Substrate Materials for Biomolecular Immobilization within Electrochemical Biosensors

Abstract
:1. Introduction
2. Au-Thiol Self-Assembled Monolayer (SAM) Formation
2.1. Overview
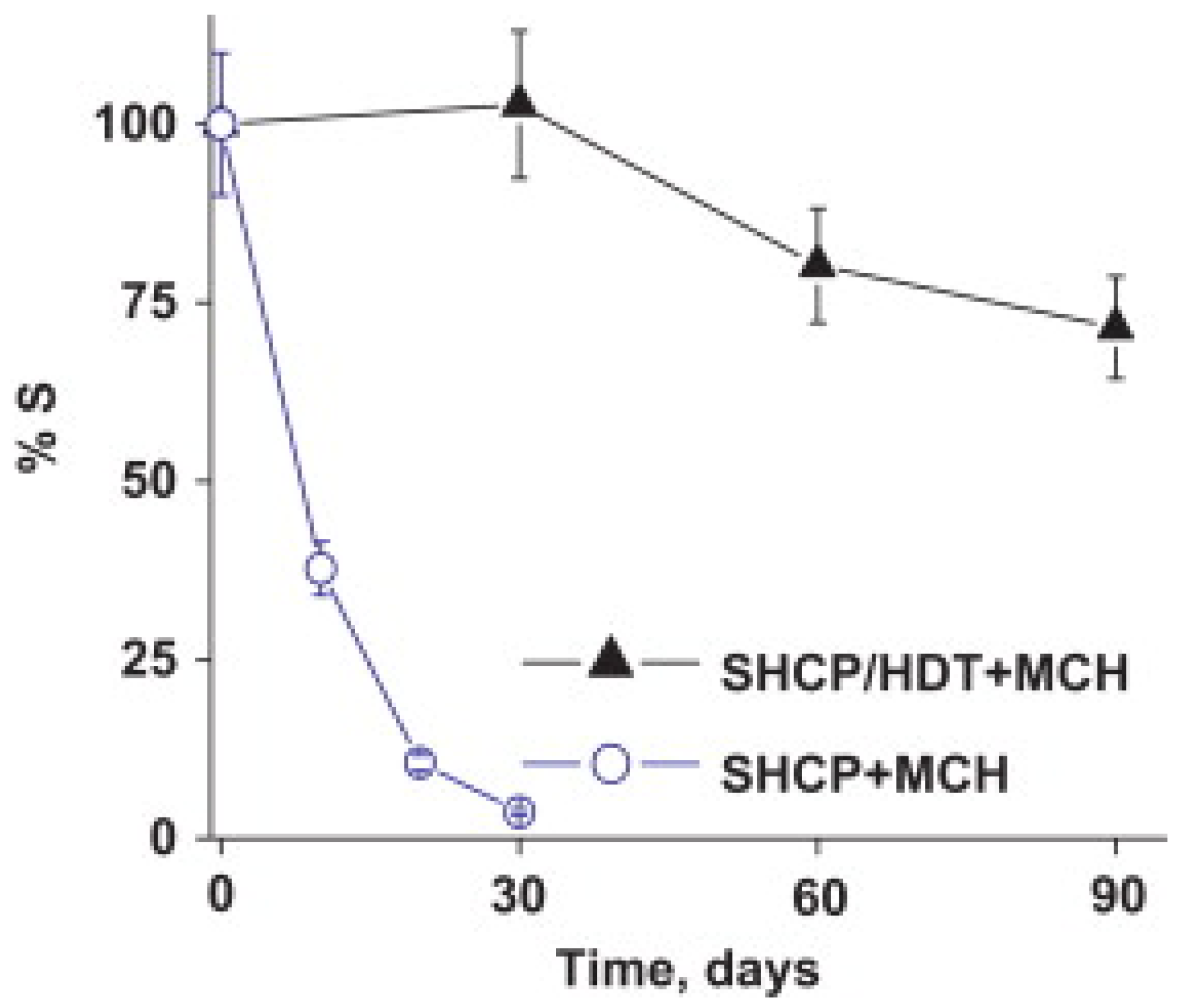
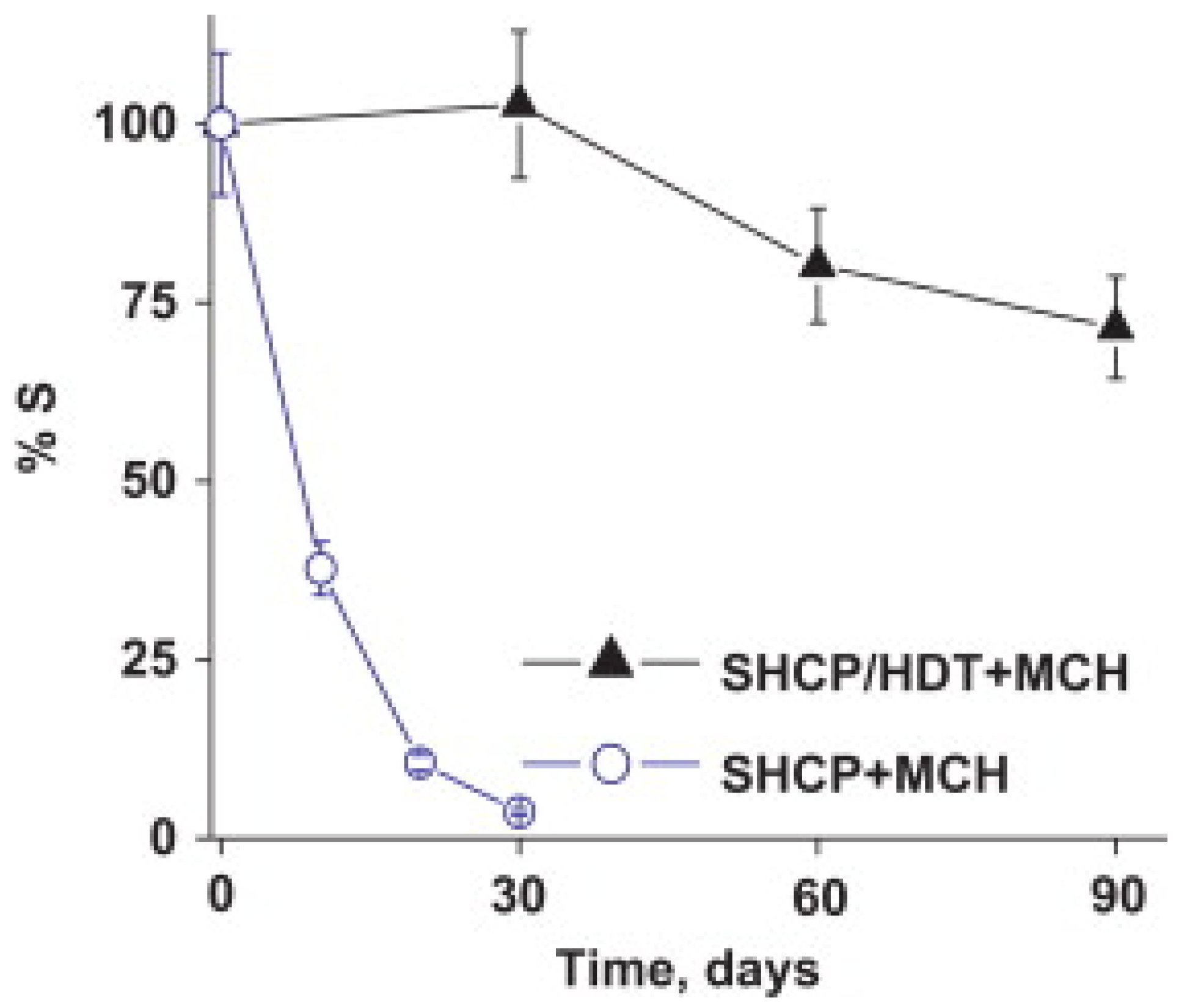
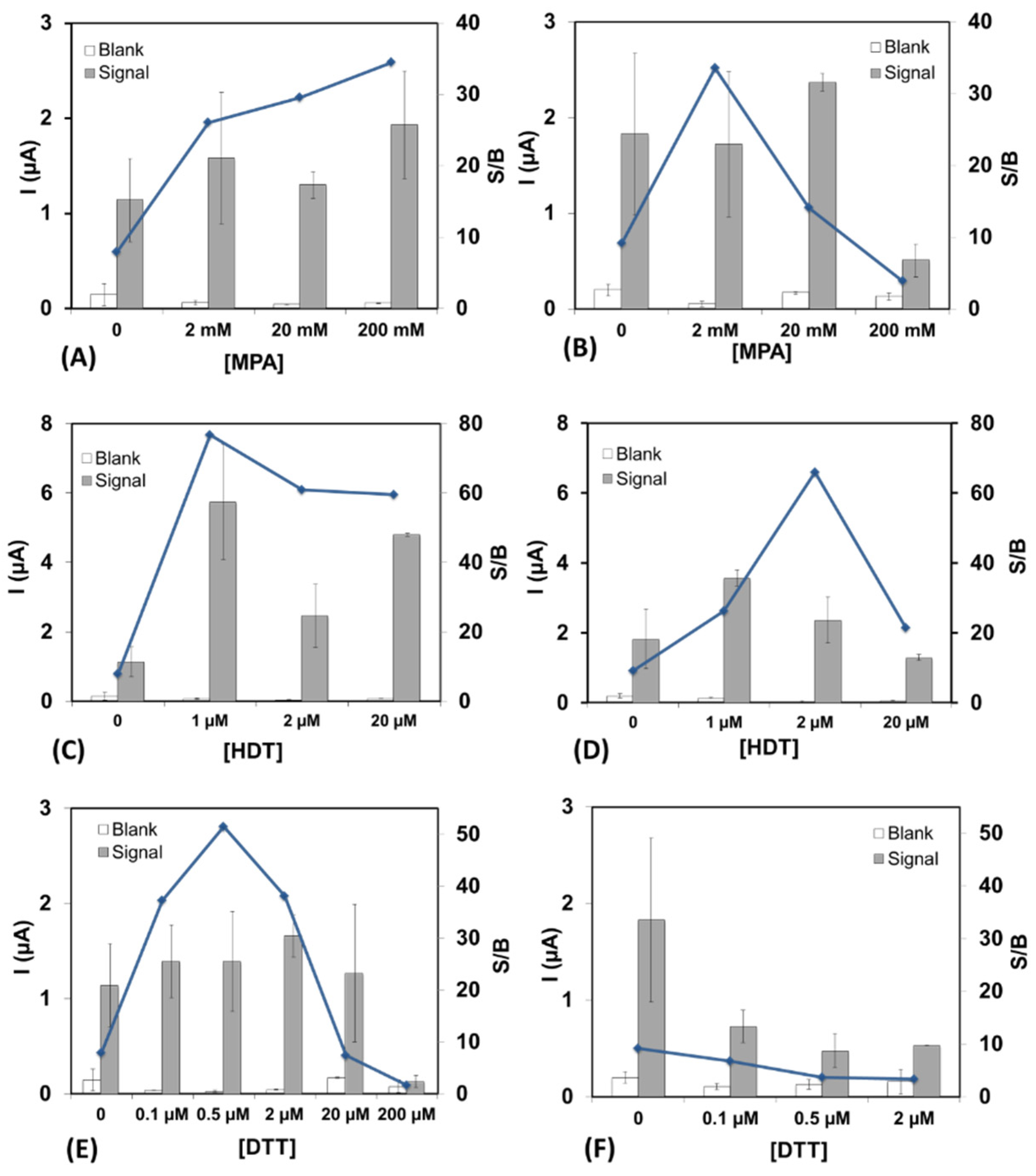
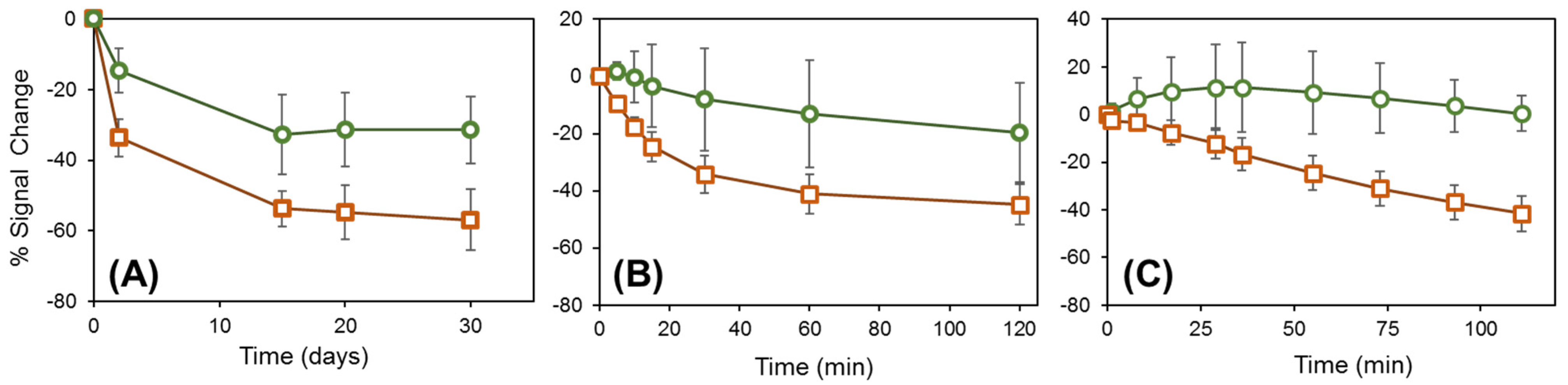
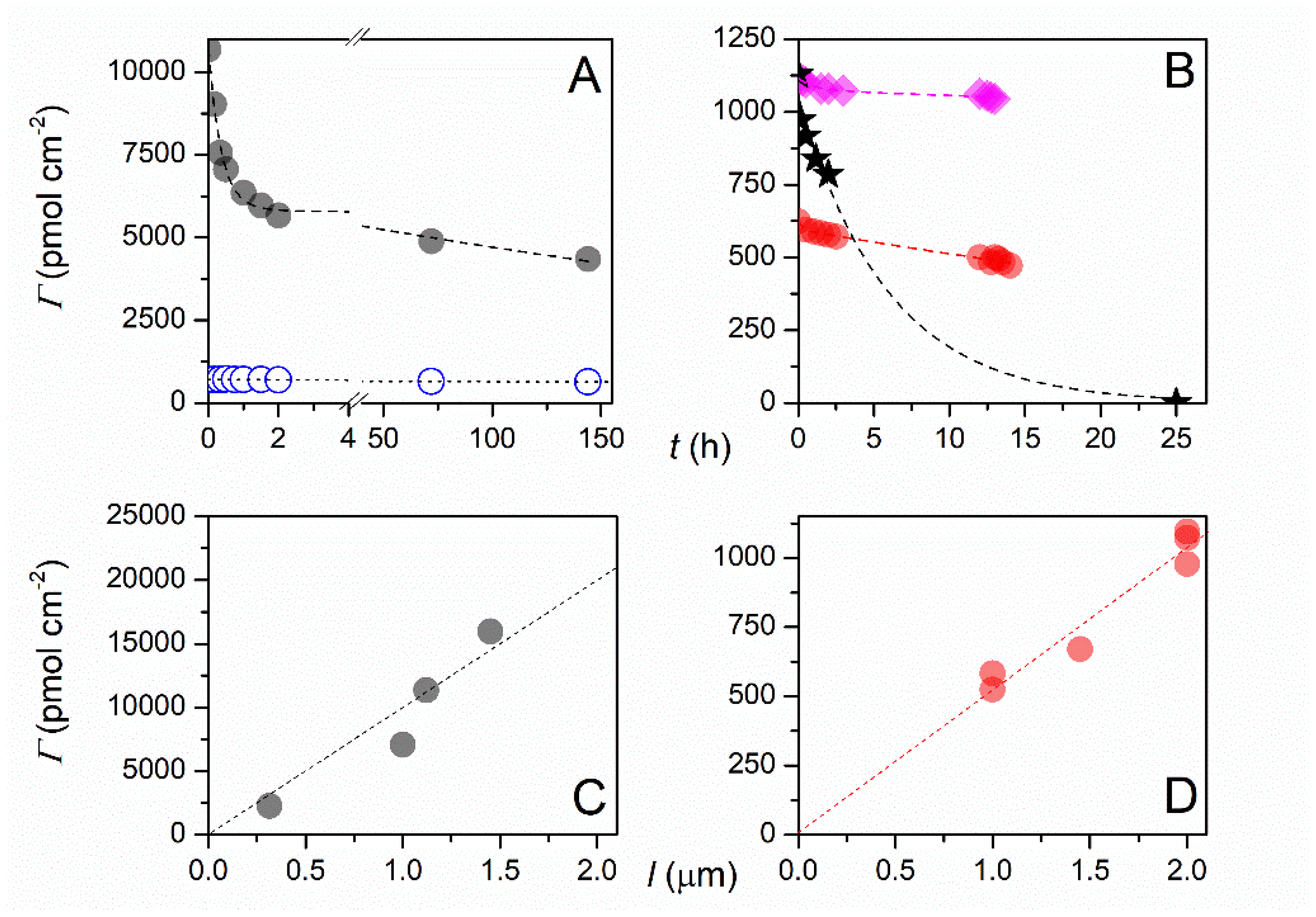
2.2. Multidentate Thiols
2.3. Other Metals
3. Other Noble Substrate Materials
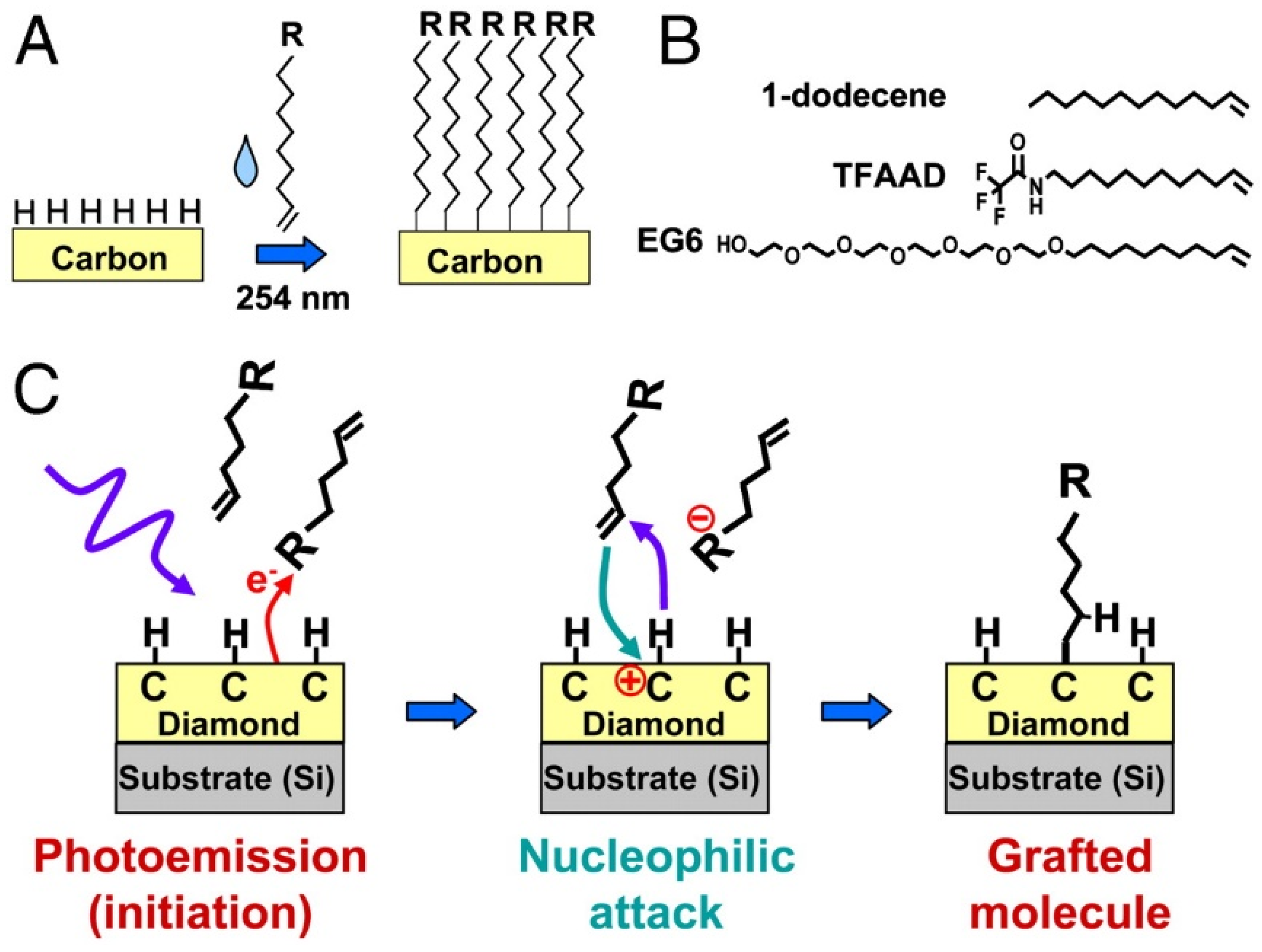
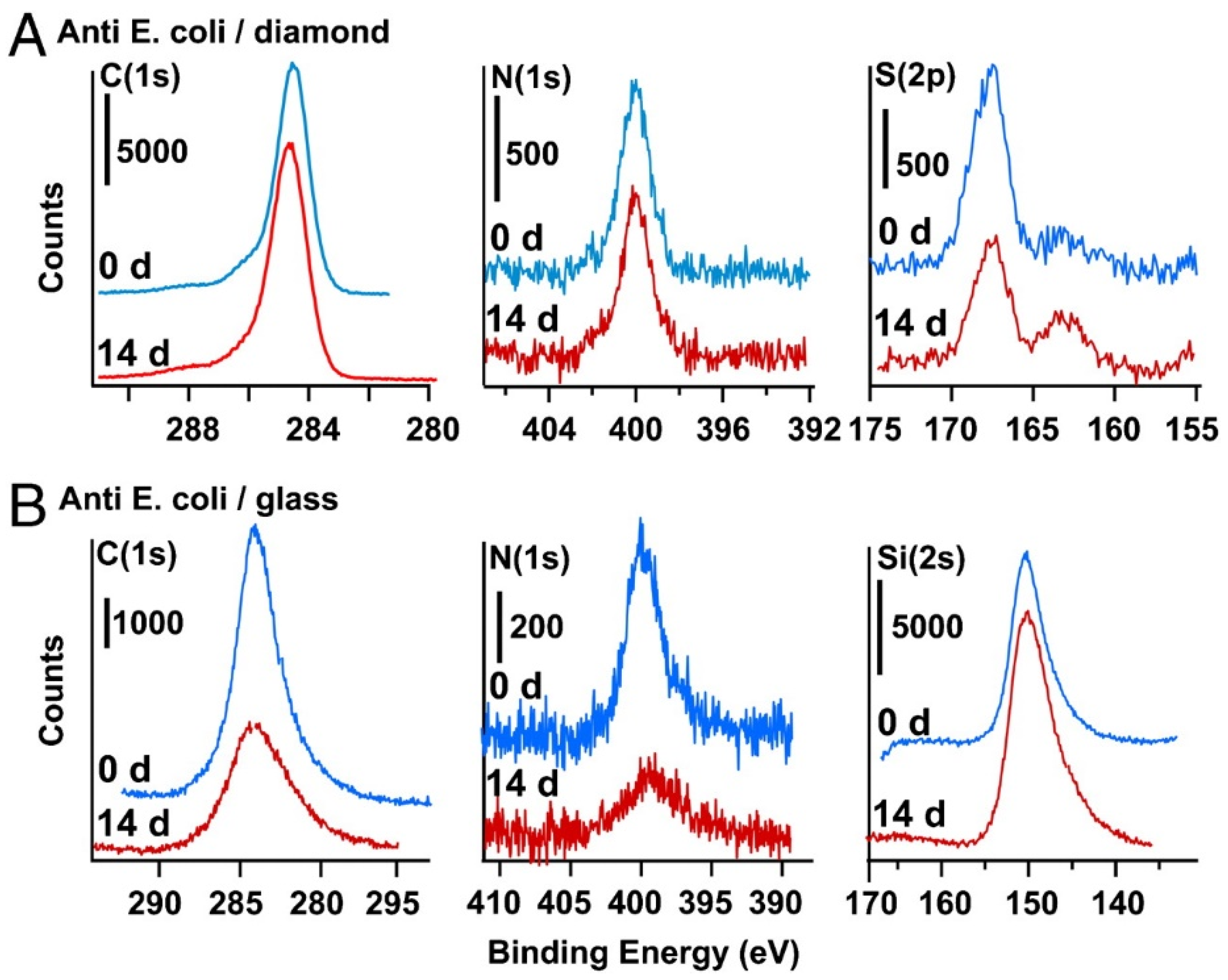
3.1. Carbon
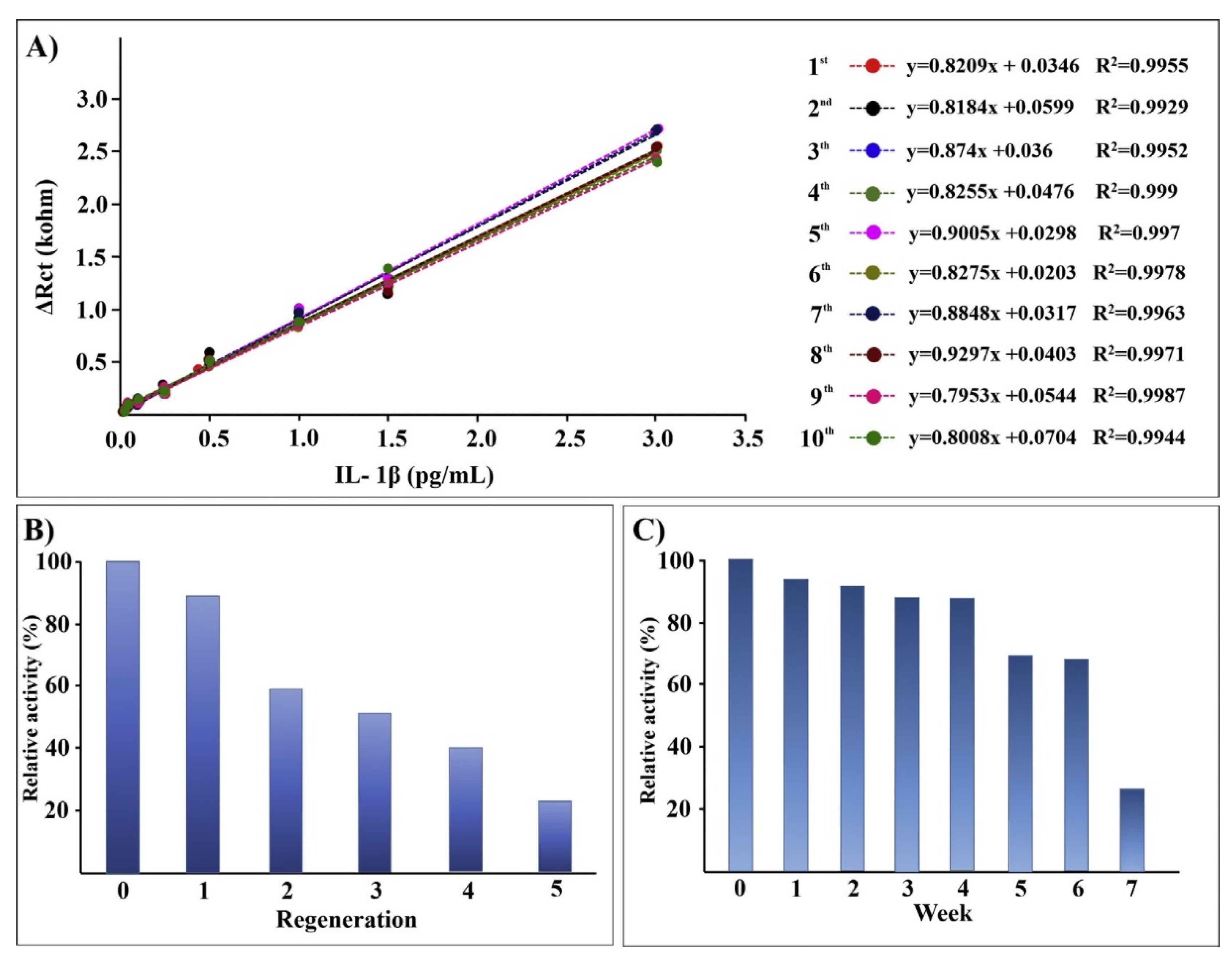
3.2. Silicon
3.3. Platinum
4. Transition Metal Dichalcogenides
5. Electrically Conductive Metal Oxides
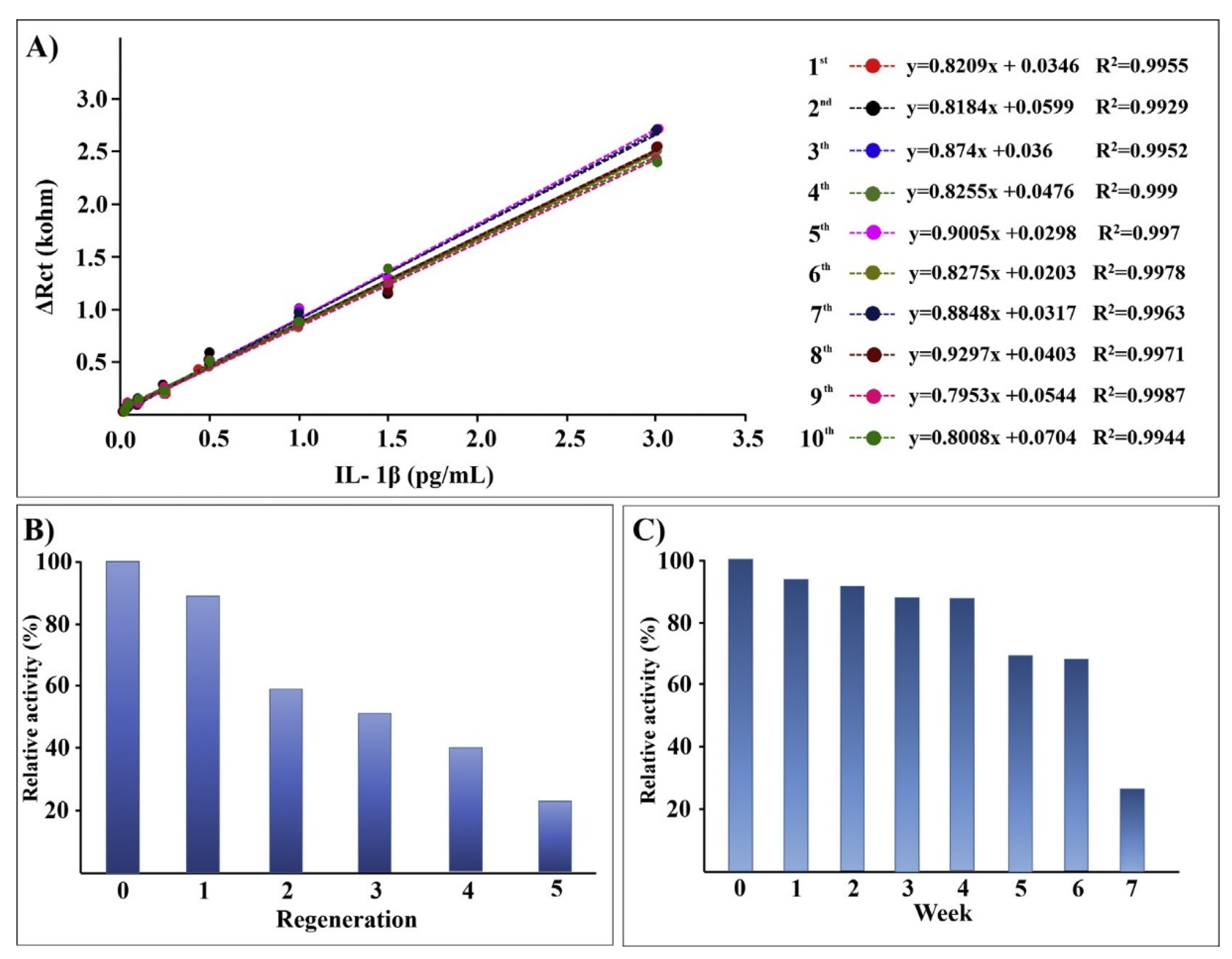
5.1. Infdium Tin Oxide (ITO)
5.2. Titanium Dioxide (TiO2)
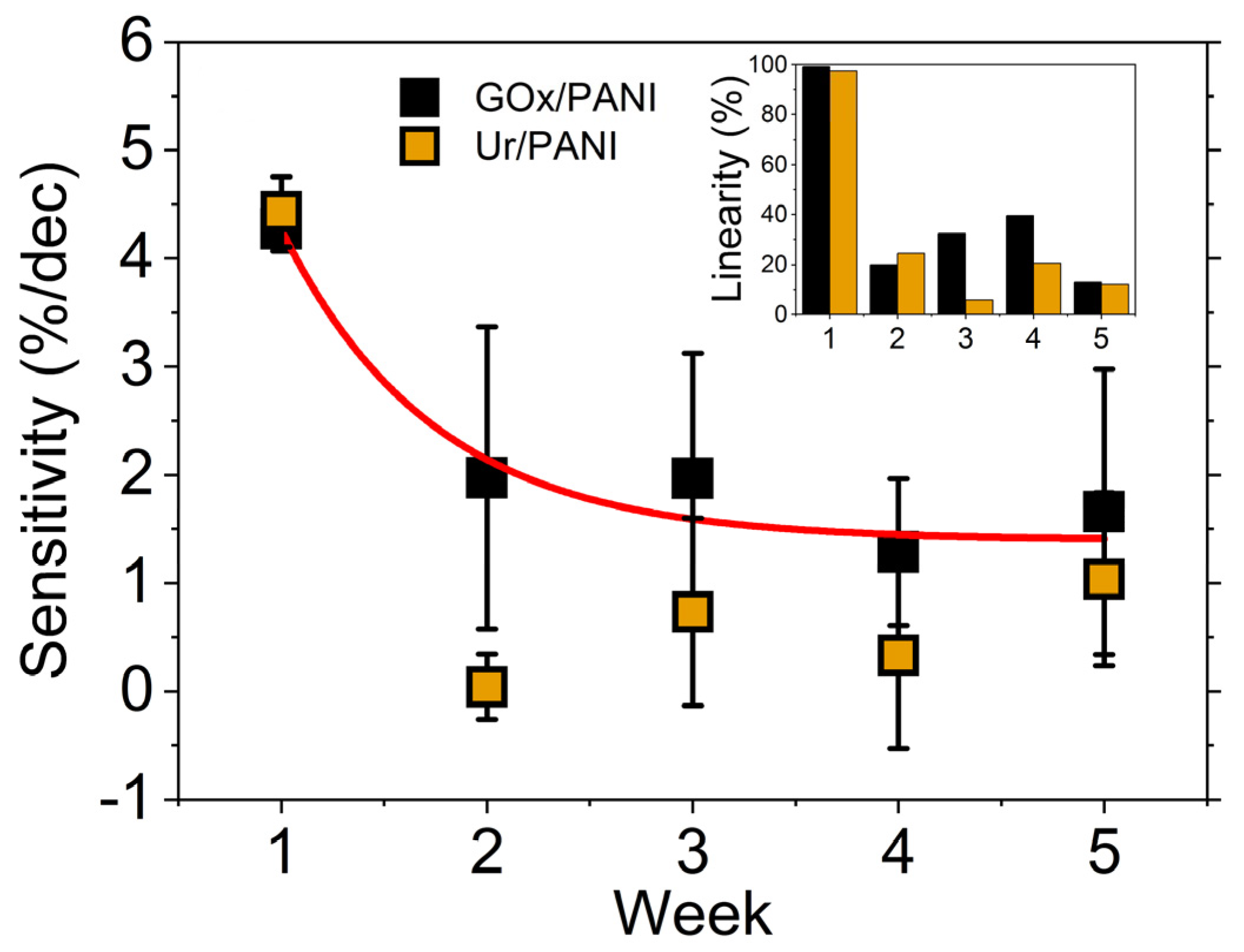
6. Electrically Conductive Polymers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | ρ (Ω-cm) | Functionalization Chemistry | Advantages | Disadvantages | Key References 2 |
|---|---|---|---|---|---|
| Au | 2.44 × 10−6 | Au-S bond | Simplicity, Cost, ULSI 3 compatible | Poor storage stability | [54,55,56,57,58,59,65,66,67,70,71,72,73,74] |
| C | 1014 (diamond) 10−2–102 (BDD) 1 10−1–10−4 (graphite) | Alkene insertion | Biocompatibility | Lack of standard substrates, Slow immobilization chemistry | [77,78,79,80] |
| Si | 2.3 × 105 (undoped) 5 × 10−3 (degenerate) | Alkene insertion | ULSI compatible | Gradual SiO2 formation | [86,87,88,89,90,91,92,93,94] |
| Pt | 1.06 × 10−5 | Pt–S bond | ULSI compatible | Not well studied | |
| ITO | 10−4 | Phospnonate | Standard substrates and immobilization chemistries | Variation in substrate properties | [120,121,122,123,124,125] |
| TiO2 | >108 | Phosphonate | Current use in biomedical implants | Very low conductivity | |
| MoS2 WS2 | - 4 - | Thiol adsorption | Rapid technological advances | Lack of functionalization chemistry | |
| Polyaniline Polypyrrole Polythiophene | 2 × 106 (HBr doped) 0.01–0.5 (doped) 0.01 (doped) | Various | Probe incorporation during film growth possible | Lack of standard electropolymerization methods | [138,139,140,141,142] |
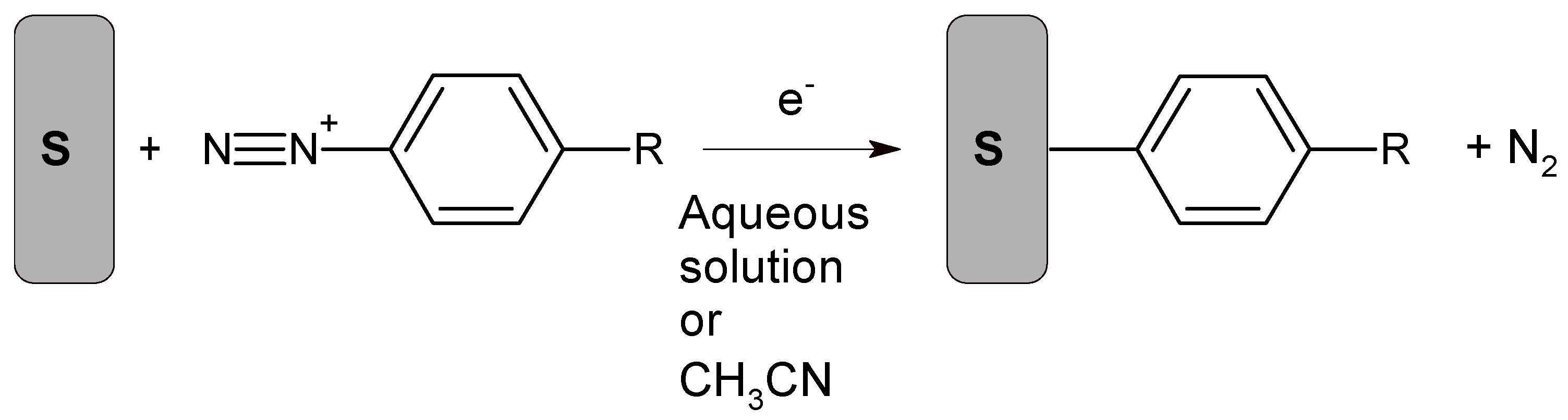
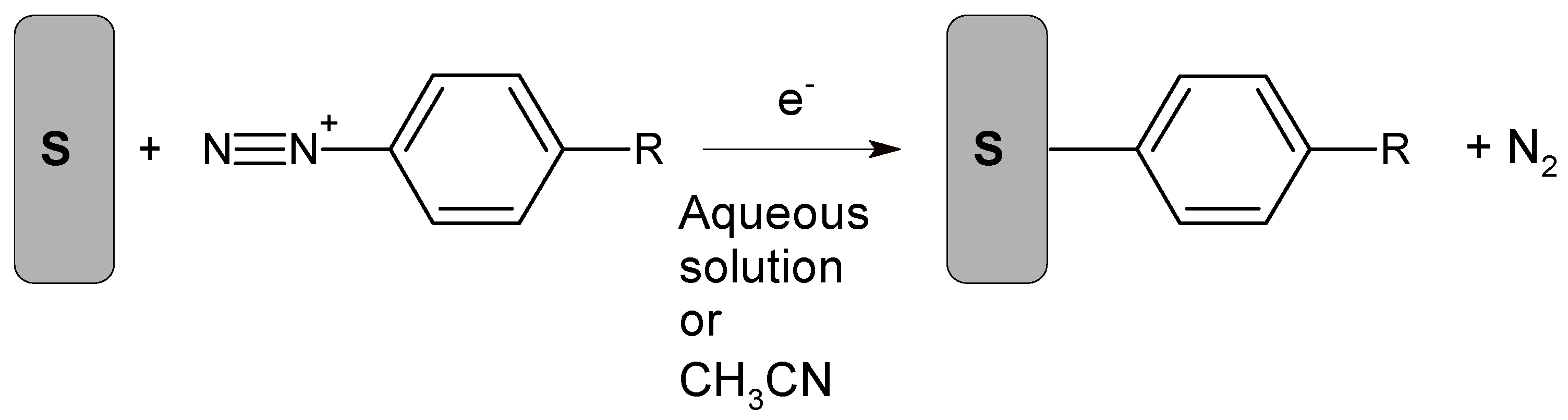
7. Aryldiazonium-Based Electrochemical Reduction
8. Conclusions and Future Directions
Funding
Conflicts of Interest
References
- Nagel, B.; Dellweg, H.; Gierasch, L.M. Glossary for chemists of terms used in biotechnology. Pure Appl. Chem. 1992, 64, 143–168. [Google Scholar] [CrossRef] [Green Version]
- Thevenot, D.R.; Toth, K.; Durst, R.A.; Wilson, G.S. Electrochemical biosensors: Recommended definitions and classification. Pure Appl. Chem. 1999, 71, 2333–2348. [Google Scholar] [CrossRef] [Green Version]
- Grieshaber, D.; MacKenzie, R.; Voros, J.; Reimhult, E. Electrochemical biosensors—Sensor principles and architectures. Sensors 2008, 8, 1400–1458. [Google Scholar] [CrossRef]
- Menon, S.; Mathew, M.R.; Sam, S.; Keerthi, K.; Kumar, K.G. Recent advances and challenges in electrochemical biosensors for emerging and re-emerging infectious diseases. J. Electroanal. Chem. 2020, 878, 114596. [Google Scholar] [CrossRef]
- Zhao, W.H.; Tian, S.L.; Huang, L.; Liu, K.; Dong, L.J.; Guo, J.H. A smartphone-based biomedical sensory system. Analyst 2020, 145, 2873–2891. [Google Scholar] [CrossRef]
- Teymourian, H.; Barfidokht, A.; Wang, J. Electrochemical glucose sensors in diabetes management: An updated review (2010–2020). Chem. Soc. Rev. 2020, 49, 7671–7709. [Google Scholar] [CrossRef]
- Heller, A.; Feldman, B. Electrochemical glucose sensors and their applications in diabetes management. Chem. Rev. 2008, 108, 2482–2505. [Google Scholar] [CrossRef] [Green Version]
- Rattu, G.; Khansili, N.; Maurya, V.K.; Krishna, V.M. Lactate detection sensors for food, clinical and biological applications: A review. Environ. Chem. Lett. 2021, 19, 1135–1152. [Google Scholar] [CrossRef]
- Bhalla, N.; Jolly, P.; Formisano, N.; Estrela, P. Introduction to biosensors. Essays Biochem. 2016, 60, 1–8. [Google Scholar]
- Bollella, P.; Katz, E. Biosensors- Recent advances and future challenges. Sensors 2020, 20, 6645. [Google Scholar] [CrossRef]
- McLamore, E.S.; Alocilja, E.; Gomes, C.; Gunasekaran, S.; Jenkins, D.; Datta, S.P.A.; Li, Y.B.; Mao, Y.; Nugen, S.R.; Reyes-De-Corcuera, J.I.; et al. FEAST of biosensors: Food, environmental and agricultural sensing technologies (FEAST) in North America. Biosens. Bioelectron. 2021, 178, 113011. [Google Scholar] [CrossRef]
- Ali, Q.; Ahmar, S.; Sohail, M.A.; Kamran, M.; Ali, M.; Saleem, M.H.; Rizwan, M.; Ahmed, A.M.; Mora-Poblete, F.; do Amaral Júnior, A.T.; et al. Research advances and applications of biosensing technology for the diagnosis of pathogens in sustainable agriculture. Environ. Sci. Pollut. Res. 2021, 28, 9002–9019. [Google Scholar] [CrossRef]
- Shen, Y.F.; Xu, L.Z.; Li, Y.B. Biosensors for rapid detection of Salmonella in food: A review. Compr. Rev. Food Sci. Food Saf. 2020, 20, 149–197. [Google Scholar] [CrossRef]
- Sun, Y.M.; Zhao, J.L.; Liang, L.J. Recent development of antibiotic detection in food and environment: The combination of sensors and nanomaterials. Microchim. Acta 2021, 188, 21. [Google Scholar] [CrossRef]
- Kim, H.J.; Jang, G.P.; Yoon, Y.D. Specific heavy metal/metalloid sensors: Current state and perspectives. Appl. Microbiol. Biotechnol. 2020, 104, 907–914. [Google Scholar] [CrossRef]
- Kalyani, N.; Goel, S.; Jaiswal, S. On-site sensing of pesticides using point-of-care biosensors: A review. Environ. Chem. Lett. 2021, 19, 345–354. [Google Scholar] [CrossRef]
- Ye, S.; Feng, S.L.; Huang, L.; Bian, S.T. Recent progress in wearable biosensors: From healthcare monitoring to sports analytics. Biosensors 2020, 10, 205. [Google Scholar] [CrossRef]
- Saylan, Y.; Akgönüllü, S.; Denizli, A. Plasmonic sensors for monitoring biological and chemical threat agents. Biosensors 2020, 10, 142. [Google Scholar] [CrossRef]
- Pöhlmann, C.; Elßner, T. Multiplex immunoassay techniques for on-site detection of security sensitive toxins. Toxins 2020, 12, 727. [Google Scholar] [CrossRef]
- Parihar, A.; Ranjan, P.; Sanghi, S.K.; Srivastava, A.K.; Khan, R. Point-of-care biosensor-based diagnosis of COVID-19 holds promise to combat current and future pandemics. ACS Appl. Bio Mater. 2020, 3, 7326–7343. [Google Scholar] [CrossRef]
- Mohankumar, P.; Ajayan, J.; Mohanraj, T.; Yasodharan, R. Recent developments in biosensors for healthcare and biomedical applications: A review. Measurement 2021, 167, 108293. [Google Scholar] [CrossRef]
- Dejous, C.; Krishnan, U.M. Sensors for diagnosis of prostate cancer: Looking beyond the prostate specific antigen. Biosens. Bioelectron. 2021, 173, 112790. [Google Scholar] [CrossRef]
- Sfragano, P.S.; Pillozzi, S.; Palchetti, I. Electrochemical and PEC platforms for miRNA and other epigenetic markers of cancer diseases: Recent updates. Electrochem. Commun. 2021, 124, 106929. [Google Scholar] [CrossRef]
- Love, J.C.; Estroff, L.A.; Kriebel, J.K.; Nuzzo, R.G.; Whitesides, G.M. Self-assembled monolayers of thiolates on metals as a form of nanotechnology. Chem. Rev. 2005, 105, 1103–1169. [Google Scholar] [CrossRef]
- Ducker, R.E.; Montague, M.Y.; Leggett, G.J. A comparative investigation of methods for protein immobilization on self-assembled monolayers using glutaraldehyde, carbodiimide, and anhydride reagents. Biointerphases 2008, 3, 59–65. [Google Scholar] [CrossRef]
- Hernandez, K.; Fernandez-Lafuente, R. Control of protein immobilization: Coupling immobilization and site-directed mutagenesis to improve biocatalyst or biosensor performance. Enzym. Microb. Technol. 2011, 48, 107–122. [Google Scholar] [CrossRef]
- Akram, M.S.; Rehman, J.U.; Hall, E.A.H. Engineered proteins for bioelectrochemistry. Annu. Rev. Anal. Chem. 2014, 7, 257–274. [Google Scholar] [CrossRef]
- Sharma, H.; Mutharasan, R. Half antibody fragments improve biosensor sensitivity without loss of selectivity. Anal. Chem. 2013, 85, 2472–2477. [Google Scholar] [CrossRef]
- Lai, R.Y. Folding- and dynamics-based electrochemical DNA sensors. Meth. Enzymol. 2017, 589, 221–252. [Google Scholar]
- Lockett, M.R.; Smith, L.M. Carbon substrates: A stable foundation for biomolecular arrays. Annu. Rev. Anal. Chem. 2015, 8, 263–285. [Google Scholar] [CrossRef]
- Raymakers, J.; Haenen, K.; Maes, W. Diamond surface functionalization: From gemstone to photoelectrochemical applications. J. Mater. Chem. C 2019, 7, 10134–10165. [Google Scholar] [CrossRef]
- Chazalviel, J.N.; Allongue, P.; Gouget-Laemmel, A.C.; de Villeneuve, C.H.; Moraillon, A.; Ozanam, F. Covalent functionalizations of silicon surfaces and their application to biosensors. Sci. Adv. Mater. 2011, 3, 332–353. [Google Scholar] [CrossRef]
- Gonçales, V.R.; Lian, J.X.; Gautam, S.; Tilley, R.D.; Gooding, J.J. Functionalized silicon electrodes in electrochemistry. Annu. Rev. Anal. Chem. 2020, 13, 135–158. [Google Scholar] [CrossRef] [Green Version]
- Aissaoui, N.; Bergaoui, L.; Landoulsi, J.; Lambert, J.F.; Boujday, S. Silane layers on silicon surfaces: Mechanism of interaction, stability, and influence on protein adsorption. Langmuir 2012, 28, 656–665. [Google Scholar] [CrossRef]
- Haensch, C.; Hoeppener, S.; Schubert, U.S. Chemical modification of self-assembled silane based monolayers by surface reactions. Chem. Soc. Rev. 2010, 39, 2323–2334. [Google Scholar] [CrossRef]
- Queffélec, C.; Petit, M.; Janvier, P.; Knight, D.A.; Bujoli, B. Surface modification using phosphonic acids and esters. Chem. Rev. 2012, 112, 3777–3807. [Google Scholar] [CrossRef]
- Aydın, E.B.; Sezgintürk, M.K. Indium tin oxide (ITO): A promising material in biosensing technology. Trends Anal. Chem. 2017, 97, 309–315. [Google Scholar] [CrossRef]
- Pan, H.M.; Gonuguntla, S.; Li, S.; Trau, D. Conjugated Polymers for Biosensor Devices. Compr. Biomater. 2017, 3, 716–754. [Google Scholar]
- Pagán, M.; Suazo, D.; del Toro, N.; Griebenow, K. A comparative study of different protein immobilization methods for the construction of an efficient nano-structured lactate oxidase-SWCNT-biosensor. Biosens. Bioelectron. 2015, 64, 138–146. [Google Scholar] [CrossRef] [Green Version]
- Kaura, K.; Sahab, S.; Tomarc, M.; Gupta, V. Influence of immobilization strategies on biosensing response characteristics: A comparative study. Enzyme Microb. Technol. 2016, 82, 144–150. [Google Scholar] [CrossRef]
- Karimi-Maleh, H.; Orooji, Y.; Karimi, F.; Alizadeh, M.; Baghayeri, M.; Rouhi, J.; Tajik, S.; Beitollahi, H.; Agarwal, S.; Gupta, V.K.; et al. A critical review on the use of potentiometric based biosensors for biomarkers detection. Biosens. Bioelectron. 2021, 184, 113252. [Google Scholar] [CrossRef]
- Kucherenko, I.S.; Soldatkin, O.O.; Dzyadevych, S.V.; Soldatkin, A.P. Electrochemical biosensors based on multienzyme systems: Main groups, advantagesand limitations—A review. Anal. Chim. Acta 2020, 1111, 114–131. [Google Scholar] [CrossRef]
- Monteiro, T.; Almeida, M.G. Electrochemical enzyme biosensors revisited: Old solutions for new problems. Crit. Rev. Anal. Chem. 2019, 49, 44–66. [Google Scholar] [CrossRef]
- Leva-Bueno, J.; Peyman, S.A.; Millner, P.A. A review on impedimetric immunosensors for pathogen and biomarker detection. Med. Microbiol. Immunol. 2020, 209, 343–362. [Google Scholar] [CrossRef] [Green Version]
- Strong, M.E.; Richards, J.R.; Torres, M.; Beck, C.M.; La Belle, J.T. Faradaic electrochemical impedance spectroscopy for enhanced analyte detection in diagnostics. Biosens. Bioelectron. 2021, 177, 112949. [Google Scholar] [CrossRef]
- Jin, H.; Gui, R.J.; Yu, J.B.; Lv, W.; Wang, Z.H. Fabrication strategies, sensing modes and analytical applications of ratiometric electrochemical biosensors. Biosens. Bioelectron. 2017, 91, 523–537. [Google Scholar] [CrossRef]
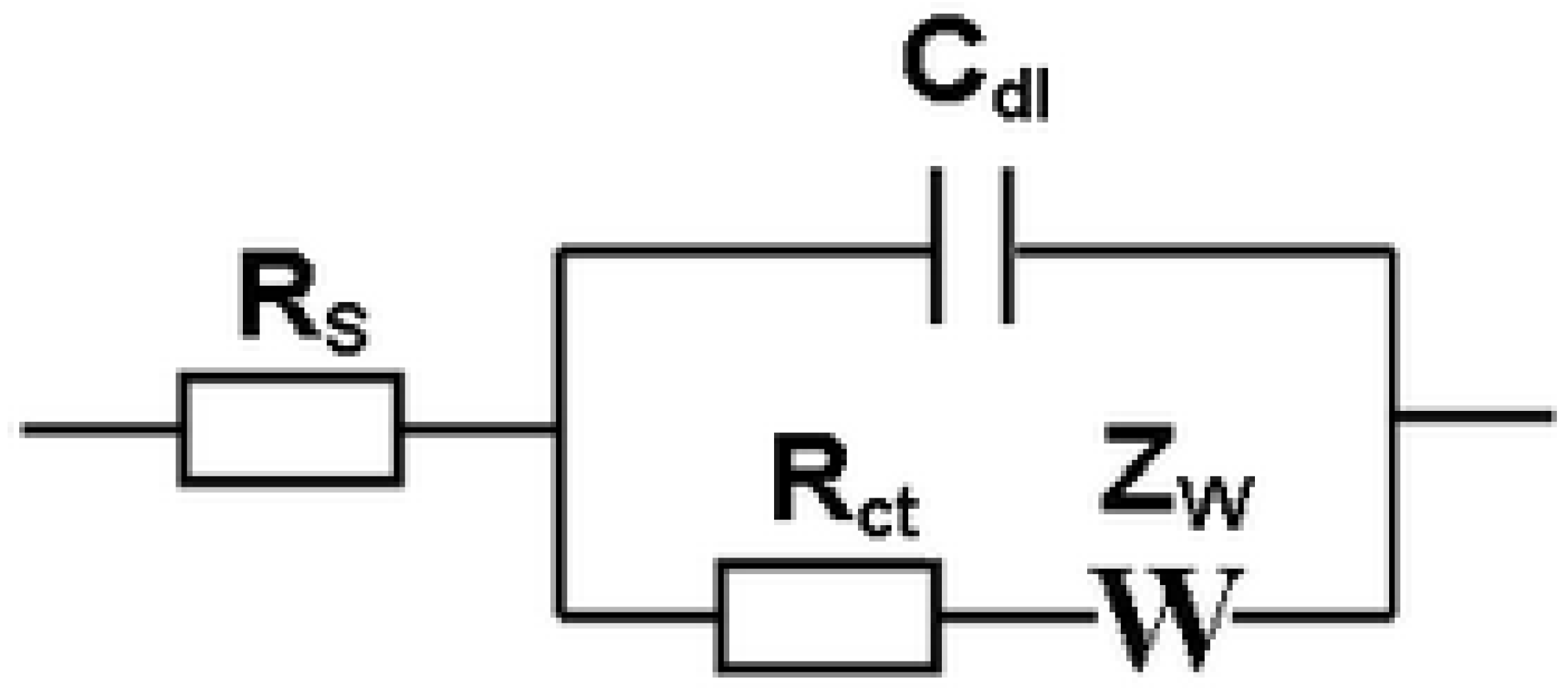
- Randles, J.E.B. Kinetics of rapid electrode reactions. Discuss. Faraday Soc. 1947, 1, 11–19. [Google Scholar] [CrossRef]
- Vericat, C.; Vela, M.E.; Benitez, G.; Carrob, P.; Salvarezza, R.C. Self-assembled monolayers of thiols and dithiols on gold: New challenges for a well-known system. Chem. Soc. Rev. 2010, 39, 1805–1834. [Google Scholar] [CrossRef]
- Patel, N.; Davies, M.C.; Hartshorne, M.; Heaton, R.J.; Roberts, C.J.; Tendler, S.J.B.; Williams, P.M. Immobilization of protein molecules onto homogeneous and mixed carboxylate-terminated self-assembled monolayers. Langmuir 1997, 13, 6485–6490. [Google Scholar] [CrossRef]
- Ma, S.; Laurent, C.V.F.P.; Meneghello, M.; Tuoriniemi, J.; Oostenbrink, C.; Gorton, L.; Bartlett, P.N.; Ludwig, R. Direct electron-transfer anisotropy of a site-specifically immobilized cellobiose dehydrogenase. ACS Catal. 2019, 9, 7607–7615. [Google Scholar] [CrossRef]
- Meneghello, M.; Al-Lolage, F.A.; Ma, S.; Ludwig, R.; Bartlett, P.N. Studying direct electron transfer by site-directed immobilization of cellobiose dehydrogenase. ChemElectroChem 2019, 6, 700–713. [Google Scholar] [CrossRef]
- Pali, M.; Suni, I.I. Impedance detection of 3-phenoxybenzoic acid comparing wholes antibodies and antibody fragments for biomolecular recognition. Electroanalysis 2018, 30, 2899–2907. [Google Scholar] [CrossRef]
- Bradbury, C.R.; Zhao, J.J.; Fermin, D.J. Distance-independent charge-transfer resistance at gold electrodes modified by thiol monolayers and metal nanoparticles. J. Phys. Chem. C 2008, 112, 10153–10160. [Google Scholar] [CrossRef]
- Kuralay, F.; Campuzano, S.; Wang, J. Greatly extended storage stability of electrochemical DNA biosensors using ternary thiolated self-assembled monolayers. Talanta 2012, 99, 155–160. [Google Scholar] [CrossRef]
- Miodek, A.; Regan, E.M.; Bhalla, N.; Hopkins, N.A.E.; Goodchild, S.A.; Estrela, P. Optimisation and characterisation of anti-fouling ternary SAM layers for impedance-based aptasensors. Sensors 2015, 15, 25015–25032. [Google Scholar] [CrossRef] [Green Version]
- Miranda-Castro, R.; Sánchez-Salcedo, R.; Suárez-Álvarez, B.; de-los-Santos-Álvarez, N.; Miranda-Ordieres, A.J.; Lobo-Castañón, M.J. Thioaromatic DNA monolayers for target-amplification-free electrochemical sensing of environmental pathogenic bacteria. Biosens. Bioelectron. 2017, 92, 162–170. [Google Scholar] [CrossRef]
- Aufartova, J.; Lopez, M.S.P.; Martin-Fernandez, B.; Lopez-Ruiz, B. Key factors of ternary monolayers to improve DNA sensors performance. Electroanalysis 2017, 29, 131–139. [Google Scholar] [CrossRef]
- Liu, Z.M.; Wang, H.Y. An antifouling interface integrated with HRP-based amplification to achieve a highly sensitive electrochemical aptasensor for lysozyme detection. Analyst 2019, 144, 5794–5801. [Google Scholar] [CrossRef]
- Fan, J.L.; Tang, Y.; Yang, W.W.; Yu, Y.S. Disposable multiplexed electrochemical sensors based on electro-triggered selective immobilization of probes for simultaneous detection of DNA and proteins. J. Mater. Chem. B 2020, 8, 7501–7510. [Google Scholar] [CrossRef]
- Srisombat, L.; Jamison, A.C.; Lee, T.R. Stability: A key issue for self-assembled monolayers on gold as thin-film coatings and nanoparticle protectants. Colloids Surf. A Physicochem. Eng. Asp. 2011, 390, 1–19. [Google Scholar] [CrossRef]
- Lavrich, D.J.; Wetterer, S.M.; Bernasek, S.L.; Scoles, G. Physisorption and chemisorption of alkanethiols and alkyl sulfides on Au(111). J. Phys. Chem. B 1998, 102, 3456–3465. [Google Scholar] [CrossRef]
- Nuzzo, R.G.; Zegarski, B.R.; Dubois, L.H. Fundamental studies of the chemisorption of organosulfur compounds on Au(111). Implications for molecular self-assembly on gold surfaces. J. Am. Chem. Soc. 1987, 109, 733–740. [Google Scholar] [CrossRef]
- Futera, Z.; Blumberger, J. Adsorption of amino acids on gold: Assessing the accuracy of the GolP-CHARMM force field and parametrization of Au−S Bonds. J. Chem. Theory Comput. 2019, 15, 613–624. [Google Scholar] [CrossRef] [Green Version]
- Chinwangso, P.; Jamison, A.C.; Lee, T.R. Multidentate adsorbates for self-assembled monolayer films. Acc. Chem. Res. 2011, 44, 511–519. [Google Scholar] [CrossRef]
- Sakata, T.; Maruyama, S.; Ueda, A.; Otsuka, H.; Miyahara, Y. Stable Immobilization of an oligonucleotide probe on a gold substrate using tripodal thiol derivatives. Langmuir 2007, 23, 2269–2272. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Pali, M.; Lee, H.J.; Lee, T.R.; Suni, I.I. Impedance biosensor incorporating a carboxylate-terminated bidentate thiol for antibody immobilization. J. Electrochem. Soc. 2016, 163, B125–B130. [Google Scholar] [CrossRef] [Green Version]
- Staderini, M.; González-Fernández, E.; Murray, A.F.; Mount, A.R.; Bradley, M. A tripod anchor offers improved robustness of peptide-based electrochemical biosensors. Sens. Actuators B 2018, 274, 662–667. [Google Scholar] [CrossRef]
- Sharma, J.; Chhabra, R.; Yan, H.; Liu, Y. A facile in situ generation of dithiocarbamate ligands for stable gold nanoparticle–oligonucleotide conjugates. Chem. Commun. 2008, 2140–2142. [Google Scholar] [CrossRef]
- Raigoza, A.F.; Kolettis, G.; Brandt, T.E.S.; Caponigri-Guerra, G.; Agostino, C.; Kandel, S.A. Coadsorption of octanethiol and dialkyldithiocarbamate on Au(111). J. Phys. Chem. C 2012, 116, 1930–1934. [Google Scholar] [CrossRef]
- Wang, L.; Wang, X.L.; Chen, X.H.; Liu, J.; Liu, S.F.; Zhao, C.Z. Development of an electrochemical DNA biosensor with the DNA immobilization based on in situ generation of dithiocarbamate ligands. Bioelectrochemistry 2012, 88, 30–35. [Google Scholar] [CrossRef]
- Lou, J.; Liu, S.S.; Tu, W.W.; Dai, Z.H. Graphene quantum dots combined with endonuclease cleavage and bidentate chelation for highly sensitive electrochemiluminescent DNA biosensing. Anal. Chem. 2015, 87, 1145–1151. [Google Scholar] [CrossRef]
- Drozd, M.; Pietrzak, M.D.; Malinowska, E. SPRi-based biosensing platforms for detection of specific DNA sequences using thiolate and dithiocarbamate assemblies. Front. Chem. 2018, 6, 173. [Google Scholar] [CrossRef]
- Niu, Y.; Matos, A.I.; Abrantes, L.M.; Viana, A.S.; Jin, G. Antibody oriented immobilization on gold using the reaction between carbon disulfide and amine groups and its application in immunosensing. Langmuir 2012, 28, 17718–17725. [Google Scholar] [CrossRef] [Green Version]
- Almeida, I.; Henriques, F.; Carvalho, M.D.; Viana, A.S. Carbon disulfide mediated self-assembly of Laccase and iron oxide nanoparticles on gold surfaces for biosensing applications. J. Colloid Interface Sci. 2017, 485, 242–250. [Google Scholar] [CrossRef]
- Procter, D.J. The synthesis of thiols, selenols, sulfides, selenides, sulfoxides, selenoxides, sulfones and selenones. J. Chem. Soc. Perkin Trans. 2000, 1, 835–871. [Google Scholar] [CrossRef]
- Qing, Z.H.; Luo, G.Y.; Xing, S.H.; Zou, Z.; Lei, Y.L.; Liu, J.W.; Yang, R.H. Pt–S Bond-Mediated nanoflares for high-fidelity intracellular applications by avoiding thiol cleavage. Angew. Chem. Int. Ed. 2020, 59, 14044–14048. [Google Scholar] [CrossRef]
- Yang, W.S.; Auciello, O.; Butler, J.E.; Cai, W.; Carlisle, J.A.; Gerbi, J.E.; Gruen, D.M.; Knickerbocker, T.; Lasseter, T.L.; Russell, J.N.; et al. DNA-modified nanocrystalline diamond thin films as stable, biologically active substrates. Nat. Mater. 2002, 1, 253–257. [Google Scholar] [CrossRef]
- Knickerbocker, T.; Strother, T.; Schwartz, M.P.; Russell, J.N.; Butler, J.; Smith, L.M.; Hamers, R.J. DNA-modified diamond surfaces. Langmuir 2003, 19, 1938–1942. [Google Scholar] [CrossRef]
- Wang, X.Y.; Landis, E.C.; Franking, R.; Hamers, R.J. Surface chemistry for stable and smart molecular and biomolecular interfaces via photochemical grafting of alkenes. Acc. Chem. Res. 2010, 43, 1205–1215. [Google Scholar] [CrossRef]
- Stavisa, C.; Clare, T.L.; Butler, J.E.; Radadia, A.D.; Carr, R.; Zeng, H.J.; King, W.P.; Carlisle, J.A.; Aksimentiev, A.; Bashir, R.; et al. Surface functionalization of thin-film diamond for highly stable and selective biological interfaces. Proc. Natl. Acad. Sci. USA 2011, 108, 983–988. [Google Scholar] [CrossRef] [Green Version]
- Migneault, I.; Dartiguenave, C.; Bertrand, M.J.; Waldron, K.C. Glutaraldehyde: Behavior in aqueous solution, reaction with proteins, and application to enzyme crosslinking. Biotechniques 2004, 37, 790–802. [Google Scholar] [CrossRef]
- Barbosa, O.; Ortiz, C.; Berenguer-Murcia, A.; Torres, R.; Rodrigues, R.C.; Fernandez-Lafuente, R. Glutaraldehyde in bio-catalysts design: A useful crosslinker and a versatile tool in enzyme immobilization. RSC Adv. 2014, 4, 1583–1600. [Google Scholar] [CrossRef] [Green Version]
- Ciampi, S.; Eggers, P.K.; Le Saux, G.; James, M.; Harper, J.B.; Gooding, J.J. Silicon (100) electrodes resistant to oxidation in aqueous solutions: An unexpected benefit of surface acetylene moieties. Langmuir 2009, 25, 2530–2539. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Suni, I.I. Antibody regeneration on degenerate Si electrodes for calibration and reuse of impedance biosensors. Sens. Bio-Sens. Res. 2016, 7, 20–24. [Google Scholar] [CrossRef] [Green Version]
- Lasmi, K.; Derder, H.; Kermad, A.; Sam, S.; Boukhalfa-Abib, H.; Belhousse, S.; Tighilt, F.Z.; Hamdani, K.; Gabouze, N. Tyrosinase immobilization on functionalized porous silicon surface for optical monitoring of pyrocatechol. Appl. Surf. Sci. 2018, 446, 3–9. [Google Scholar] [CrossRef]
- Aschl, T.; Frison, G.; Moraillon, A.; Ozanam, F.; Allongue, P.; Gouget-Laemmel, A.C. Insights into the ochratoxin A/aptamer interactions on a functionalized silicon surface by Fourier transform infrared and UV–vis studies. Langmuir 2020, 36, 13908–13917. [Google Scholar] [CrossRef]
- Fopasea, R.; Paramasivam, S.; Kale, P.; Paramasivan, B. Strategies, challenges and opportunities of enzyme immobilization on porous silicon for biosensing applications. J. Environ. Chem. Eng. 2020, 8, 104266. [Google Scholar] [CrossRef]
- Peiris, C.R.; Ciampi, S.; Dief, E.M.; Zhang, J.Y.; Canfield, P.J.; Le Brun, A.P.; Kosov, D.S.; Reimers, J.R.; Darwish, N. Spontaneous S–Si bonding of alkanethiols to Si(111)–H: Towards Si–molecule–Si circuits. Chem. Sci. 2020, 11, 5246–5256. [Google Scholar] [CrossRef]
- Dief, E.M.; Vogel, Y.B.; Peiris, C.R.; Le Brun, A.P.; Gonçales, V.R.; Ciampi, S.; Reimers, J.R.; Darwish, N. Covalent linkages of molecules and proteins to Si−H surfaces formed by disulfide reduction. Langmuir 2020, 36, 14999–15009. [Google Scholar] [CrossRef]
- Singh, V.; Rawal, V.; Lakhanpal, S.; Jain, P.; Dahiya, S.; Tripathi, C.C. Immobilized bacteriophage used for specific detection of E. coli using electrochemical impedance sensing. Int. J. Pharm. Sci. Res. 2015, 6, 3913–3919. [Google Scholar]
- Sidhu, R.K.; Cavallaro, N.D.; Pola, C.C.; Danyluk, M.D.; McLamore, E.S.; Gomes, C.L. Planar interdigitated aptasensor for flow-through detection of Listeria spp. in hydroponic lettuce growth media. Sensors 2020, 20, 5773. [Google Scholar]
- Ucar, A.; González-Fernández, E.; Staderini, M.; Avlonitis, N.; Murray, A.F.; Bradley, M.; Mount, A.R. Miniaturisation of a peptide-based electrochemical protease activity sensor using platinum microelectrodes. Analyst 2020, 145, 975–982. [Google Scholar] [CrossRef] [Green Version]
- Husain, S.; Gupta, R.; Kumar, A.; Kumar, P.; Behera, N.; Brucas, R.; Chaudhary, S.; Svedlindh, P. Emergence of spin–orbit torques in 2D transition metal dichalcogenides: A status update. Appl. Phys. Rev. 2020, 7, 041312. [Google Scholar] [CrossRef]
- Li, H.; Tao, L.; Xu, J.B. Intrinsic memristive mechanisms in 2D layered materials for high-performance memory. J. Appl. Phys. 2021, 129, 050902. [Google Scholar] [CrossRef]
- Tebyetekerwa, M.; Zhang, J.; Xu, Z.; Truong, T.N.; Yin, Z.Y.; Lu, Y.R.; Ramakrishna, S.; Macdonald, D.; Nguyen, H.T. Mechanisms and applications of steady-state photoluminescence spectroscopy in two-dimensional transition-metal dichalcogenides. ACS Nano 2020, 14, 14579–14604. [Google Scholar] [CrossRef]
- Zhu1, Y.; Sun, X.Q.; Tang, Y.L.; Fu, L.; Lu, Y.R. Two-dimensional materials for light emitting applications: Achievement, challenge and future perspectives. Nano Res. 2021, 14, 1912–1936. [Google Scholar] [CrossRef]
- Wang, C.X.; Zhang, L.Y.; Zhang, Z.W.; Zhao, R.Z.; Zhao, D.Y.; Ma, R.Z.; Yin, L.W. Layered materials for supercapacitors and batteries: Applications and challenges. Prog. Mater. Sci. 2021, 118, 100763. [Google Scholar]
- Li, Y.H.; Wang, M.L.; Yi, Y.Y.; Lu, C.; Dou, S.X.; Sun, J.Y. Metallic transition metal dichalcogenides of Group VIB: Preparation, stabilization, and energy applications. Small 2021, 17, 2005573. [Google Scholar] [CrossRef]
- Kukkar, M.; Mohanta, G.C.; Tuteja, S.K.; Kumar, P.; Bhadwal, A.S.; Samaddar, P.; Kim, K.H.; Deep, A. A comprehensive review on nano-molybdenum disulfide/DNA interfaces as emerging biosensing platforms. Biosens. Bioelectron. 2018, 107, 244–258. [Google Scholar] [CrossRef]
- Meng, S.; Zhang, Y.Y.; Wang, H.; Wang, L.; Kong, T.T.; Zhang, H.; Meng, S. Recent advances on TMDCs for medical diagnosis. Biomaterials 2021, 269, 120471. [Google Scholar] [CrossRef]
- Rahman, M.T.; Kumar, R.; Kumar, M.; Qiao, Q.Q. Two-dimensional transition metal dichalcogenides and their composites for lab-based sensing applications: Recent progress and future outlook. Sens. Actuators A 2021, 318, 112517. [Google Scholar] [CrossRef]
- Xiao, M.Y.; Wei, S.; Chen, J.J.; Tian, J.Y.; Brooks, C.L.; Marsh, E.N.G.; Chen, Z. Molecular mechanisms of interactions between monolayered transition metal dichalcogenides and biological molecules. J. Am. Chem. Soc. 2019, 141, 9980–9988. [Google Scholar] [CrossRef]
- Bertolazzi, S.; Gobbi, M.; Zhao, Y.; Backes, C.; Samorı, P. Molecular chemistry approaches for tuning the properties of two-dimensional transition metal dichalcogenides. Chem. Soc. Rev. 2018, 47, 6845–6888. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.; Pak, J.; Chung, S.; Lee, T. Recent advances in interface engineering of transition-metal dichalcogenides with organic molecules and polymers. ACS Nano 2019, 13, 9713–9734. [Google Scholar] [CrossRef] [Green Version]
- Joshi, Y.V.; Ghosh, P.; Venkataraman, P.S.; Delgass, W.N.; Thomson, K.T. Electronic descriptors for the adsorption energies of sulfur-containing molecules on Co/MoS2 using DFT calculations. J. Phys. Chem. C 2009, 113, 9698–9709. [Google Scholar] [CrossRef]
- Kukkar, M.; Tuteja, S.K.; Sharma, A.L.; Kumar, V.; Paul, A.K.; Kim, K.H.; Sabherwal, P.; Deep, A. A new electrolytic synthesis method for few-layered MoS2 nanosheets and their robust biointerfacing with reduced antibodies. ACS Appl. Mater. Interfaces 2016, 8, 16555–16563. [Google Scholar] [CrossRef]
- Giang, H.; Pali, M.; Fan, L.; Suni, I.I. Impedance biosensing atop MoS2 thin films with Mo-S bond formation to antibody fragments created by disulphide bond reduction. Electroanalysis 2019, 31, 957–965. [Google Scholar] [CrossRef]
- Kinnamon, D.; Ghanta, R.; Lin, K.C.; Muthukumar, S.; Prasad, S. Portable biosensor for monitoring cortisol in low-volume perspired human sweat. Sci. Rep. 2017, 7, 13312. [Google Scholar] [CrossRef] [Green Version]
- Chiu, N.F.; Lin, T.L. Affinity capture surface carboxyl-functionalized MoS2 sheets to enhance the sensitivity of surface plasmon resonance immunosensors. Talanta 2018, 185, 174–181. [Google Scholar] [CrossRef]
- Palomar, Q.; Gondran, C.; Lellouche, J.P.; Cosnier, S.; Holzinger, M. Functionalized tungsten disulfide nanotubes for dopamine and catechol detection in a tyrosinase-based amperometric biosensor design. J. Mater. Chem. B 2020, 8, 3566–3573. [Google Scholar] [CrossRef]
- Raichman, D.; Strawser, D.A.; Lellouche, J.P. Covalent functionalization/polycarboxylation of tungsten disulfide inorganic nanotubes (INTs-WS2). Nano Res. 2015, 8, 1454–1463. [Google Scholar] [CrossRef]
- Suh, J.; Park, T.E.; Lin, D.Y.; Fu, D.Y.; Park, J.; Jung, H.J.; Chen, Y.B.; Ko, C.; Jang, C.; Sun, Y.H.; et al. Doping against the native propensity of MoS2: Degenerate hole doping by cation substitution. Nano Lett. 2014, 14, 6976–6982. [Google Scholar] [CrossRef]
- Tedstone, A.A.; Lewis, D.J.; O’Brien, P. Synthesis, properties, and applications of transition metal-doped layered transition metal dichalcogenides. Chem. Mater. 2016, 28, 1965–1974. [Google Scholar] [CrossRef]
- Singh, S.; Singh, A.K. Origin of n-type conductivity of monolayer MoS2. Phys. Rev. B 2019, 99, 121201. [Google Scholar] [CrossRef]
- Li, M.G.; Yao, J.D.; Wu, X.X.; Zhang, X.C.; Xing, B.; Niu, X.Y.; Yan, X.Y.; Yu, Y.; Liu, Y.L.; Wang, Y.W. P-type Doping in large-area monolayer MoS2 by chemical vapor deposition. ACS Appl. Mater. Interfaces 2020, 12, 6276–6282. [Google Scholar] [CrossRef]
- Liu, K.L.; Luo, P.; Han, W.; Yang, S.J.; Zhou, S.S.; Li, H.Q.; Zhai, T.Y. Approaching ohmic contact to two-dimensional semiconductors. Sci. Bull. 2019, 64, 1426–1435. [Google Scholar] [CrossRef] [Green Version]
- Arnfinnsdottir, N.B.; Chapman, C.A.; Bailey, R.C.; Aksnes, A.; Stokke, B.T. Impact of silanization parameters and antibody immobilization strategy on binding capacity of photonic ring resonators. Sensors 2020, 20, 3163. [Google Scholar] [CrossRef]
- Chockalingam, M.; Darwish, N.; Le Saux, G.; Gooding, J.J. Importance of the indium tin oxide substrate on the quality of self-assembled monolayers formed from organophosphonic acids. Langmuir 2011, 27, 2545–2552. [Google Scholar] [CrossRef]
- Chen, X.; Luais, E.; Darwish, N.; Ciampi, S.; Thordarson, P.; Gooding, J.J. Studies on the effect of solvents on self-assembled monolayers formed from organophosphonic acids on indium tin oxide. Langmuir 2012, 28, 9487–9495. [Google Scholar] [CrossRef]
- Aydın, E.B.; Sezgintürk, M.K. A sensitive and disposable electrochemical immunosensor for detection of SOX2, a biomarker of cancer. Talanta 2017, 172, 162–170. [Google Scholar] [CrossRef]
- Gündogdu, A.; Aydın, E.B.; Sezgintürk, M.K. A novel electrochemical immunosensor based on ITO modified by carboxyl-ended silane agent for ultrasensitive detection of MAGE-1 in human serum. Anal. Biochem. 2017, 537, 84–92. [Google Scholar] [CrossRef]
- Demirbakan, B.; Sezgintürk, M.K. A novel electrochemical immunosensor based on disposable ITO-PET electrodes for sensitive detection of PAK 2 antigen. J. Electroanal. Chem. 2019, 848, 113304. [Google Scholar] [CrossRef]
- Barreda-Garcıa, S.; Miranda-Castro, R.; de-los-Santos-Alvarez, N.; Miranda-Ordieres, A.J.; Lobo-Castanon, M.J. Solid-phase helicase dependent amplification and electrochemical detection of Salmonella on highly stable oligonucleotide-modified ITO electrodes. Chem. Commun. 2017, 53, 9721–9724. [Google Scholar] [CrossRef]
- Aydın, E.B.; Sezgintürk, M.K. A disposable and ultrasensitive ITO based biosensor modified by 6-phosphonohexanoic acid for electrochemical sensing of IL-1β in human serum and saliva. Anal. Chim. Acta 2018, 1039, 41–50. [Google Scholar] [CrossRef]
- Aydın, E.B.; Sezgintürk, M.K. An impedimetric immunosensor for highly sensitive detection of IL-8 in human serum and saliva samples: A new surface modification method by 6-phosphonohexanoic acid for biosensing applications. Anal. Biochem. 2018, 554, 44–52. [Google Scholar] [CrossRef]
- Herranz-Diez, C.; Mas-Moruno, C.; Neubauer, S.; Kessler, H.; Gil, F.J.; Pegueroles, M.; Manero, J.M.; Guillem-Marti, J. Tuning mesenchymal stem cell response onto titanium−niobium−hafnium alloy by recombinant fibronectin fragments. ACS Appl. Mater. Interfaces 2016, 8, 2517–2525. [Google Scholar] [CrossRef]
- Ritz, U.; Nusselt, T.; Sewing, A.; Ziebart, T.; Kaufmann, K.; Baranowski, A.; Rommens, P.M.; Hofmann, A. The effect of different collagen modifications for titanium and titanium nitrite surfaces on functions of gingival fibroblasts. Clin. Oral Investig. 2017, 21, 255–265. [Google Scholar] [CrossRef]
- Rezvanian, P.; Daza, R.; López, P.A.; Ramos, M.; González-Nieto, D.; Elices, M.; Guinea, G.V.; Pérez-Rigueiro, J. Enhanced biological response of AVS-functionalized Ti-6Al-4V alloy through covalent immobilization of collagen. Sci. Rep. 2018, 8, 3337. [Google Scholar] [CrossRef] [Green Version]
- Sharan, J.; Koul, V.; Dinda, A.K.; Kharbanda, O.P.; Lale, S.V.; Duggal, R.; Mishra, M.; Gupta, G.; Singh, M.P. Bio-functionalization of grade V titanium alloy with type I human collagen for enhancing and promoting human periodontal fibroblast cell adhesion–an in-vitro study. Colloids Surf. B 2018, 161, 1–9. [Google Scholar] [CrossRef]
- Mantzila, A.G.; Prodromidis, M.I. Performance of impedimetric biosensors based on anodically formed Ti/TiO2 electrodes. Electroanalysis 2005, 17, 1878–1885. [Google Scholar] [CrossRef]
- Mantzila, A.G.; Prodromidis, M.I. Development and study of anodic Ti/TiO2 electrodes and their potential use as impedimetric immunosensors. Electrochim. Acta 2006, 51, 3537–3542. [Google Scholar] [CrossRef]
- Sheppard, L.R.; Bak, T.; Nowotny, J. Electrical properties of niobium-doped titanium dioxide. 1. Defect disorder. J. Phys. Chem. B 2006, 110, 22447–22454. [Google Scholar] [CrossRef]
- Xu, W.L.; Schultz, T.; Koch, N.; Pinna, N. Niobium-doped titanium dioxide with high dopant contents for enhanced lithium-ion storage. ChemElectroChem 2020, 7, 4016–4023. [Google Scholar] [CrossRef]
- Dorow-Gerspach, C.; Mergel, D.; Wuttig, M. Metal-like conductivity in undoped TiO2−x: Understanding an unconventional transparent conducting oxide. Thin Solid Films 2019, 669, 1–7. [Google Scholar] [CrossRef]
- Dorow-Gerspach, C.; Mergel, D.; Wuttig, M. Effects of different amounts of Nb doping on electrical, optical and structural properties in sputtered TiO2−x films. Crystals 2021, 11, 301. [Google Scholar] [CrossRef]
- Tajik, S.; Beitollahi, H.; Nejad, F.G.; Shoaie, I.S.; Khalilzadeh, M.A.; Asl, M.S.; Le, Q.V.; Zhang, K.Q.; Jang, H.W.; Shokouhimehr, M. Recent developments in conducting polymers: Applications for electrochemistry. RSC Adv. 2020, 10, 37834–37856. [Google Scholar] [CrossRef]
- Luong, J.H.T.; Narayan, T.; Solanki, S.; Malhotra, B.D. Recent advances of conducting polymers and their composites for electrochemical biosensing applications. J. Funct. Biomater. 2020, 11, 71. [Google Scholar] [CrossRef]
- Wen, Y.P.; Li, D.; Xu, J.K.; Wang, X.Q.; He, H.H. Electrosynthesis of poly(thiophene-3-acetic acid) film in ionic liquids for covalent immobilization of biologically active species. Int. J. Polym. Mater. 2013, 62, 437–443. [Google Scholar] [CrossRef]
- Mello, H.J.N.P.D.; Bueno, P.R.; Mulato, M. Comparing glucose and urea enzymatic electrochemical and optical biosensors based on polyaniline thin films. Anal. Methods 2020, 12, 4199–4210. [Google Scholar]
- Mello, H.J.N.P.D.; Mulato, M. Enzymatically functionalized polyaniline thin films produced with one-step electrochemical immobilization and its application in glucose and urea potentiometric biosensors. Biomed. Microdevices 2020, 22, 22. [Google Scholar] [CrossRef]
- Gokce, Z.G.; Akalın, P.; Kok, F.N.; Sarac, A.S. Impedimetric DNA biosensor based on polyurethane/poly(m-anthranilicacid) nanofiber. Sens. Actuators B 2018, 254, 719–726. [Google Scholar] [CrossRef]
- Niyomdechaa, S.; Limbut, W.; Numnuam, A.; Kanatharana, P.; Charlermroj, R.; Karoonuthaisiri, N.; Thavarungkul, P. Phage-based capacitive biosensor for Salmonella detection. Talanta 2018, 188, 658–664. [Google Scholar] [CrossRef]
- Hetemi, D.; Noël, V.; Pinson, J. Grafting of diazonium salts on surfaces: Application to biosensors. Biosensors 2020, 10, 4. [Google Scholar] [CrossRef] [Green Version]
- Pilan, L. Tailoring the performance of electrochemical biosensors based on carbon nanomaterials via aryldiazonium electrografting. Bioelectrochemistry 2021, 138, 107697. [Google Scholar] [CrossRef]
- Anariba, F.; DuVall, S.H.; McCreery, R.L. Mono- and multilayer formation by diazonium reduction on carbon surfaces monitored with atomic force microscopy “scratching”. Anal. Chem. 2003, 75, 3837–3844. [Google Scholar] [CrossRef]
- López, I.; Cesbron, M.; Levillain, E.; Breton, T. Diazonium grafting control through a redox cross-reaction: Elucidation of the mechanism involved when using 2,2-diphenylpicrylhydrazyl as an inhibitor. ChemElectroChem 2018, 5, 1197–1202. [Google Scholar] [CrossRef]
- Pichereau, I.; Lopez, I.; Cesbron, M.; Dabos-Seignon, S.; Gautier, C.; Breton, T. Controlled diazonium electrografting driven by overpotential reduction: A general strategy to prepare ultrathin layers. Chem. Commun. 2019, 55, 455–457. [Google Scholar] [CrossRef]
- Tahara, K.; Kubo, Y.; Lindner, B.; Hashimoto, S.; Hirose, S.; Brown, A.; Hirsch, B.; Daukiya, L.; De Feyter, S.; Tobe, Y. Steric and electronic effects of electrochemically generated aryl radicals on grafting of the graphite surface. Langmuir 2019, 35, 2089–2098. [Google Scholar] [CrossRef]
- Wu, T.; Lankshear, E.R.; Downard, A.J. Simultaneous electro-click and electrochemically mediated polymerization reactions for one-pot grafting from a controlled density of anchor sites. ChemElectroChem 2019, 6, 5149–5154. [Google Scholar] [CrossRef]
- González, M.C.R.; Brown, A.; Eyley, S.; Thielemans, W.; Mali, K.S.; De Feyter, S. Self-limiting covalent modification of carbon surfaces: Diazonium chemistry with a twist. Nanoscale 2020, 12, 18782–18789. [Google Scholar] [CrossRef]
- Mattiuzzi, A.; Lenne, Q.; Padilha, J.C.; Troian-Gautier, L.; Leroux, Y.R.; Jabin, I.; Lagrost, C. Strategies for the formation of monolayers from diazonium salts: Unconventional grafting media, unconventional building blocks. Front. Chem. 2020, 8, 559. [Google Scholar] [CrossRef]
- Breton, T.; Downard, A.J. Controlling grafting from aryldiazonium salts: A review of methods for the preparation of monolayers. Aust. J. Chem. 2017, 70, 960–972. [Google Scholar] [CrossRef]
- Hapiot, P.; Lagrost, C.; Leroux, Y.R. Molecular nano-structuration of carbon surfaces through reductive diazonium salts grafting. Curr. Opin. Electrochem. 2018, 7, 103–108. [Google Scholar] [CrossRef]
- Chu, X.S.; Yousaf, A.; Li, D.O.; Tang, A.A.; Debnath, A.; Ma, D.; Green, A.A.; Santos, E.J.G.; Wang, Q.H. Direct covalent chemical functionalization of unmodified two-dimensional molybdenum disulfide. Chem. Mater. 2018, 30, 2112–2128. [Google Scholar] [CrossRef]
- Li, D.O.; Chu, X.S.; Wang, Q.H. Reaction kinetics for the covalent functionalization of two-dimensional MoS2 by aryl diazonium salts. Langmuir 2019, 35, 5693–5701. [Google Scholar] [CrossRef]
- Li, D.O.; Gilliam, M.S.; Chu, X.S.; Yousaf, A.; Guo, Y.Q.; Green, A.A.; Wang, Q.H. Covalent chemical functionalization of semiconducting layered chalcogenide nanosheets. Mol. Syst. Des. Eng. 2019, 4, 962–973. [Google Scholar] [CrossRef]
- Park, Y.H.; Shin, S.H.; An, Y.J.; Ahn, J.G.; Shin, G.B.; Ahn, C.Y.; Bang, J.W.; Baik, J.Y.; Kim, Y.S.; Jung, J.H.; et al. Tunable optical transition in 2H-MoS2 via direct electrochemical engineering of vacancy defects and surface S−C Bonds. ACS Appl. Mater. Interfaces 2020, 12, 40870–40878. [Google Scholar] [CrossRef]
- Lihter, M.; Graf, M.; Iveković, D.; Zhang, M.; Shen, T.H.; Zhao, Y.F.; Macha, M.; Tileli, V.; Radenovic, A. Electrochemical functionalization of selectively addressed MoS2 nanoribbons for sensor device fabrication. ACS Appl. Nano Mater. 2021, 4, 1076–1084. [Google Scholar] [CrossRef]
- Daukiya, L.; Teyssandier, J.; Eyley, S.; Kazzi, S.E.; González, M.C.R.; Pradhan, B.; Thielemans, W.; Hofkens, J.; De Feyter, S. Covalent functionalization of molybdenum disulfide by chemically activated diazonium salts. Nanoscale 2021, 13, 2972–2981. [Google Scholar] [CrossRef]
- Nazemi, Z.; Shams, E.; Amini, M.K. Construction of a biointerface for glucose oxidase through diazonium chemistry and electrostatic self-assembly technique. J. Solid State Electrochem. 2016, 20, 429–438. [Google Scholar] [CrossRef]
- Martínez-García, G.; Agüí, L.; Yáñez-Sedeño, P.; Pingarrón, J.M. Multiplexed electrochemical immunosensing of obesity-related hormones at grafted graphene-modified electrodes. Electrochim. Acta 2016, 202, 209–215. [Google Scholar] [CrossRef]
- Dulac, M.; Melet, A.; Harris, K.D.; Limoges, B.; Galardon, E.; Balland, V. An optical H2S biosensor based on the chemoselective Hb-I protein tethered to a transparent, high surface area nanocolumnar electrode. Sens. Actuators B 2019, 290, 326–335. [Google Scholar] [CrossRef] [Green Version]
- Kuo, T.M.; Shen, M.Y.; Huang, S.Y.; Li, Y.K.; Chuang, M.C. Facile fabrication of a sensor with a bifunctional interface for logic analysis of the New Delhi metallo-β-lactamase (NDM)-coding gene. ACS Sens. 2016, 1, 124–130. [Google Scholar] [CrossRef]
- Mousavisani, D.Z.; Raoof, J.B.; Turner, A.P.F.; Ojani, R.; Mak, W.C. Label-free DNA sensor based on diazonium immobilization for detection of DNA damage in breast cancer 1 gene. Sens. Actuators B 2018, 264, 59–66. [Google Scholar] [CrossRef]
- Payne, N.A.; Mauzeroll, J. Identifying nanoscale pinhole defects in nitroaryl layer with scanning electrochemical cell microscopy. ChemElectroChem 2019, 6, 5439–5445. [Google Scholar] [CrossRef]
- Diao, P.; Jiang, D.L.; Cui, X.L.; Gu, D.P.; Tong, R.T.; Zhong, B. Studies of structural disorder of self-assembled thiol monolayers on gold by cyclic voltammetry and AC impedance. J. Electroanal. Chem. 1994, 464, 61–67. [Google Scholar] [CrossRef]
- Diao, P.; Jiang, D.L.; Cui, X.L.; Gu, D.P.; Tong, R.T.; Zhong, B. Unmodified supported lipid/thiol bilayers: Studies of structural disorder and conducting mechanism by cyclic voltammetry and AC impedance. Bioelectrochem. Bioenerg. 1999, 48, 469–475. [Google Scholar] [CrossRef]
- Diao, P.; Guo, M.; Jiang, D.L.; Jia, Z.B.; Cui, X.L.; Gu, D.P.; Tong, R.T.; Zhong, B. Fractional coverage of defects in self-assembled thiol monolayers on gold. J. Electroanal. Chem. 2000, 480, 59–63. [Google Scholar] [CrossRef]
- Diao, P.; Guo, M.; Tong, R.T. Characterization of defects in the formation process of self-assembled thiol monolayers by electrochemical impedance spectroscopy. J. Electroanal. Chem. 2001, 495, 98–105. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suni, I.I. Substrate Materials for Biomolecular Immobilization within Electrochemical Biosensors. Biosensors 2021, 11, 239. https://doi.org/10.3390/bios11070239
Suni II. Substrate Materials for Biomolecular Immobilization within Electrochemical Biosensors. Biosensors. 2021; 11(7):239. https://doi.org/10.3390/bios11070239
Chicago/Turabian StyleSuni, Ian Ivar. 2021. "Substrate Materials for Biomolecular Immobilization within Electrochemical Biosensors" Biosensors 11, no. 7: 239. https://doi.org/10.3390/bios11070239
APA StyleSuni, I. I. (2021). Substrate Materials for Biomolecular Immobilization within Electrochemical Biosensors. Biosensors, 11(7), 239. https://doi.org/10.3390/bios11070239