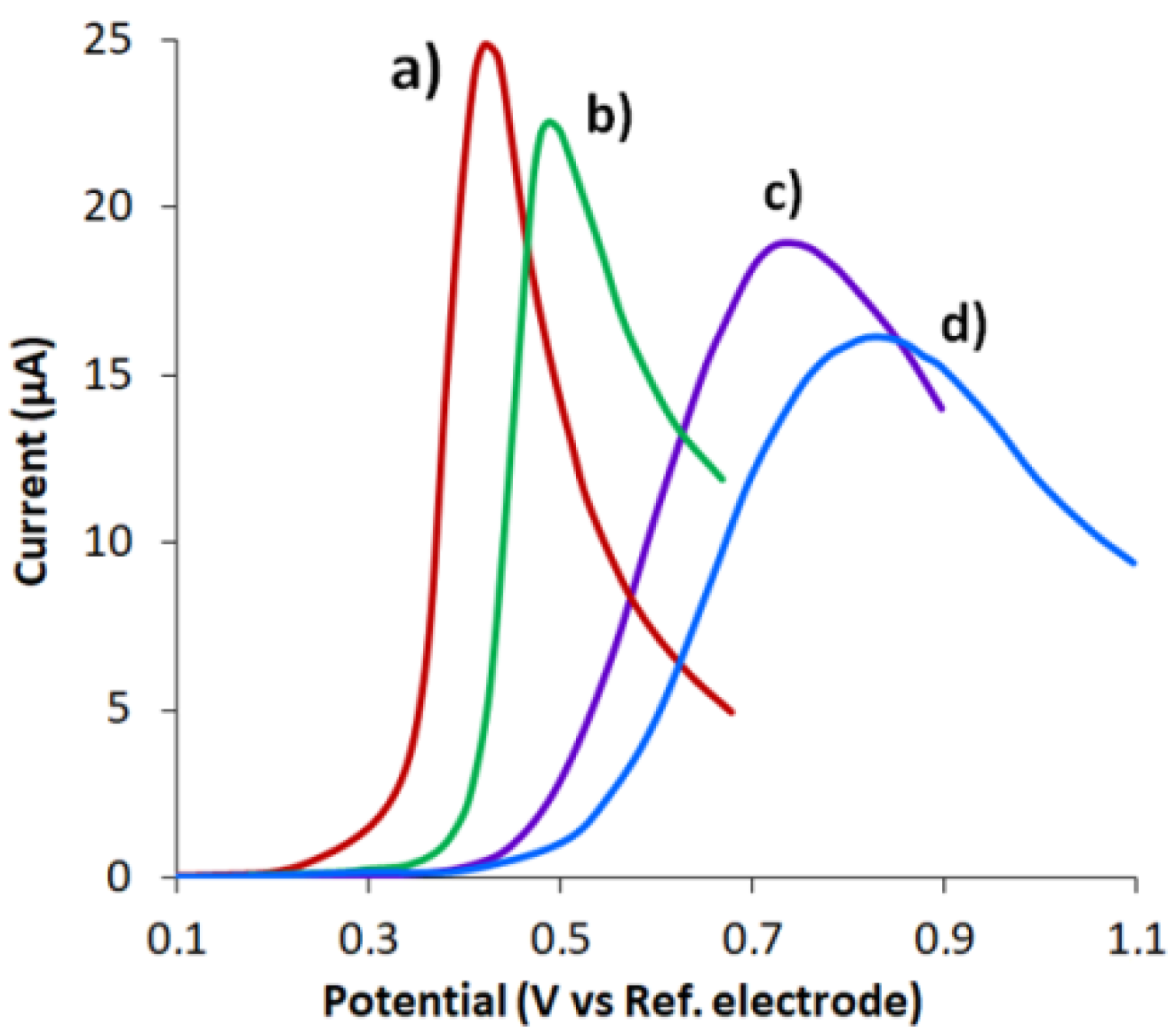
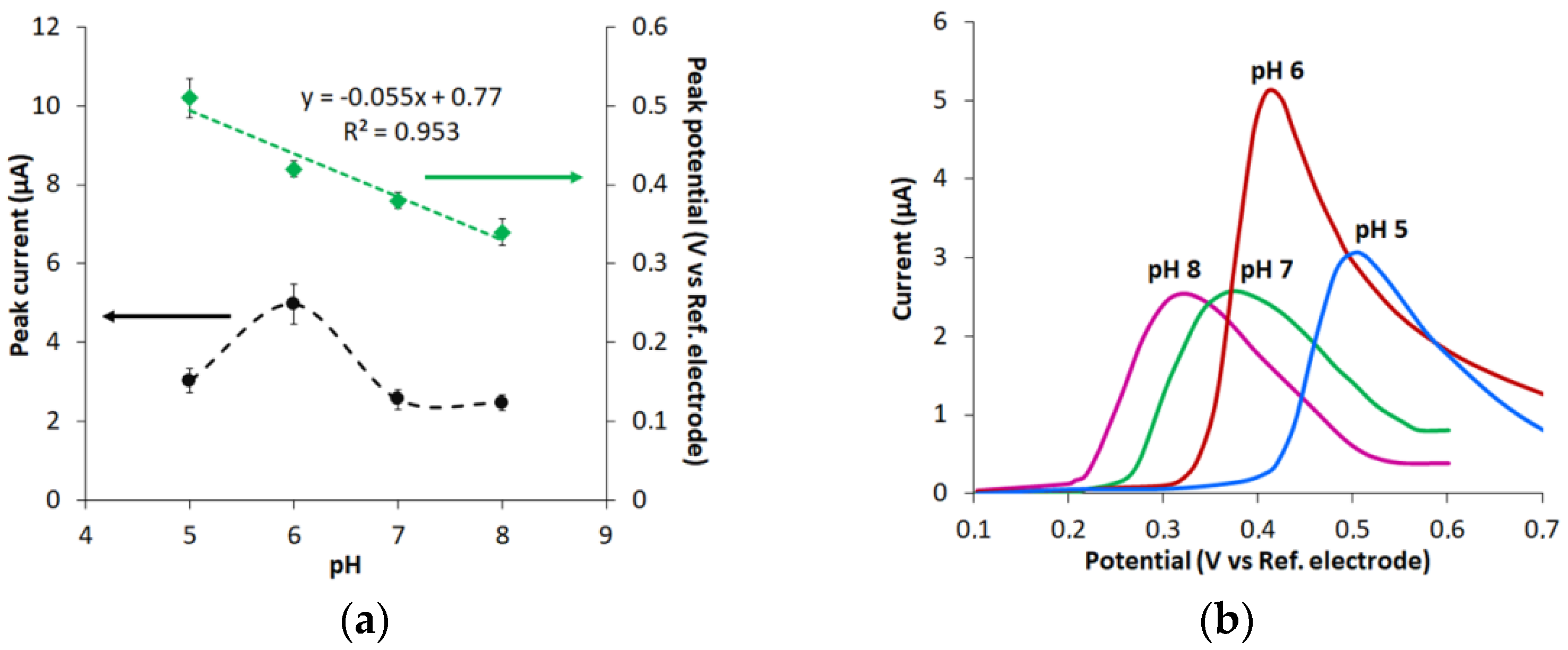
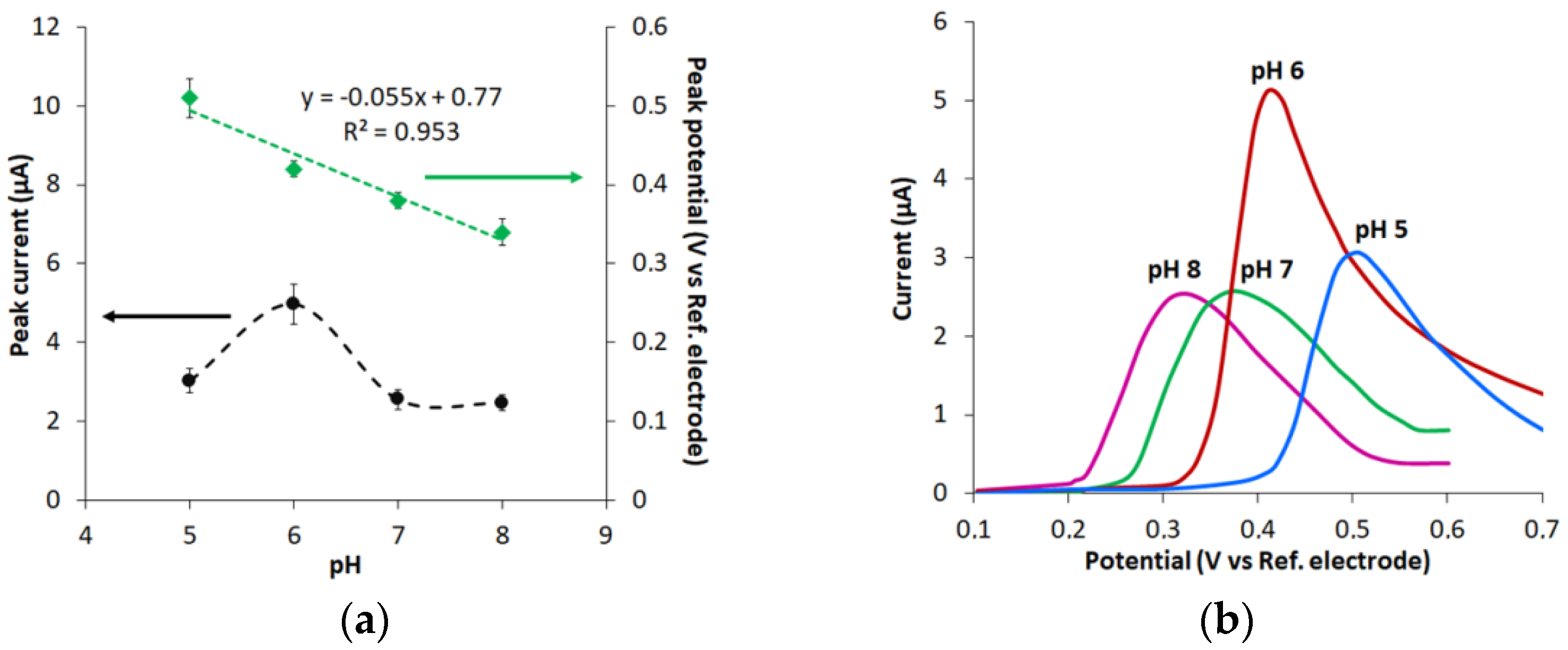
3.3. Effect of pH
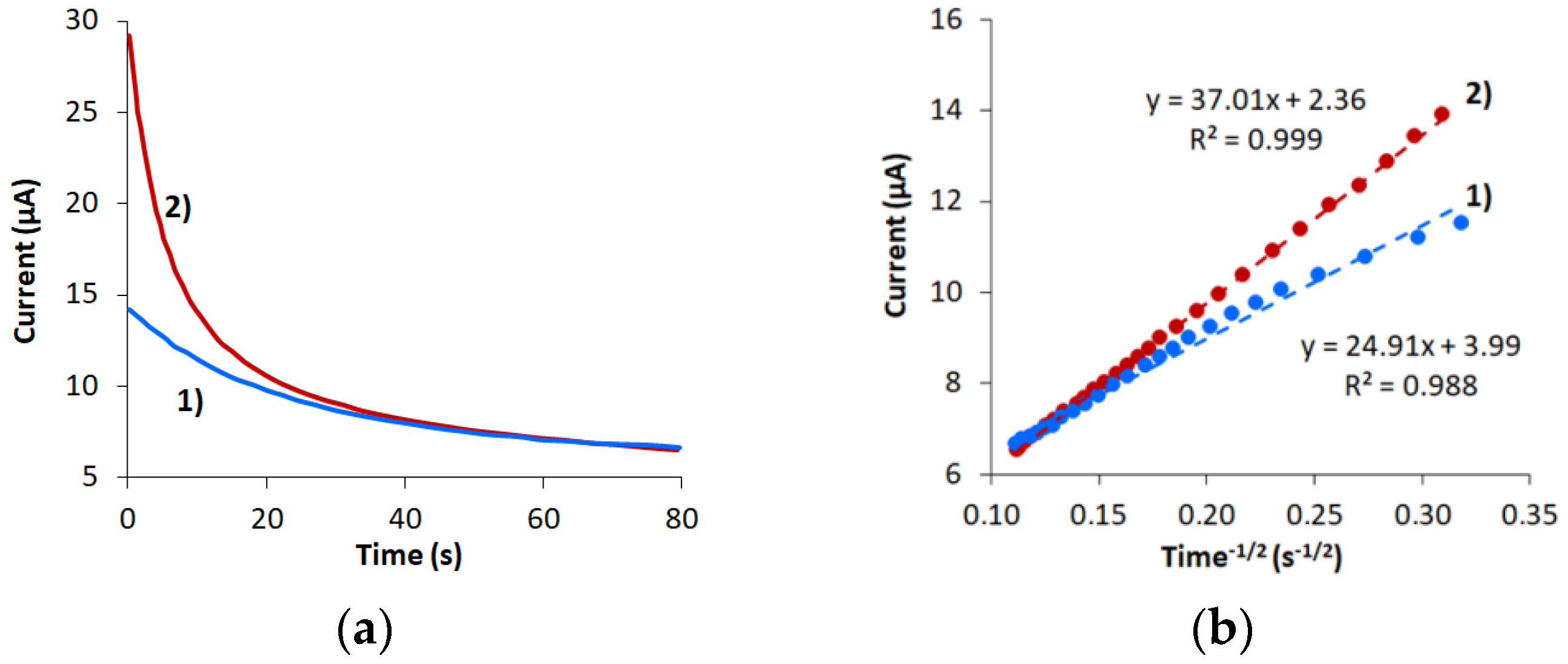
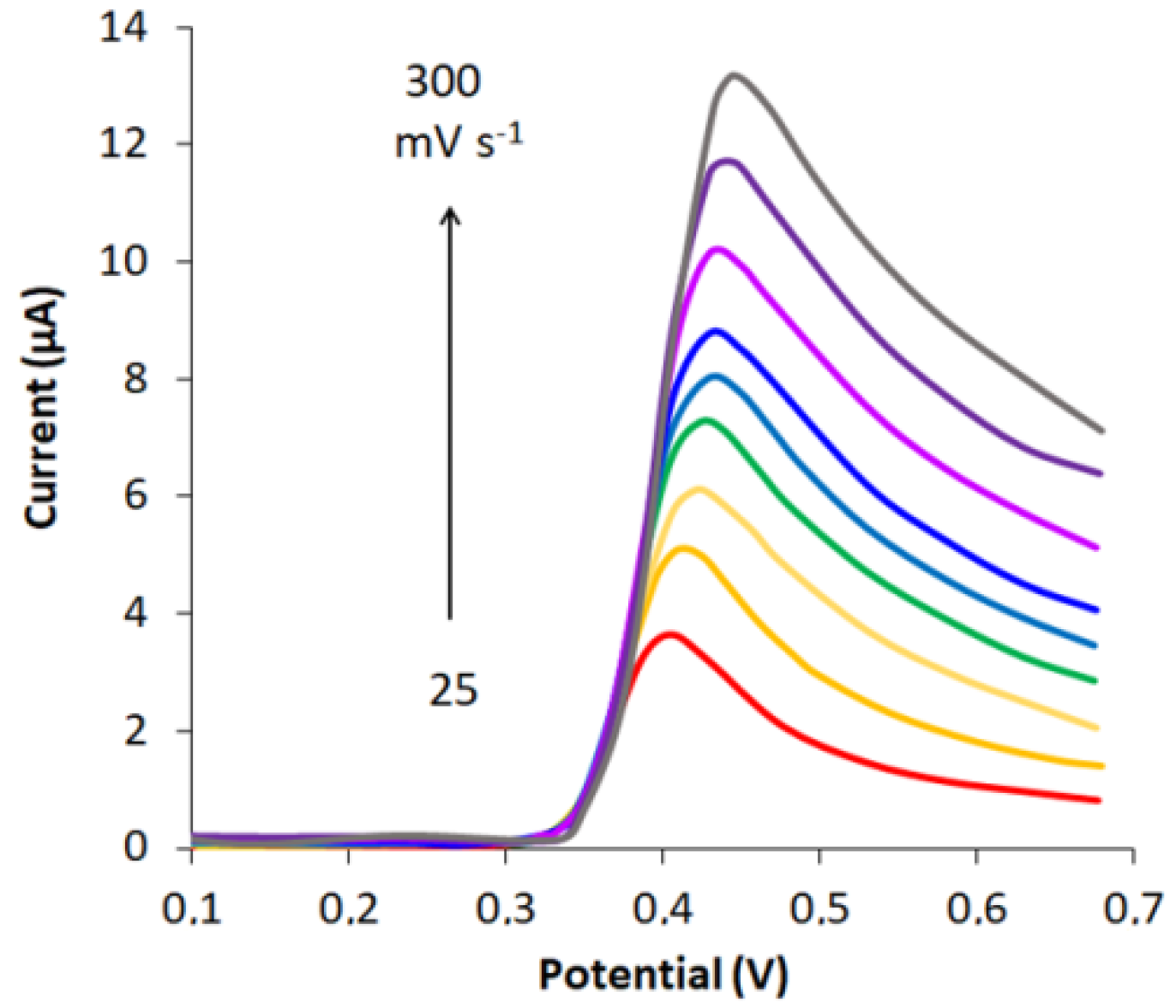
3.4. Effect of Potential Scan Rate
3.5. Analytical Characteristics of CVEact
3.6. Salivary UA Determination
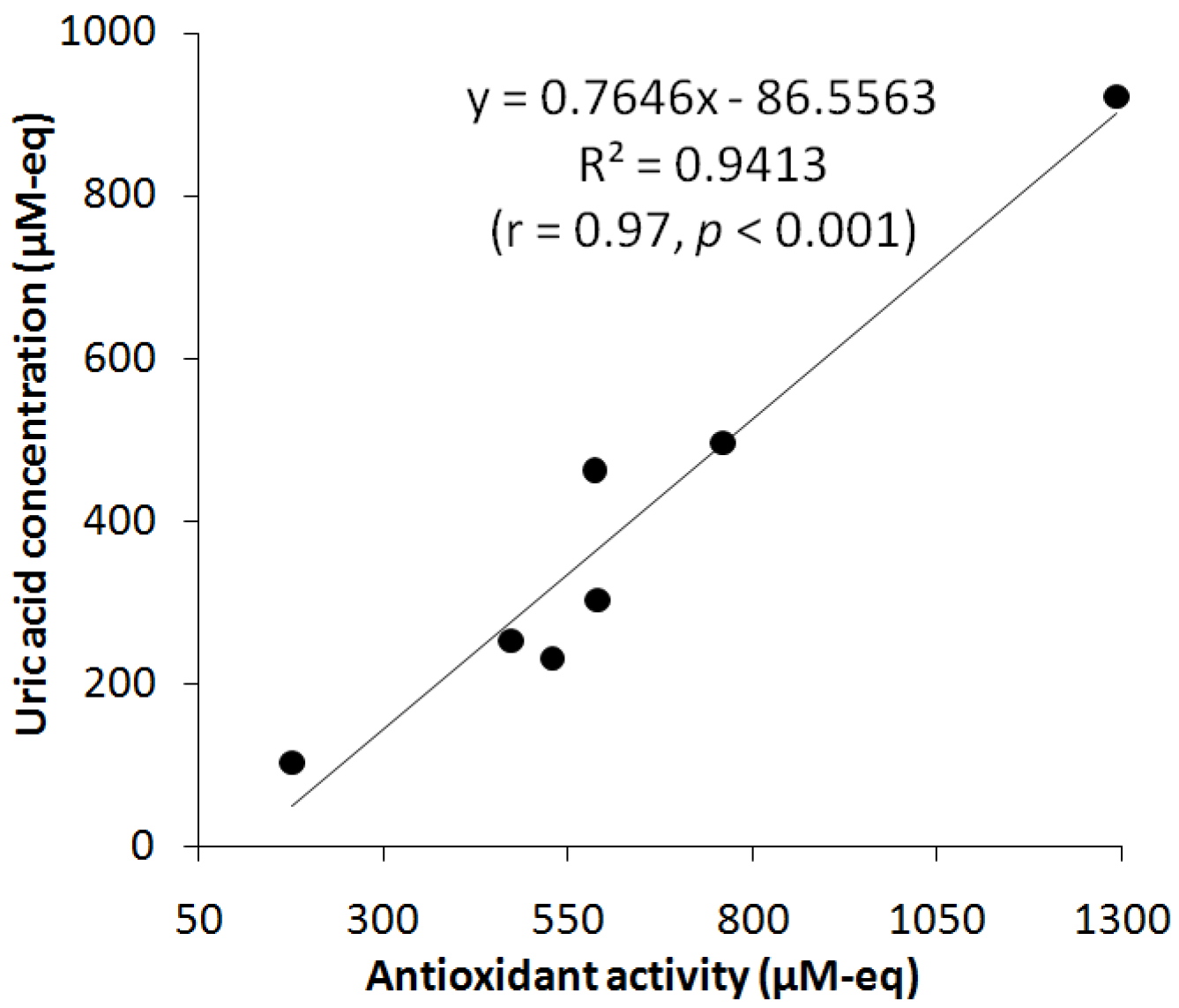
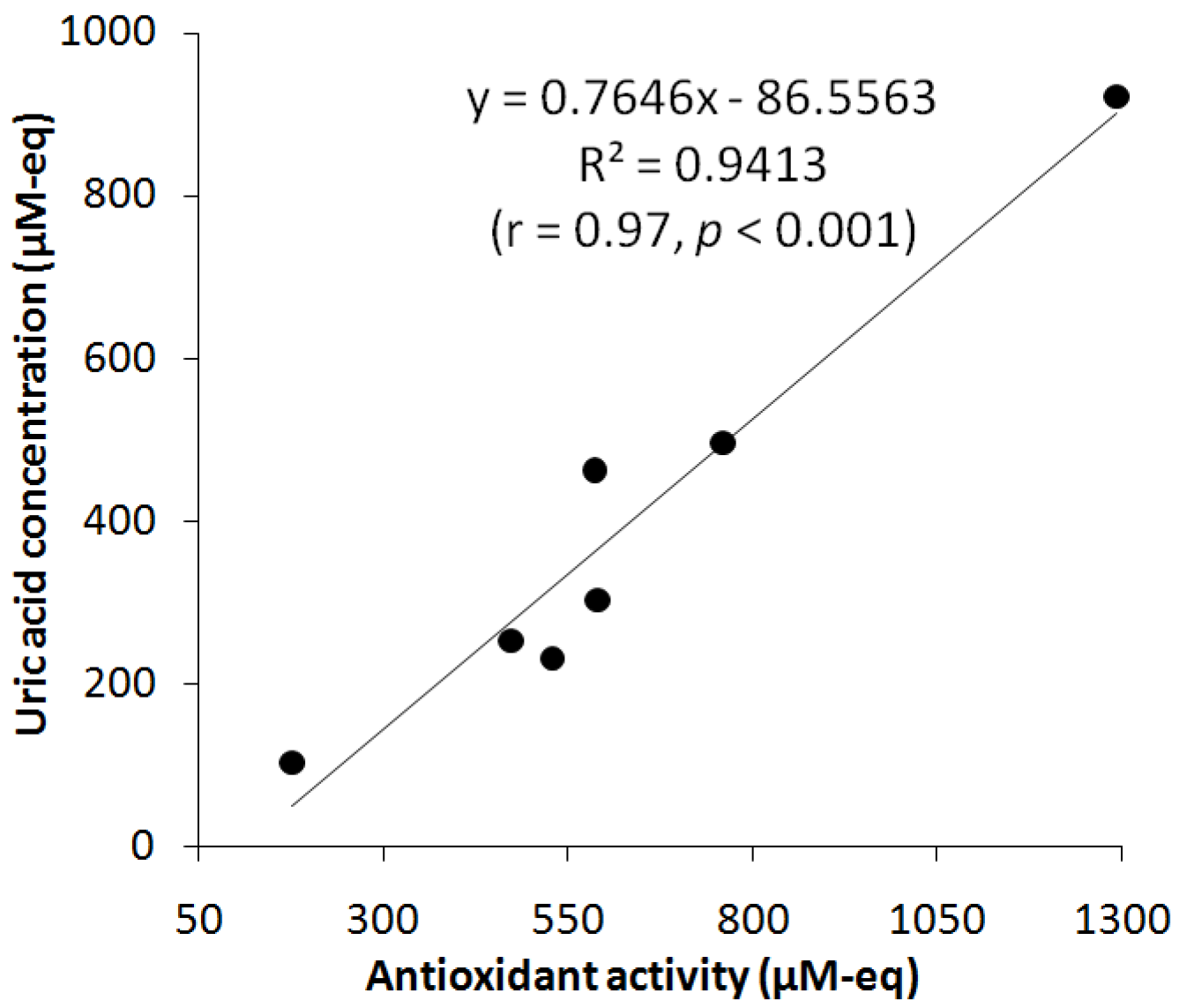
3.7. Study of UA and АОА Correlation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vernerová, A.; Kujovská Krčmová, L.; Melichar, B.; Švec, F. Non-invasive determination of uric acid in human saliva in the diagnosis of serious disorders. Clin. Chem. Lab. Med. 2021, 59, 797–812. [Google Scholar] [CrossRef] [PubMed]
- Choromańska, K.; Choromańska, B.; Dąbrowska, E.; Bączek, W.; Myśliwiec, P.; Dadan, J.; Zalewska, A. Saliva of obese patients – Is it different? Postepy Hig. Med. Dosw. 2015, 69, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Bakhtiari, S.; Toosi, P.; Samadi, S.; Bakhshi, M. Assessment of uric acid level in the saliva of patients with oral lichen planus. Med. Princ. Pract. 2017, 26, 57–60. [Google Scholar] [CrossRef]
- Riis, J.L.; Bryce, C.I.; Matin, M.J.; Stebbins, J.L.; Kornienko, O.; van Huisstede, L.; Granger, D.A. The validity, stability, and utility of measuring uric acid in saliva. Biomark. Med. 2018, 12, 583–596. [Google Scholar] [CrossRef]
- Bilancio, G.; Cavallo, P.; Lombardi, C.; Guarino, E.; Cozza, V.; Giordano, F.; Palladino, G.; Cirillo, M. Saliva for assessing creatinine, uric acid, and potassium in nephropathic patients. BMC Nephrol. 2019, 20, 242. [Google Scholar] [CrossRef]
- Wu, W.-C.; Chen, H.-Y.T.; Lin, S.-C.; Chen, H.-Y.; Chen, F.-R.; Chang, H.-T.; Tseng, F.-G. Nitrogen-doped carbon nanodots prepared from polyethylenimine for fluorometric determination of salivary uric acid. Microchim. Acta 2019, 186, 166. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Luo, Y.; Zhou, C.-Y.; Peng, A.; Liu, J.-Y. A sensitive and accurate method to simultaneously measure uric acid and creatinine in human saliva by using LC-MS/MS. Bioanalysis 2017, 9, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Honeychurch, K.C. The determination of uric acid in human saliva by liquid chromatography with electrochemical detection. J. Anal. Bioanal. Sep. Tech. 2017, 2, 47–51. [Google Scholar] [CrossRef]
- Wang, X.; Lu, J.; Tang, X.; Qiu, P. Colorimetric detection of uric acid with high sensitivity using Cu2O@Ag nanocomposites. Chem. Afr. 2020, 3, 749–758. [Google Scholar] [CrossRef]
- Guan, Y.; Chu, Q.; Ye, J. Determination of uric acid in human saliva by capillary electrophoresis with electrochemical detection: Potential application in fast diagnosis of gout. Anal. Bioanal. Chem. 2004, 380, 913–917. [Google Scholar] [CrossRef]
- Chu, Q.C.; Lin, M.; Geng, C.H.; Ye, J.N. Determination of uric acid in human saliva and urine using miniaturized capillary electrophoresis with amperometric detection. Chromatographia 2007, 65, 179–184. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.X.; Xue, W.; Zhou, Y.J.; Duan, D.D.; Ding, Y.P.; Zhang, R.Z. A flexible and highly selective nonenzymatic uric acid sensor based on free-standing carbon fiber. Funct. Mater. 2020, 27, 218–223. [Google Scholar] [CrossRef]
- Stozhko, N.; Bukharinova, M.; Galperin, L.; Brainina, K. A nanostructured sensor based on gold nanoparticles and nafion for determination of uric acid. Biosensors 2018, 8, 21. [Google Scholar] [CrossRef]
- Wang, X.; Chen, S.; Tang, X.; Linc, D.; Qiu, P. Ultrasensitive detection of uric acid in serum of patients with gout by a new assay based on Pt@Ag nanoflowers. RSC Adv. 2019, 9, 36578–36585. [Google Scholar] [CrossRef]
- Erden, P.E.; Kılıç, E. A review of enzymatic uric acid biosensors based on amperometric detection. Talanta 2013, 107, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi, D.; Whitcombe, M.J.; Davis, F.; Sharma, P.S.; Prasad, B.B. Electrochemical detection of uric acid in mixed and clinical samples: A review. Electroanalysis 2011, 23, 305–320. [Google Scholar] [CrossRef]
- Ratautaite, V.; Samukaite-Bubniene, U.; Plausinaitis, D.; Boguzaite, R.; Balciunas, D.; Ramanaviciene, A.; Neunert, G.; Ramanavicius, A. Molecular imprinting technology for determination of uric acid. Int. J. Mol. Sci. 2021, 22, 5032. [Google Scholar] [CrossRef] [PubMed]
- Kaya, S.I.; Kurbanoglu, S.; Ozkan, S.A. Nanomaterials-based nanosensors for the simultaneous electrochemical determination of biologically important compounds: Ascorbic acid, uric acid, and dopamine. Crit. Rev. Anal. Chem. 2019, 49, 101–125. [Google Scholar] [CrossRef]
- Shi, W.; Li, J.; Wu, J.; Wie, Q.; Chen, C.; Bao, N.; Yu, C.; Gu, H. An electrochemical biosensor based on multi-wall carbon nanotube–modified screen-printed electrode immobilized by uricase for the detection of salivary uric acid. Anal. Bioanal. Chem. 2020, 412, 7275–7283. [Google Scholar] [CrossRef]
- Cinková, K.; Kianičková, K.; Stanković, D.M.; Vojs, M.; Martond, M.; Švorc, L. The doping level of boron-doped diamond electrodes affects the voltammetric sensing of uric acid. Anal. Methods 2018, 10, 991–996. [Google Scholar] [CrossRef]
- Buledi, J.A.; Ameen, S.; Memon, S.A.; Fatima, A.; Solangi, A.R.; Mallah, A.; Karimi, F.; Malakmohammadi, S.; Agarwal, S.; Gupta, V.K. An improved non-enzymatic electrochemical sensor amplified with CuO nanostructures for sensitive determination of uric acid. Open Chem. 2021, 19, 481–491. [Google Scholar] [CrossRef]
- Thanh, T.S.; Qui, P.T.; Tu, N.T.T.; Toan, T.T.T.; Hoa, T.T.B.; Son, L.V.T.; Nguyen, D.M.; Tuyen, T.N.; Khieu, D.Q. Electrochemical determination of uric acid in urine by using zeolite imidazolate framework-11 modified electrode. J. Nanomater. 2021, 2021, 9914062. [Google Scholar] [CrossRef]
- Bhatt, P.; Goe, A. Carbon fibres: Production, properties and potential use. Mat. Sci. Res. India 2017, 14, 52–57. [Google Scholar] [CrossRef]
- Kumar, N.; Gangwar, A.K.; Devi, K.S. Carbon fibers in biomedical applications. In Recent Developments in the Field of Carbon Fibers; Khanna, R., Cayumil, R., Eds.; Intech Open: London, UK, 2018; pp. 728–1157. [Google Scholar] [CrossRef]
- Torrinha, A.; Morais, S. Electrochemical (bio)sensors based on carbon cloth and carbon paper: An overview. TrAC Trends Anal. Chem. 2021, 142, 116324. [Google Scholar] [CrossRef]
- Stozhko, N.Y.; Bukharinova, M.A.; Khamzina, E.I.; Tarasov, A.V.; Sokolkov, S.V. Film carbon veil-based electrode modified with Triton X-100 for nitrite determination. Chemosensors 2020, 8, 78. [Google Scholar] [CrossRef]
- Zhu, W.; Zhang, Y.; Gong, J.; Ma, Y.; Sun, J.; Li, T.; Wang, J. Surface engineering of carbon fiber paper toward exceptionally high-performance and stable electrochemical nitrite sensing. ACS Sens. 2019, 4, 2980–2987. [Google Scholar] [CrossRef]
- Brainina, K.Z.; Bukharinova, M.A.; Stozhko, N.Y.; Sokolkov, S.V.; Tarasov, A.V.; Vidrevich, M.B. Electrochemical sensor based on a carbon veil modified by phytosynthesized gold nanoparticles for determination of ascorbic acid. Sensors 2020, 20, 1800. [Google Scholar] [CrossRef] [PubMed]
- Torrinha, Á.; Martins, M.; Tavares, M.; Delerue-Matos, C.; Morais, S. Carbon paper as a promising sensing material: Characterization and electroanalysis of ketoprofen in wastewater and fish. Talanta 2021, 226, 122111. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.V.; Khamzina, E.I.; Bukharinova, M.A.; Stozhko, N.Y. Flexible potentiometric sensor system for non-invasive determination of antioxidant activity of human skin: Application for evaluating the effectiveness of phytocosmetic products. Chemosensors 2021, 9, 76. [Google Scholar] [CrossRef]
- Benjamin, O.; Silcock, P.; Beauchamp, J.; Buettner, A.; Everett, D.W. Volatile release and structural stability of b-lactoglobulin primary and multilayer emulsions under simulated oral conditions. Food Chem. 2013, 140, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Brainina, K.Z.; Varzakova, D.P.; Kazakov, Y.E.; Vidrevich, M.B. Noninvasive electrochemical antioxidant activity estimation: Saliva analysis. Biointerface Res. Appl. Chem. 2018, 8, 3381–3387. [Google Scholar]
- Burns, D.T.; Danzer, K.; Townshend, A. Use of the terms “recovery” and “apparent recovery” in analytical procedures (IUPAC Recommendations 2002). Pure Appl. Chem. 2002, 74, 2201–2205. [Google Scholar] [CrossRef]
- Zhu, W.; Zhang, X.; Yin, Y.; Qin, Y.; Zhang, J.; Wang, Q. In-situ electrochemical activation of carbon fiber paper for the highly efficient electroreduction of concentrated nitric acid. Electrochim. Acta 2018, 291, 328–334. [Google Scholar] [CrossRef]
- Kakhki, R.M. A review to recent developments in modification of carbon fiber electrodes. Arab. J. Chem. 2019, 12, 1783–1794. [Google Scholar] [CrossRef]
- Tukimin, N.; Abdullah, J.; Sulaiman, Y. Electrodeposition of poly(3,4-ethylenedioxythiophene)/reduced graphene oxide/manganese dioxide for simultaneous detection of uric acid, dopamine and ascorbic acid. J. Electroanal. Chem. 2018, 820, 74–81. [Google Scholar] [CrossRef]
- Wang, Z.; Han, Y.; Zeng, Y.; Qie, Y.; Wang, Y.; Zheng, D.; Lu, X.; Tong, Y. Activated carbon fiber paper with exceptional capacitive performance as a robust electrode for supercapacitors. J. Mater. Chem. A 2016, 4, 5828–5833. [Google Scholar] [CrossRef]
- Wei, Y.; Xu, Z.; Wang, S.; Liu, Y.; Zhang, D.; Fang, Y. One-step preparation of carbon quantum dots-reduced graphene oxide nanocomposite–modified glass carbon electrode for the simultaneous detection of ascorbic acid, dopamine, and uric acid. Ionics 2020, 26, 5817–5828. [Google Scholar] [CrossRef]
- Motshakeri, M.; Travas-Sejdic, J.; Phillips, A.R.J.; Kilmartin, P.A. Rapid electroanalysis of uric acid and ascorbic acid using a poly(3,4-ethylenedioxythiophene)-modified sensor with application to milk. Electrochim. Acta 2018, 265, 184–193. [Google Scholar] [CrossRef]
- Shekh, M.I.; Amirian, J.; Du, B.; Kumar, A.; Sharma, G.; Stadler, F.J.; Song, J. Electrospun ferric ceria nanofibers blended with MWCNTs for high-performance electrochemical detection of uric acid. Ceram. Int. 2020, 46, 9050–9064. [Google Scholar] [CrossRef]
- Golovanov, S.A.; Sivkov, A.V.; Polikarpova, A.M.; Drozhzheva, V.V.; Andryuhin, M.I.; Prosyannikov, M.Y. Metabolic risk factors and formation of urinary stones. Study III: Effect of urine pH. Exp. Clin. Urol. 2018, 1, 84–90. (In Russian) [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; John Wiley & Sons Inc: Hoboken, NJ, USA, 2001. [Google Scholar]
- Shibasaki, K.; Kimura, M.; Ikarashi, R.; Yamaguchi, A.; Watanabe, T. Uric acid concentration in saliva and its changes with the patients receiving treatment for hyperuricemia. Metabolomics 2012, 8, 484–491. [Google Scholar] [CrossRef]
- Mäkilä, E.; Kirveskari, P. A study of ascorbic acid in human saliva. Arch. Oral. Biol. 1969, 14, 1285–1292. [Google Scholar] [CrossRef]
- Huang, X.; Shi, W.; Li, J.; Bao, N.; Yu, C.; Gu, H. Determination of salivary uric acid by using poly(3,4-ethylenedioxythipohene) and graphene oxide in a disposable paper-based analytical device. Anal. Chim. Acta 2020, 1103, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Azeredo, N.F.B.; Gonçalves, J.M.; Rossini, P.O.; Araki, K.; Wang, J.; Angnes, L. Uric acid electrochemical sensing in biofluids based on Ni/Zn hydroxide nanocatalyst. Microchim. Acta 2020, 187, 379. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Berriozabal, M.; Galicia, L.; Gutiérrez-Granados, S.; Cortes, J.S.; Herrasti, P. Selective electrochemical determination of uric acid in the presence of ascorbic acid using a carbon paste electrode modified with β-Cyclodextrin. Electroanalysis 2008, 20, 1678–1683. [Google Scholar] [CrossRef]
- Cardoso, R.M.; Silva, P.R.L.; Lima, A.P.; Rocha, D.P.; Oliveira, T.C.; do Prado, T.M.; Fava, E.L.; Fatibello-Filho, O.; Richter, E.M.; Muñoz, R.A.A. 3D-Printed graphene/polylactic acid electrode for bioanalysis: Biosensing of glucose and simultaneous determination of uric acid and nitrite in biological fluids. Sens. Actuators B 2020, 307, 1276212. [Google Scholar] [CrossRef]
- Liao, C.; Mak, C.; Zhang, M.; Chan, H.L.; Yan, F. Flexible organic electrochemical transistors for highly selective enzyme biosensors and used for saliva testing. Adv. Mater. 2015, 27, 676–681. [Google Scholar] [CrossRef]
- Kim, J.; Imani, S.; de Araujo, W.R.; Warchall, J.; Valdés-Ramírez, G.; Paixão, T.R.L.C.; Mercier, P.P.; Wang, J. Wearable salivary uric acid mouthguard biosensor with integrated wireless electronics. Biosens. Bioelectron. 2015, 74, 1061–1068. [Google Scholar] [CrossRef]
- Kudo, H.; Takagi, Y. Electrochemical biosensor for simplified determination of salivary uric acid. Sens. Mater. 2018, 30, 1187–1195. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, Y.; Zhang, M.; Sun, T.; Li, K.; Han, S.; Chen, H.-J. Novel portable sensing system with integrated multifunctionality for accurate detection of salivary uric acid. Biosensors 2021, 11, 242. [Google Scholar] [CrossRef]
- Yang, Y.; Song, Y.; Bo, X.; Min, J.; Pak, O.S.; Zhu, L.; Wang, M.; Tu, J.; Kogan, A.; Zhang, H.; et al. A laser-engraved wearable sensor for sensitive detection of uric acid and tyrosine in sweat. Nat. Biotechnol. 2020, 38, 217–224. [Google Scholar] [CrossRef]
- Murugan, N.; Chan-Park, M.B.; Sundramoorthy, A.K. Electrochemical detection of uric acid on exfoliated nanosheets of graphitic-like carbon nitride (g-C3N4) based sensor. J. Electrochem. Soc. 2019, 166, B3163–B3170. [Google Scholar] [CrossRef]
- Shi, Y.-M.; Mei, L.; Zhang, J.-X.; Hu, K.; Zhang, X.; Li, Z.-Z.; Miao, M.-S.; Li, X.-M. Synthesis of zinc tetraaminophthalocyanine functionalized graphene nanosheets as an enhanced material for sensitive electrochemical determination of uric acid. Electroanalysis 2020, 32, 1507–1515. [Google Scholar] [CrossRef]
- Sohouli, E.; Khosrowshahi, E.M.; Radi, P.; Naghian, E.; Rahimi-Nasrabadi, M.; Ahmadi, F. Electrochemical sensor based on modified methylcellulose by graphene oxide and Fe3O4 nanoparticles: Application in the analysis of uric acid content in urine. J. Electroanal. Chem. 2020, 877, 114503. [Google Scholar] [CrossRef]
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and clinical significance of biomarkers of oxidative stress in humans. Oxid. Med. Cell. Longev. 2017, 2017, 6501046. [Google Scholar] [CrossRef] [PubMed]
- Tóthová, L.; Kamodyová, N.; Červenka, T.; Celec, P. Salivary markers of oxidative stress in oral diseases. Front. Cell. Infect. Microbiol. 2015, 5, 73. [Google Scholar] [CrossRef] [PubMed]
- Kazakov, Y.; Tarasov, A.; Alyoshina, L.; Brainina, K. Interplay between antioxidant activity, health and disease. Biointerface Res. Appl. Chem. 2020, 10, 4893–4901. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrode | Fiber | Binder | |||
|---|---|---|---|---|---|
| Weight (C), % | Weight (O), % | Weight (N), % | Weight (C), % | Weight (O), % | |
| CVE | 95.96 | 0.34 | 3.70 | 96.02 | 3.98 |
| CVEact (1.0 V) | 96.22 | 0.30 | 3.48 | 96.88 | 3.12 |
| CVEact (1.6 V) | 94.56 | 3.51 | 1.93 | 96.45 | 3.86 |
| CVEact (2.0 V) | 74.23 | 18.88 | 6.89 | 96.05 | 3.95 |
| Electrode | Rs *, Ohm | Rct **, kOhm | W, μMhos1/2 |
|---|---|---|---|
| CVE | 82.6 ± 1.1 | 8.21 ± 0.17 | 387 ± 17 |
| CVEact (1.0 V) | 67.8 ± 1.0 | 6.45 ± 0.14 | 524 ± 22 |
| CVEact (1.6 V) | 58.7 ± 1.4 | 3.36 ± 0.11 | 844 ± 31 |
| CVEact (2.0 V) | 53.1 ± 0.6 | 2.96 ± 0.09 | 690 ± 23 |
| Electrode 1 | Modifier 2 | Electrolyte 3 | LR 4, μM | LOD 5, μM | Method 6 | Sample | Reference |
|---|---|---|---|---|---|---|---|
| CVEact | – | PB pH 6.0 | 0.09–700 | 0.05 | LSV | Saliva | This work |
| CSPE | MWCNTs/UO | ASal. pH 6.8 | 5–1000 | 0.33 | CA | Saliva | [19] |
| BDDE | – | BRB pH 2.25 | 8–1000 | 7.7 | DPV | Urine | [20] |
| GCE | CuO-NFs | PBS pH 7.4 | 1–351 | 0.6 | CV | Urine | [21] |
| GCE | ZIF-11 | BRB pH 7.0 | 50–540 | 0.48 | DP-ASV | Urine | [22] |
| ITO | PEDOT–GO | ASal. pH 6.8 | 2–1000 | 0.75 | DPV | Saliva | [45] |
| CSPE | Ni0.75Zn0.25(OH)2-NPs | ASw. pH 5.0, ASal. pH 6.7 | 20–170, 200–2000 | 0.023, 0.023 | CA, CV | Sweat, saliva | [46] |
| CPE | β-CD | AB pH 5.0 | 10–170 | 4.5 | A | Urine, saliva | [47] |
| G-PLA-3DPE | – | BRB pH 2.0 | 0.5–250 | 0.02 | BIA-MPA | Saliva | [48] |
| Pt | GF-Nf/PANI/GO-UO | PBS pH 7.4 | 3–300 | 3 | A | Saliva | [49] |
| PB-CSPE | UO/PPD | ASal. pH 6.7 | 0–1000 | n/a | CA | Saliva | [50] |
| CSPE | Os-HRP/UO | PBS pH 7.4 | 10–400 | n/a | CA | Saliva | [51] |
| CSPE | Cr-Au/UO | PBS pH 7.3 | 0–500 | n/a | CA | Saliva | [52] |
| GSPE | – | AB pH 4.6 | 3–40 | 0.74 | DPV | Sweat | [53] |
| GCE | g-C3N4-NSs | PBS pH 7.4 | 100–1000 | 4.45 | DPV | Urine | [54] |
| GCE | ZnPc-rGO-UO-Nf | PB* pH 3.1 | 0.5–100 | 0.15 | DPV | Urine | [55] |
| GCE | MC-GO-Fe3O4-NPs | PB pH 7.0 | 0.5–140 | 0.17 | DPV | Urine | [56] |
| Sample | Found in Saliva, μM | RSD, % | Added, μM | Found in Saliva with Additive, μM | Found Additive, μM | R, % |
|---|---|---|---|---|---|---|
| Artificial saliva | - | - | 200 | - | 203 ± 7 | 102 |
| Sample 1 | 51 ± 5 | 3.9 | 100 | 147 ± 17 | 96 ± 13 | 96 |
| Sample 2 | 115 ± 5 | 1.7 | 100 | 216 ± 9 | 101 ± 5 | 101 |
| Sample 3 | 126 ± 12 | 3.7 | 100 | 223 ± 33 | 98 ± 9 | 98 |
| Sample 4 | 151 ± 9 | 2.4 | 100 | 254 ± 8 | 103 ± 5 | 103 |
| Sample 5 | 230 ± 20 | 3.4 | 100 | 336 ± 20 | 105 ± 6 | 105 |
| Sample 6 | 248 ± 14 | 2.3 | 300 | 553 ± 38 | 305 ± 10 | 102 |
| Sample 7 | 461 ± 11 | 1.0 | 500 | 956 ± 24 | 495 ± 17 | 99 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bukharinova, M.A.; Stozhko, N.Y.; Novakovskaya, E.A.; Khamzina, E.I.; Tarasov, A.V.; Sokolkov, S.V. Developing Activated Carbon Veil Electrode for Sensing Salivary Uric Acid. Biosensors 2021, 11, 287. https://doi.org/10.3390/bios11080287
Bukharinova MA, Stozhko NY, Novakovskaya EA, Khamzina EI, Tarasov AV, Sokolkov SV. Developing Activated Carbon Veil Electrode for Sensing Salivary Uric Acid. Biosensors. 2021; 11(8):287. https://doi.org/10.3390/bios11080287
Chicago/Turabian StyleBukharinova, Maria A., Natalia Yu. Stozhko, Elizaveta A. Novakovskaya, Ekaterina I. Khamzina, Aleksey V. Tarasov, and Sergey V. Sokolkov. 2021. "Developing Activated Carbon Veil Electrode for Sensing Salivary Uric Acid" Biosensors 11, no. 8: 287. https://doi.org/10.3390/bios11080287
APA StyleBukharinova, M. A., Stozhko, N. Y., Novakovskaya, E. A., Khamzina, E. I., Tarasov, A. V., & Sokolkov, S. V. (2021). Developing Activated Carbon Veil Electrode for Sensing Salivary Uric Acid. Biosensors, 11(8), 287. https://doi.org/10.3390/bios11080287