1. Introduction
In 1959, the World Health Organization (WHO) defined zoonoses as diseases that are naturally transmittable between humans and other vertebrates [
1]. Up to 60% of human infectious diseases are thought to have a zoonotic origin [
2]. Due to the growth of the human population, the demand for food products such as meat, milk and eggs, and the intensification of production is increasing. By this, the risk of zoonotic pathogens contaminating food and causing severe health effects is also increasing [
3]. Campylobacteriosis was the most common zoonosis reported in 2019 with approximately 221,000 cases in the European Union, followed by salmonellosis (88,000 cases), infections with shiga toxin-producing
Escherichia coli (STEC) (8000 cases), and yersiniosis (7000 cases) [
4]. In 99% of yersiniosis cases, the agent was
Yersinia enterocolitica [
5].
The genus
Yersinia (family
Enterobacteriaceae) comprises 19 species, including the human and animal pathogens
Y. enterocolitica,
Y. pestis, and
Y. pseudotuberculosis [
6].
Y. enterocolitica is a Gram-negative bacillus, with a coccoid to rod-shaped morphology. This psychrophilic species can grow at temperatures within the range 0–45 °C, with optimal growth at 25–28 °C.
Y. enterocolitica is motile at 25 °C, but loses motility at 37 °C [
7,
8]. The heterogeneous strains of
Y. enterocolitica are assigned to six biotypes on the basis of their biochemical features (1A, 1B, and 2–5) and comprise more than 70 serotypes depending on their O-lipopolysaccharide determinants [
9,
10]. Generally, biotype 1B strains are highly virulent, whereas biotypes 2–5 have comparably low virulence. Strains of biotype 1A are generally non-virulent. Virulence is conferred by the plasmid pVY and virulence-associated chromosomal genes [
11]. Nevertheless, some biotype 1A strains cause yersiniosis even though they lack these virulence components [
6].
Y. enterocolitica is usually transferred to humans via the consumption of contaminated raw or undercooked pork, or by direct contact with contaminated carcasses [
12]. Yersiniosis is an acute form of gastroenteritis that often includes symptoms such as fever and intestinal inflammation with watery or occasionally bloody diarrhea [
7]. Pigs are the main reservoir, carrying this pathogen in their tonsils, lymphatic tissues, and intestine. As infected pigs are asymptomatic, the bacteria usually were not detected until the infected tissue is exposed during slaughter and processing, allowing the pathogen to spread over the surface of the carcasses [
12,
13]. Especially due to the devastating health effects, a regular testing for zoonotic pathogens is extremely important. However, rapid and reliable (molecular) detection methods for analysis of meat are not yet available [
13].
The conventional detection of
Y. enterocolitica requires microbial cultivation. An isolation step is necessary, consisting of selective enrichment followed by plating onto selective agar medium. Presumptive colonies are identified by biochemical and pathogenic phenotype. Depending on the choice of enrichment strategy, isolation may take from 48 h up to several weeks [
14,
15,
16]. The conventional method is therefore time-consuming, laborious, and makes it difficult to detect low numbers of pathogens in the presence of abundant background flora also capable of growing on selective media [
16].
The polymerase chain reaction (PCR) allows the genotypic characterization of
Y. enterocolitica by amplifying the virulence-associated genes. Multiplex PCRs have been developed, and more sensitive detection can be achieved by real-time PCR [
6,
12,
13,
17,
18,
19]. PCR is useful for preliminary screening because it enables fast and sensitive analysis, but it requires expensive equipment and skilled personnel. Establishing reliable assays is not trivial since the presence of inhibitors in untreated samples often leads to false-negative results. Additionally, false-positive results may arise due to the detection of dead cells [
15]. Immuno-based methods such as the enzyme-linked immunosorbent assay (ELISA) or lateral flow assays for whole cell detection are rare. ELISA formats detecting
Y. enterocolitica directly are usually serotype-specific and are not well suited for sensitive detection of a bacterial infection [
9]. Indirect diagnostic assays were developed to detect antibodies against
Y. enterocolitica in animal blood and fecal samples [
20,
21]. However, this only allows the detection of
Y. enterocolitica-specific antibodies, which only permits a conclusion about (a possible past) yersiniosis. Hence, these methods are not applicable for the detection of an actual bacterial contamination, e.g., in food samples. By this, current commercially available ELISA-based methods are not feasible for efficient analysis and detection of food contamination with
Y. enterocolitica.
To address the limitations of conventional detection methods and current molecular assays, we developed a new approach for the rapid and sensitive detection of bacteria using quorum sensing (QS) signaling molecules. QS is generally known as a bacterial cell-cell communication process to coordinate gene expression depending on population density. In contrast to low cell density (LCD) populations, at which individual gene expression is favored, the synchronous expression of multiple genes is beneficial at high cell density (HCD) populations. QS-controlled processes include antibiotic production, sporulation, bioluminescence, biofilm formation, and virulence factor secretion. The QS system is based on signaling molecules called autoinducers (AIs), which are produced and secreted by cells. After reaching a local threshold concentration at HCD, the AIs trigger a signal cascade that modulates gene expression [
22,
23].
The AIs are divided into three major groups: AI-1 (AI-1), AI-2 (AI-2), and peptide-based AIs (AIPs) [
24]. AIPs are a heterogeneous group of modified oligopeptides used by Gram-positive bacteria mainly for intraspecies communication [
25]. AI-2 is a group of 4,5-dihydroxy-2,3-pentanedione (DPD) derivatives that can rapidly cyclize into furanone counterparts. AI-2 compounds are produced by a wide range of Gram-positive and Gram-negative bacteria and can be used to communicate between cells of the same or different species [
26]. AI-1 compounds are
N-Acyl homoserine lactone-based molecules (AHLs), consisting of a homoserine lactone ring moiety and an acyl side chain, used for intraspecies communication in Gram-negative bacteria [
27]. AHL specificity is determined by the length, substitution, and saturation of the acyl side chain [
28].
Y. enterocolitica produces the AHLs
N-hexanoyl-L-homoserine lactone (HHL) and
N-(3-oxohexanoyl)-L-homoserine lactone (OHHL) [
29]. Therefore, we focused on the detection of AI-1 in this study.
The best-characterized QS system as a paradigm of intraspecies communication among Gram-negative bacteria is the LuxI/R system of the bioluminescent bacterium
Vibrio fischeri. The generation of bioluminescence is regulated by the LuxI and LuxR proteins. LuxI is an AHL synthase that produces OHHL. The
luxI gene is normally expressed at a basal level, resulting in the production of small quantities of OHHL [
30]. AHLs are amphipathic molecules that can pass the cell membrane by diffusion [
28]. As the cell population grows, the intracellular and extracellular concentration of AHL increases. When it reaches a local threshold concentration, the cytoplasmic LuxR protein binds to OHHL, and the resulting complex functions as a transcriptional activator, inducing the expression of
luxI and the
lux operon (
luxCDABEG). LuxI therefore initiates a positive feedback loop by synthesizing the ligand that binds to LuxR and induces the synthesis of more LuxI. The increase in OHHL levels also triggers the expression of the
lux operon in adjacent cells [
30,
31]. The structural genes of the
lux operon generate bioluminescence via a luciferase-catalyzed oxidation reaction [
32].
Within the last decades, the application of bacterial biosensor techniques has advanced the study of AHLs (AI-1) in QS systems and enables a simple method for qualitative and quantitative detection of AHLs [
33,
34,
35]. The bacterial biosensor, which does not produce AHLs by itself, carries a sensor plasmid that encodes the
luxR gene or the gene of a LuxR homologue, the cognate promoter, as well as a reporter gene for the detection. The presence of a compatible AHL finally induces the expression of the reporter gene whose product is detectable by, for example, bioluminescence [
33]. In a previous study, several sensor plasmids (pSB401, pSB403, pSB406, pSB1075) were constructed by linking
luxR together with the promoter
luxI’, or homologues as
lasR and
rhlR with corresponding promoters, to the
lux operon (
luxCDABE) of
Photorhabdus luminescens [
36]. This
lux operon offers a non-destructive real-time detection as a reporter gene and a higher temperature stability than the
lux operon of
V. fischeri, which is relevant for expression in recombinant
E. coli at 37 °C [
36,
37]. Additionally, the induction of bioluminescence formation by activating AHLs was demonstrated [
36].
In contrast, physico-chemical methods such as GC-MS or LC-MS/MS are frequently used for identification and quantification of AHLs due to their high specificity. However, these devices are highly expensive and need highly trained staff for handling, running, and maintaining the equipment. Furthermore, for method development highly trained staff are required, and sensitivity can vary greatly depending on method parameters, machine settings, and sample preparation. As demonstrated by Bauer and colleagues (2016), GC-MS methods, e.g., showed LODs of just 3.2 to 6.2 µM of single AHLs [
38]. Targeted LC–MS/MS methods showed LODs of 0.23 to 1.51 nM [
39]. Untargeted LC-MS/MS methods that are capable of detecting and identifying known and unknown AHL entities of a sample showed LODs of 0.6 to 229.6 nM [
40]. Whole cell Biosensor approaches, as recently reviewed by Miller and Gilmore (2020), reach higher sensitivities, typically in the low nM and pM ranges [
35].
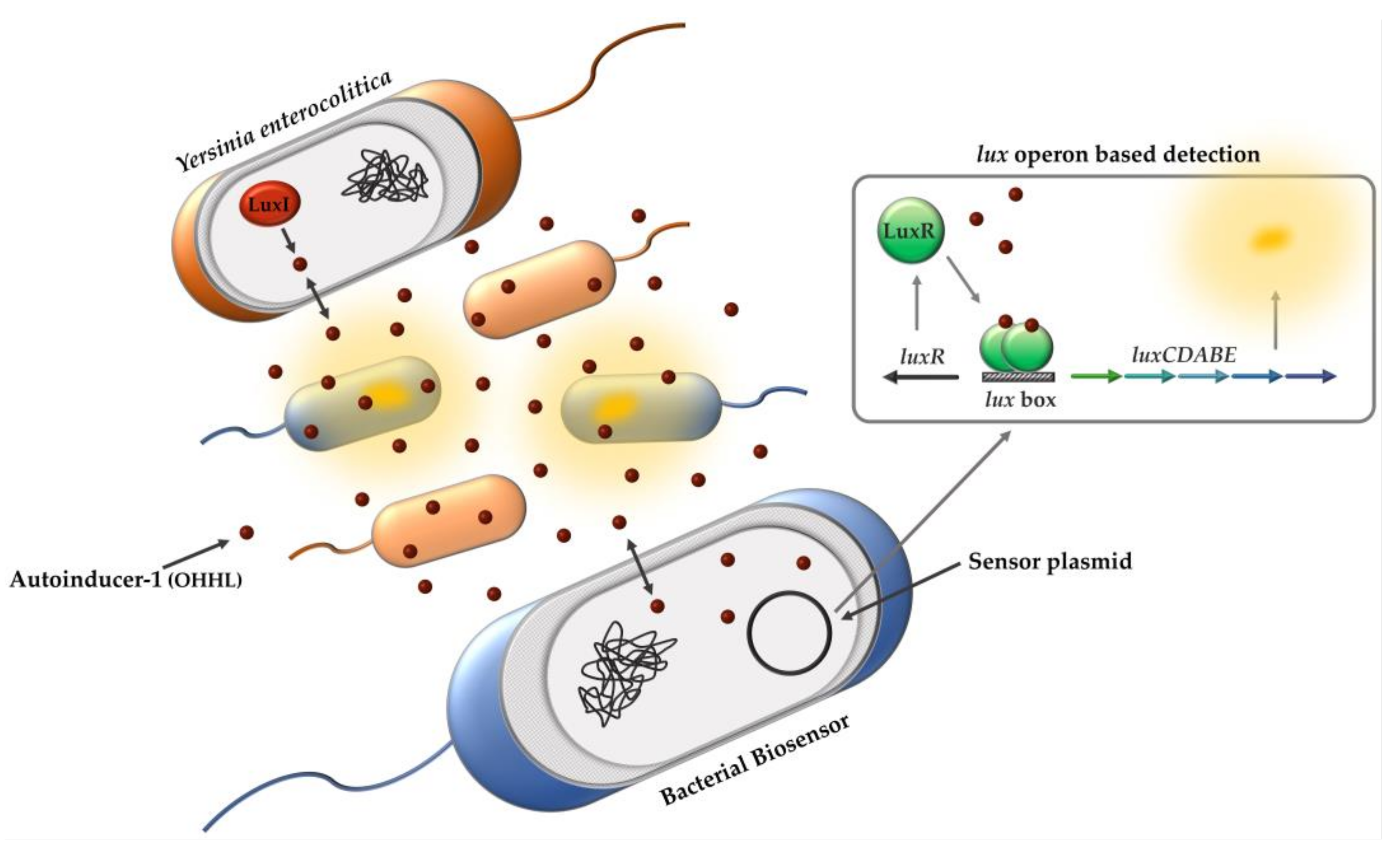
To overcome the limitations of conventional cultivation methods, we established a new whole-cell biosensor approach that enables faster, more sensitive, and more reliable detection of
Y. enterocolitica based on QS. Therefore, we used the
V. fischeri LuxI/R system for detection of
Y. enterocolitica by monitoring the production of its AHLs using a bacterial biosensor carrying a modified sensor plasmid derived from pSB401 [
36]. The presence of OHHL in a sample induces the
lux operon by binding the LuxR protein, inducing bioluminescence and indicating whether the sample is contaminated with
Y. enterocolitica (
Figure 1).
We first investigated the functionality of the biosensor in the presence of synthetic OHHL. We proved the ability to recognize AHLs produced by Y. enterocolitica and simultaneously tracked the AHL production of this pathogen via LC-MS/MS analysis for comparison. Finally, biosensor and pathogen were combined in an assay approach to achieve the highest sensitivity possible.
2. Materials and Methods
2.1. Sensor Plasmid pMA-RQ_luxR_lux
The customized sensor plasmid pMA-RQ_luxR_lux was synthesized by GeneArt (Thermo Fisher Scientific, Waltham, MA, USA). It was based on the sequence of pSB401 [
36], which encodes a fusion construct combining the
Vibrio fischeri luxRI’ and
Photorhabdus luminescens luxCDABE sequences. The fusion construct was joined to the pMA-RQ backbone, which contains an ampicillin-resistance gene and a ColE1 origin of replication (
Figure S1).
2.2. Generation of the pMA Biosensor
The pMA-RQ_luxR-lux sensor plasmid was transformed into chemically competent Escherichia coli strain BL21 (DE3) cells (New England Biolabs, Frankfurt am Main, Germany) according to the manufacturer’s protocol. Aliquots of 50 µL and 100 µL and the pellet were plated on lysogeny broth (LB) selection plates (10 g L−1 tryptone, 5 g L−1 yeast extract, 10 g L−1 NaCl, 15 g L−1 agar; pH 7.4) containing 100 µg mL−1 ampicillin. After an overnight incubation at 37 °C, successfully transformed cells were isolated on further selection plates and incubated overnight (37 °C). A colony was then used for preparation of fresh overnight cultures of the biosensor.
2.3. Luminescence Bioassay Procedure
An overnight culture in 5 mL selective LB was prepared with a clone from the master plate and incubated overnight at 37 °C, shaking at 160 rpm. On the next day, 25 mL of selective LB broth was inoculated with the overnight culture at a 1:1000 ratio. The biosensor culture was cultivated at 37 °C, shaking at 160 rpm, until the culture reached OD600 = 0.4. The culture was directly used for the luminescence assay. We mixed 20 µL of sample (synthetic AI standard concentrations or sterile culture supernatants) with 180 µL of the biosensor culture in C8 LockWell Lumi white 96-well plates (Thermo Fisher Scientific, Massachusetts, USA). For background measurement, the corresponding buffer or medium was used as blank. Kinetic measurements were carried out for 5 h at 30 °C recording luminescence signals every 20 min using a CLARIOstar plate reader (BMG Labtech, Ortenberg, Germany) with the following set parameters: top optic, 11.0 mm focal height, 10 s measuring interval time, 10 s orbital shaking at 200 rpm before measurement.
2.4. Preparation of Synthetic AI Standard
HHL and OHHL were purchased from Merck (Darmstadt, Germany). Stock solutions with a concentration of 20 mM were prepared in DMSO and stored at −20 °C. On the day of the bioassay, the stock was freshly diluted with sterile phosphate-buffered saline (PBS; 137 µM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, 1.5 mM KH2PO4; pH 7.4) to concentrations ranging from 0.3 nM up to 200 nM and used as standard concentrations for calibration in the luminescence bioassay.
2.5. Cultivation of Y. enterocolitica
Y. enterocolitica strain DSM 11503 (biovar 3, serovar 0:9) was obtained from the DSMZ-German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany). Overnight cultures were prepared by inoculating 5 mL tryptic soy broth (TSB, 17 g L
−1 casein peptone, 3 g L
−1 soy peptone, 2.5 g L
−1 d(+) glucose, 5 g L
−1 NaCl, 2.5 g L
−1 K
2HPO
4; pH 7.3) with 5 µL of a thawed
Y. enterocolitica glycerol stock. For experimental cultures, selective CIN medium was inoculated with the overnight culture at a ratio of 1:1000. The selective medium was derived from the CIN agar formulation as previously described [
41]. Agar and color indicators were omitted from the preparation (17 g L
−1 gelatin peptone, 1.5 g L
−1 casein peptone, 1.5 g L
−1 animal tissue peptone, 2 g L
−1 yeast extract, 20 g L
−1 mannitol, 0.5 g L
−1 sodium deoxycholate, 0.5 g L
−1 sodium cholate, 1 g L
−1 sodium chloride, 2 g L
−1 sodium pyruvate, 0.01 g L
−1 magnesium sulfate, 0.004 g L
−1 irgasan, 0.4 g L
−1 cefsulodin, 0.25 g L
−1 novobiocin; pH 7.4). All cultures were incubated at 28 °C, with shaking at 160 rpm.
2.5.1. Procedure of Growth Curve Analysis
A growth curve of
Y. enterocolitica was recorded by inoculating 300 mL selective CIN medium with an overnight culture (1:1000) and taking 12 mL samples at regular intervals, starting at t = 0 and ending at 24 h. The optical density was measured at 600 nm (OD
600) to determine cell growth. Culture supernatants were generated from 10 mL aliquots (
Section 2.5.2), and the cell number was estimated from the OD
600 on the basis of a
Yersinia-specific correlation factor determined in the context of this study by plating experiments. An OD
600 value of 1.0 corresponds to 2.84
10
8 ± 3.9%
Y. enterocolitica cells mL
−1. The estimated cell concentration
X was plotted semi-logarithmically against the cultivation time. The specific growth rate µ was determined by linear regression in the exponential phase. The specific growth rate µ of
Y. enterocolitica and the doubling time t
D was calculated using the following equations [
42]:
2.5.2. Preparation of Bacterial Culture Supernatants
Sterile culture supernatants were prepared from 10 mL samples taken during the growth curve experiment (
Section 2.5.1). Bacterial cells were pelleted by centrifugation (400×
g for 5 min). The supernatants were filtered using a 0.2 µm syringe sterile filter (Carl Roth, Karlsruhe, Germany) and subsequently stored at −80 °C.
2.6. Extraction of AHLs
AHLs were extracted from sterile culture supernatants by liquid-liquid extraction (LLE) in pro-analysis-grade ethyl acetate (Carl Roth, Karlsruhe, Germany) containing 1% (v/v) analytical grade anhydrous acetic acid (Merck, Darmstadt, Germany). For this, acidified ethyl acetate was added to collected supernatant (1:3 v/v) and vortexed for 1 min. Afterwards, the organic phase was collected, and the remaining aqueous phase was extracted again as described. In total, five extraction cycles were performed. Pooled organic phases were concentrated by evaporation before drying using a SpeedVac vacuum concentrator (Eppendorf, Hamburg, Germany) for 1 h at RT. For LC-MS/MS analysis, the residue was reconstituted in 200 µL acetonitrile (LC-MS-grade, Carl Roth, Karlsruhe, Germany). To determine the performance and recovery rates achieved using the LLE protocol, we spiked the culture supernatants with 100 ng of OHHL and HHL as additional samples.
2.7. Analysis of Extracted AHLs Using LC-MS/MS
AHL extracts were passed through a Claristep 0.2 µm regenerated cellulose filter with an area of 0.7 cm2 (Sartorius, Göttingen, Germany) into autosampler vials with inserts. The samples were separated by HPLC on a Curosil-PFP 3 µm 150 × 3.0 mm column (Phenomenex, Aschaffenburg, Germany) using the Agilent 1200 HPLC system with a diode array detector set to 190–400 nm. The mobile phases were (A) degassed ddH2O containing 5 mM LiChropur ammonium acetate (Merck), 0.1% (v/v) HPLC-grade trifluoroacetic acid (Merck, Darmstadt, Germany), and 0.1% (v/v) ACS-grade formic acid (Carl Roth) and (B) ultra-LC-MS/MS-grade acetonitrile (Carl Roth, Karlsruhe, Germany) containing 0.1% (v/v) trifluoroacetic acid. We injected 10 µL samples and separated them by gradient flow at a constant flow rate of 0.4 mL min−1. The gradient was held at 100% A for 2 min before changing to 100% B over a 12 min linear gradient, followed by a 2 min hold step at 100% B before returning to 100% A over 4 min, followed by a final re-equilibration for 5 min at 100% A. For calibration, stock solutions of OHHL and HHL (2 mg mL−1, dissolved in DSMO) were thawed and diluted with acetonitrile to the following concentrations: 0.01, 0.05, 0.1, 0.15, 0.3, 0.5 µg mL−1.
AHL extracts were passed through a Claristep 0.2 µm regenerated cellulose filter with an area of 0.7 cm2 (Sartorius, Göttingen, Germany) into autosampler vials with inserts. The samples were separated by HPLC on a Curosil-PFP 3 µm 150 × 3.0 mm column (Phenomenex, Aschaffenburg, Germany) using the Agilent 1200 HPLC system with a diode array detector set to 190–400 nm. The mobile phases were (A) degassed ddH2O containing 5 mM LiChropur ammonium acetate (Merck), 0.1% (v/v) HPLC-grade trifluoroacetic acid (Merck, Darmstadt, Germany), and 0.1% (v/v) ACS-grade formic acid (Carl Roth) and (B) ultra-LC-MS/MS-grade acetonitrile (Carl Roth, Karlsruhe, Germany) containing 0.1% (v/v) trifluoroacetic acid. We injected 10 µL samples and separated them by gradient flow at a constant flow rate of 0.4 mL min−1. The gradient was held at 100% A for 2 min before changing to 100% B over a 12 min linear gradient, followed by a 2 min hold step at 100% B before returning to 100% A over 4 min, followed by a final re-equilibration for 5 min at 100% A. For calibration, stock solutions of OHHL and HHL (2 mg mL−1, dissolved in DSMO) were thawed and diluted with acetonitrile to the following concentrations: 0.01, 0.05, 0.1, 0.15, 0.3, 0.5 µg mL−1.
The HPLC fractions were analyzed on an API 3000/3200 Q TRAP device (Applied Biosystems, Darmstadt, Germany) equipped with an electro spray ionization probe. Mass scans were carried out in positive ion mode with the following parameters: curtain gas (30 AU), collision gas (medium), ion spray voltage (5500 V), source temperature (550 °C), ion source gas 1 (40), ion source gas 2 (50). AHLs were detected in two multiple reaction monitoring (MRM) experiments, one based upon the selective 102.1 Da lactone ring fragment shown previously to be well suited for AHL detection [
43], and the other ion unique to the AHL under investigation. The optimized parameters for each AHL are described in the
supplemental methods (Table S1).
2.8. Co-Cultivation of Y. enterocolitca and pMA Biosensor
Overnight cultures of the biosensor and
Y. enterocolitica were prepared as described in
Section 2.3 and 2.5. On the next day, 25 mL of CIN medium without cefsulodin, irgasan, or novobiocin was inoculated with the biosensor overnight culture at a ratio of 1:1000, and 20 mL of selective CIN medium was inoculated with the
Yersinia overnight culture at the same ratio. The biosensor culture was incubated at 37 °C, shaking at 160 rpm, until an OD
600 reached 0.4, at which point the culture was directly used in the bioassay. The
Yersinia culture was incubated at 28 °C, shaking at 160 rpm, and was diluted to defined OD
600 values once the OD
600 had reached or slightly exceeded 0.05. These defined values were based on cell numbers previously selected for this study. The corresponding OD
600 was estimated using the specific correlation factor (
Section 2.5.1).
Similar to the method described in
Section 2.3, 180 µL biosensor culture was mixed with 20 µL culture sample in each well of a C8 LockWell Lumi white 96-well plate (Thermo Fisher Scientific, Waltham, MA, USA). Kinetic measurement was carried out for 20 h at 30 °C, with detection of the luminescence signal every 20 min. The following parameters were set for the top optic measurement: 11.0 mm focal height, 10 s measuring interval, 10 s orbital shaking at 200 rpm before measurement, 0.1 s settling time. Optimized assays were conducted using the bottom optic measurement in a white 96-well microplate with a clear bottom and lid (Berthold Technologies, Bad Wildbad, Germany). Here, the focal height was set to −2.6 mm.
4. Discussion
We established and demonstrated a concept for rapid and sensitive detection of Y. enterocolitica by using QS. Our results proved the feasibility of a real-time detection by monitoring the production of the pathogen’s AHLs using the pMA biosensor. Aiming to overcome the limitations of the conventional detection methods, we successfully adapted a co-cultivation approach to keep microbial enrichment and the simultaneous detection of Y. enterocolitica by the pMA biosensor as short and easy as possible.
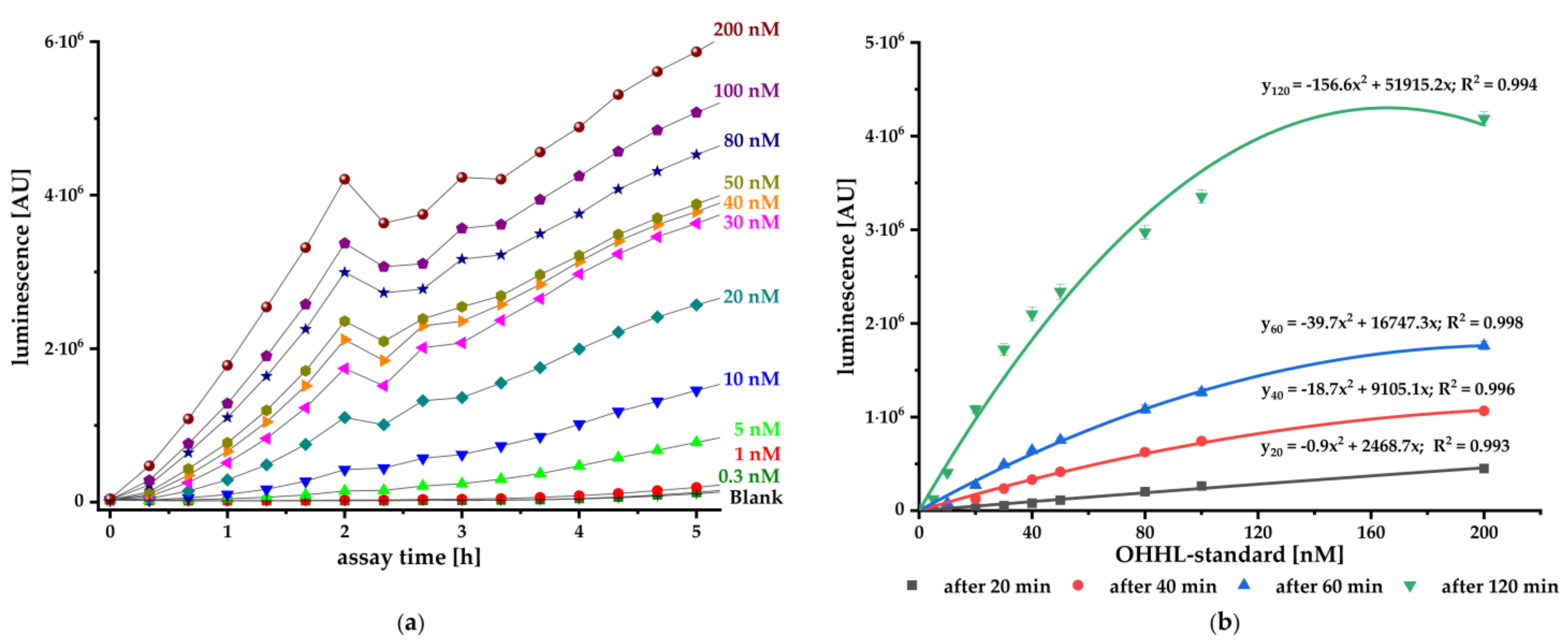
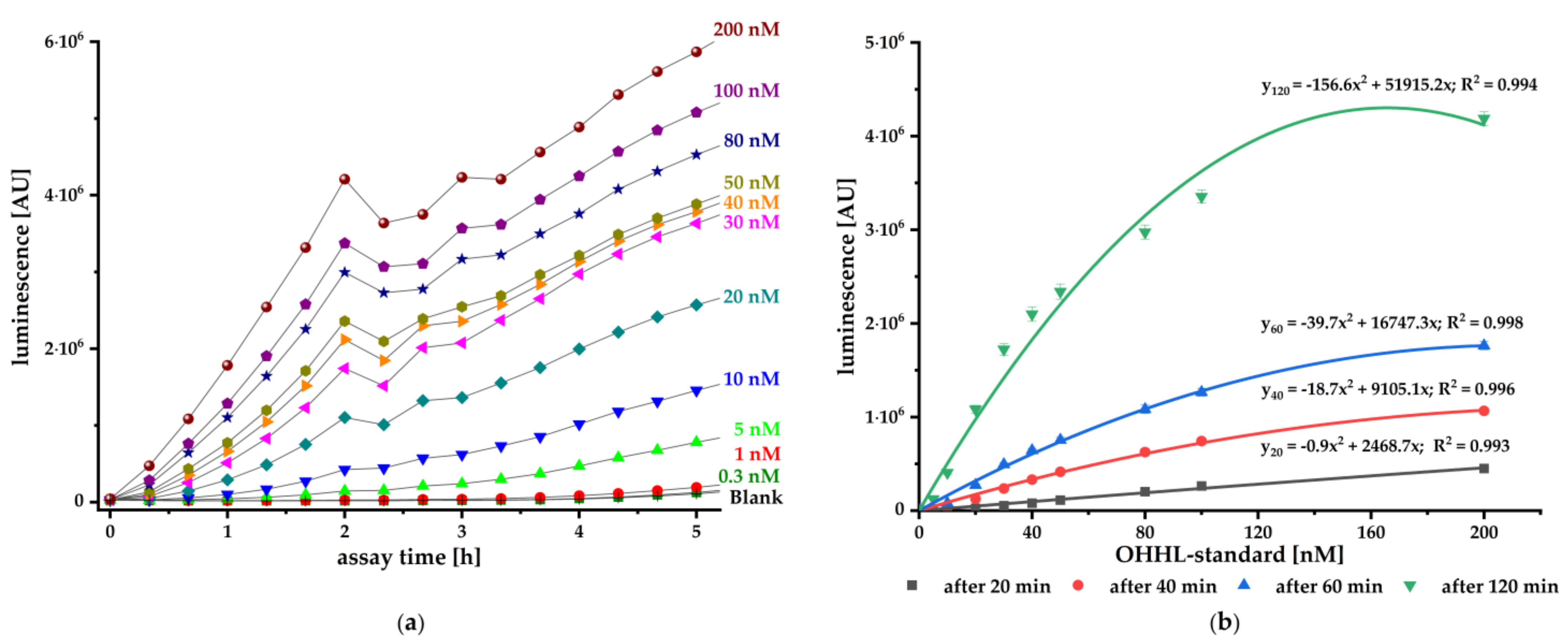
To validate the pMA biosensor, we carried out kinetic luminescence assays using different standard concentrations of synthetic AI. The biosensor responded concentration-dependent to the authentic AHL and provides reliable and sensitive signals from 20 min onwards (
Figure 2). It is particularly suitable for fast detection of small amounts OHHL with a sensitivity ranging from 0.50 nM up to 0.17 nM (LoD). This biosensor is at least as sensitive as the published constructs (pSB401, pSB403, pSB406, and pSB1075) with sensitivity to different AHLs in the pM and nM range [
36].
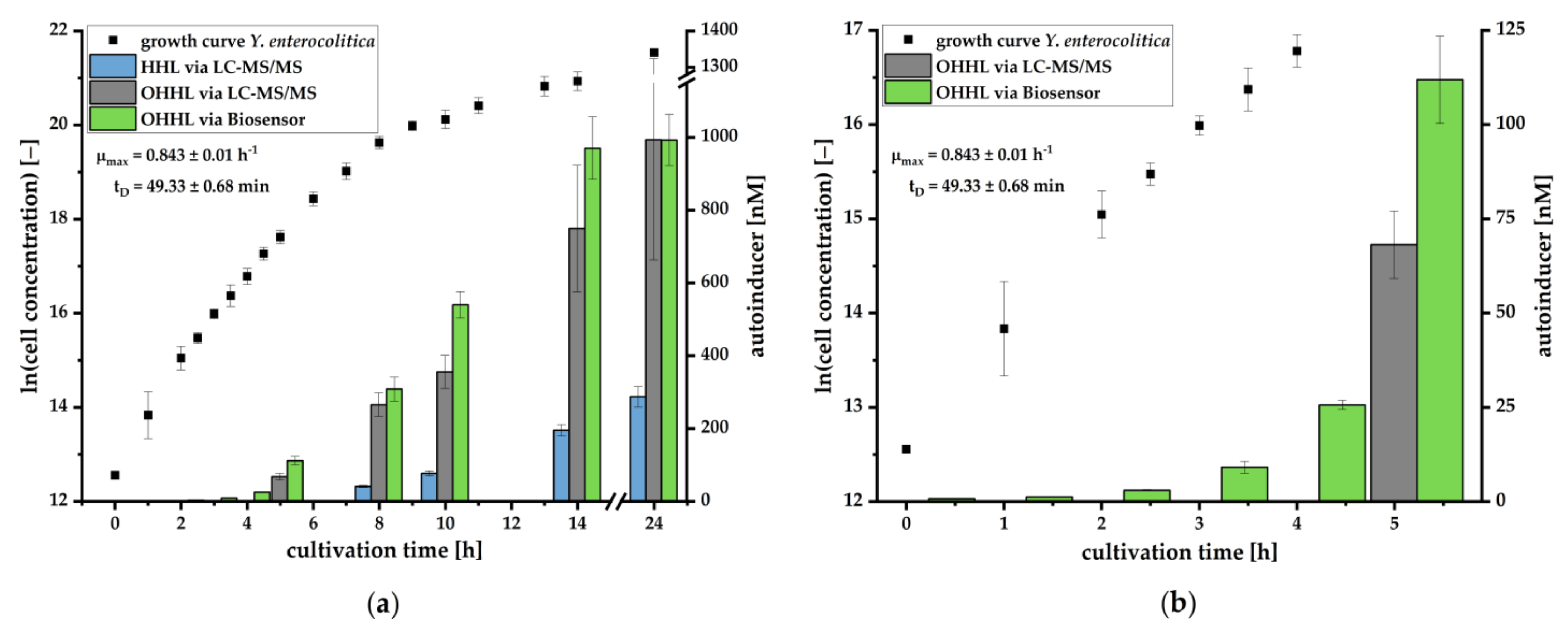
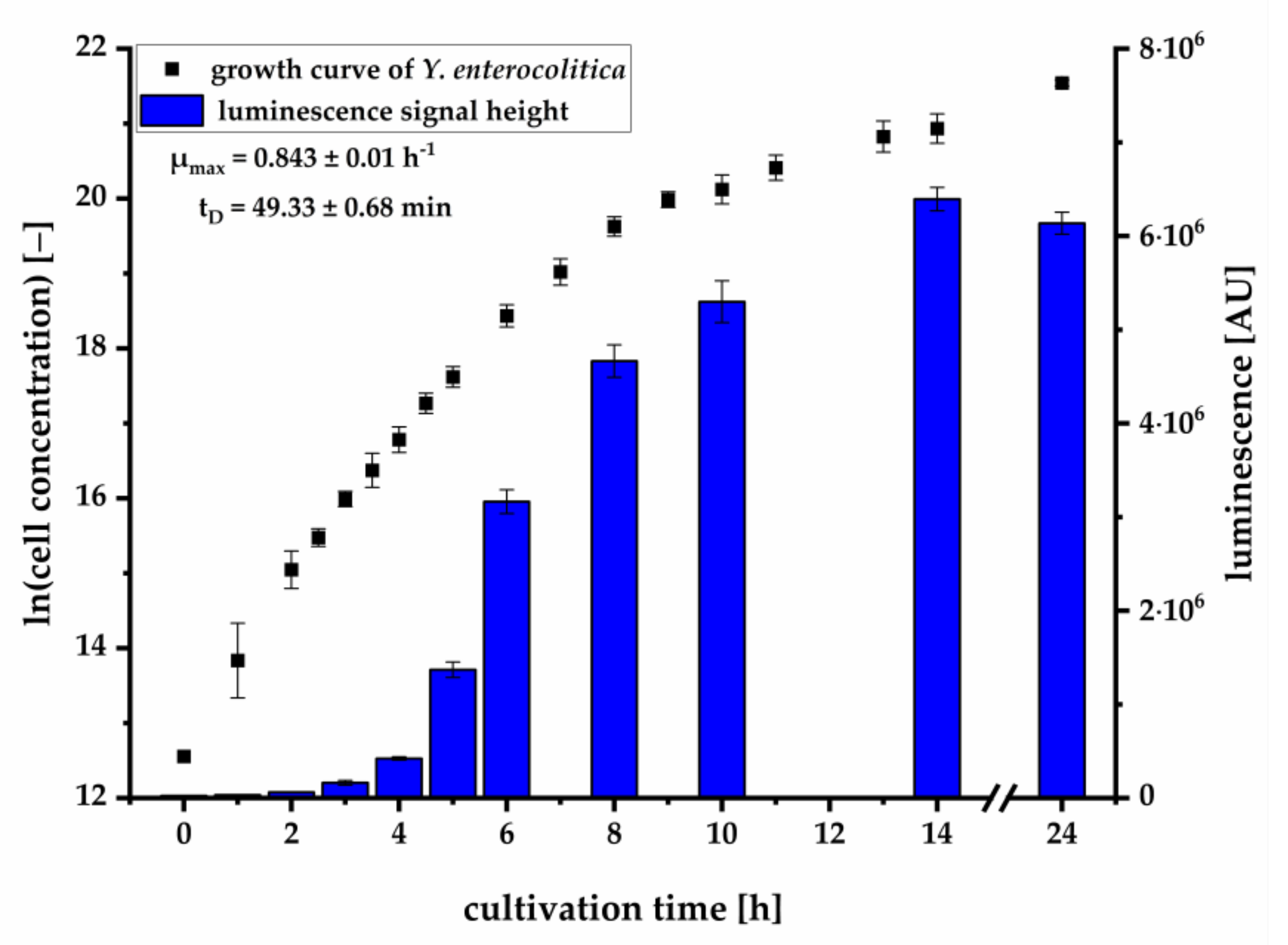
We could further demonstrate that the pMA biosensor is able to respond to AHLs in culture supernatants of
Y. enterocolitica (
Figure 3) without being suppressed by compounds of selective media. The increase of the luminescence signal in relation to the growth of the pathogen demonstrates the characteristic accumulation of AHLs during cell growth. To investigate which AHL is produced during cell growth and is mainly responsible for induction of the
lux cassette, we analyzed culture supernatants using LC-MS/MS. Here, we successfully verified the presence and production of OHHL and HHL during growth of
Y. enterocolitica. OHHL is the cognate AI of LuxR and activates the
lux gene expression most effectively [
36]. HHL differs from OHHL by the absence of a carbonyl-group at the C3-atom in the side chain. This OHHL analogue binds with less efficiency to LuxR and induces the
lux mechanism with less activity as OHHL. A previous study demonstrated that HHL inhibited the binding of OHHL to LuxR by about 10% when molar ratio was 1:1 of OHHL and HHL [
45]. Our study revealed that
Y. enterocolitica produced 3-6 fold less HHL than OHHL. Thus, an inhibition effect is negligible, confirming that the
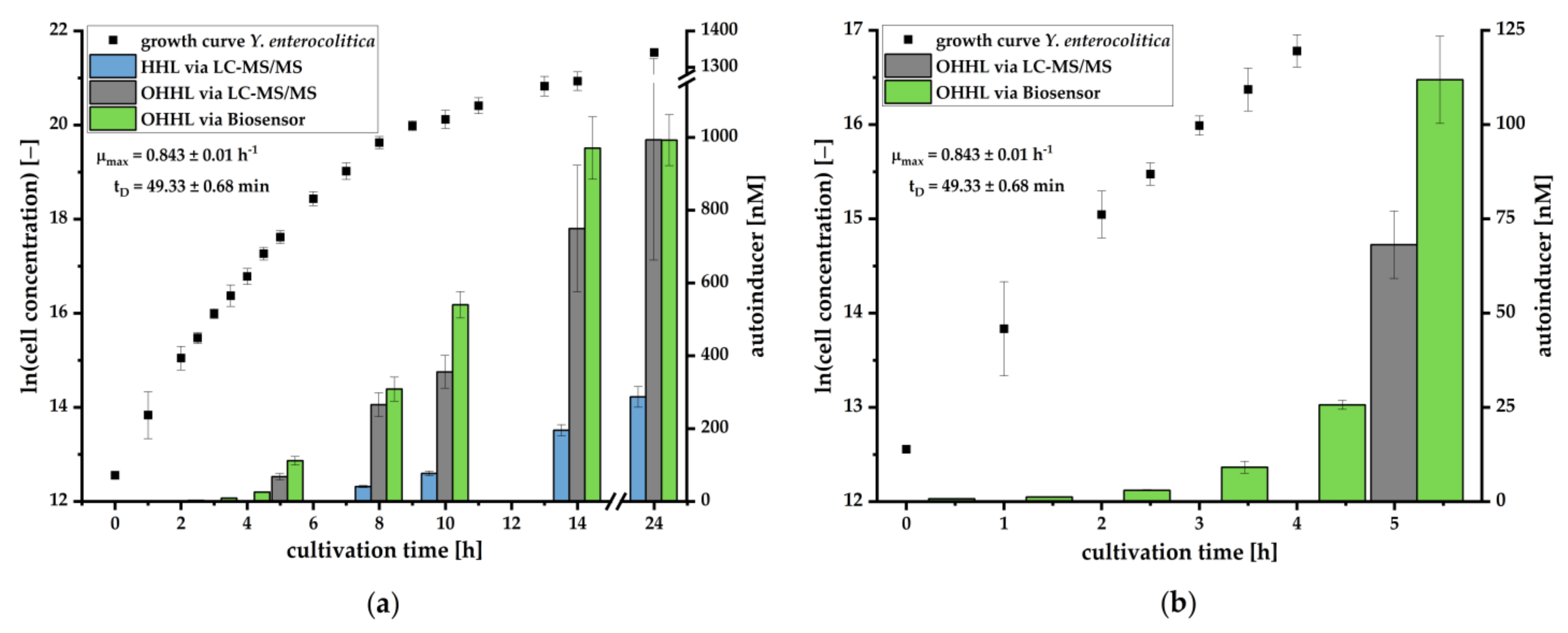
lux mechanism is primarily induced by OHHL. This condition allows for quantification of OHHL in culture supernatants by the pMA biosensor. Determined concentrations correlated in great manner with concentrations quantified by LC-MS/MS (
Figure 4). The quantification by the pMA biosensor revealed a higher sensitivity than our LC-MS/MS-based analytical method, which by itself revealed a higher sensitivity than published methods demonstrating the ability for
Y. enterocolitica detection in early stages of cultivation (compare [
40]). Further research is necessary to determine whether and to what extent other AHLs may interact with the
lux promoter. To generate data to the specificity or selectivity of the analytical procedure, we would need to investigate the selectivity of the selective medium and to determine which other bacteria could also produce OHHL and induce luminescent responses. This is especially important in terms of determining how specific our bacterial sensor is and what kind of selective enrichment steps need to be employed.
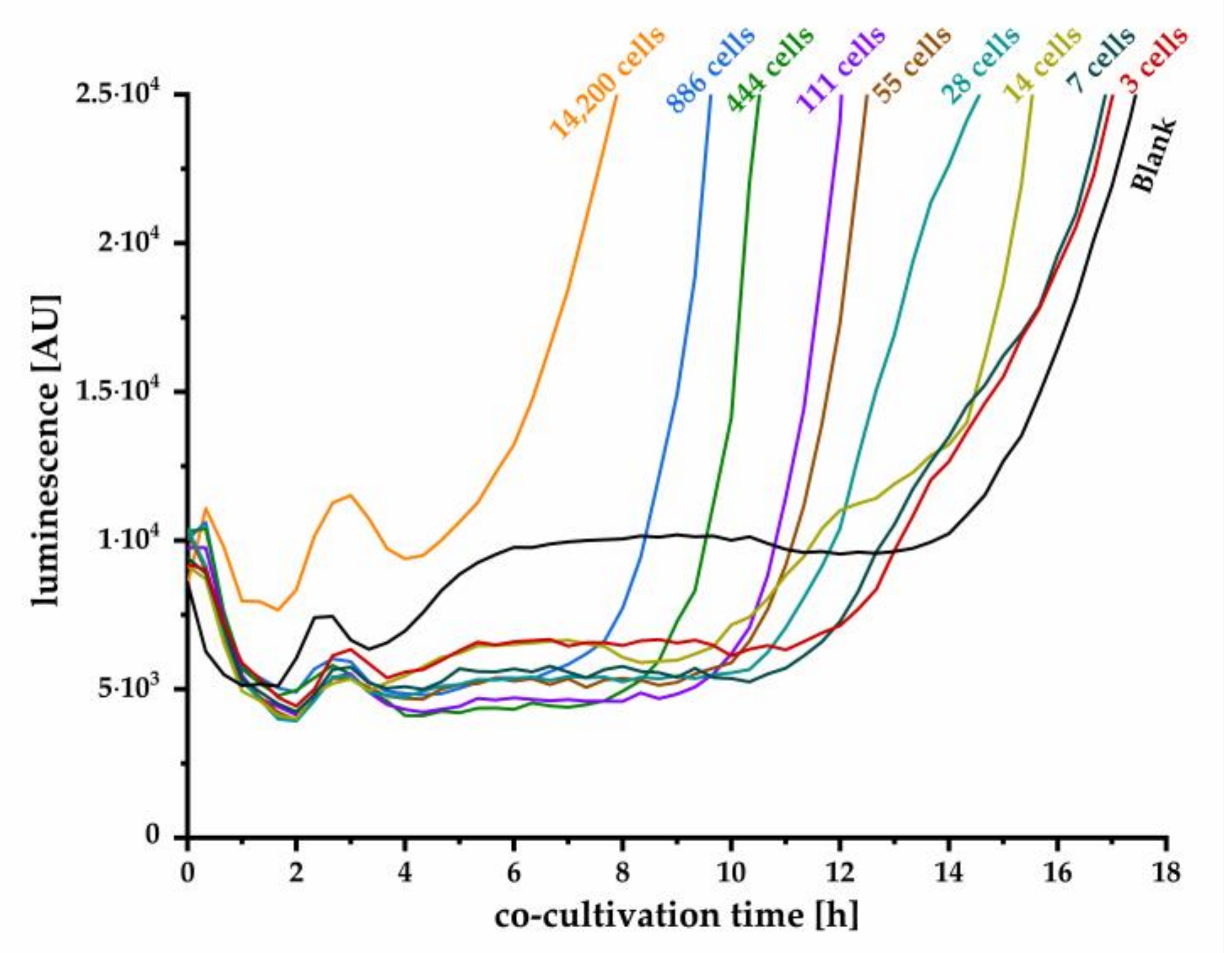
To date, several approaches have been developed using bacterial biosensors to detect and quantitatively estimate different kinds of bacteria on the basis of their secreted autoinducers [
35]. However, sensitivity was not high enough to detect very low cell numbers. Hence, by applying the pMA biosensor in a co-cultivation approach, we could establish a real-time detection of
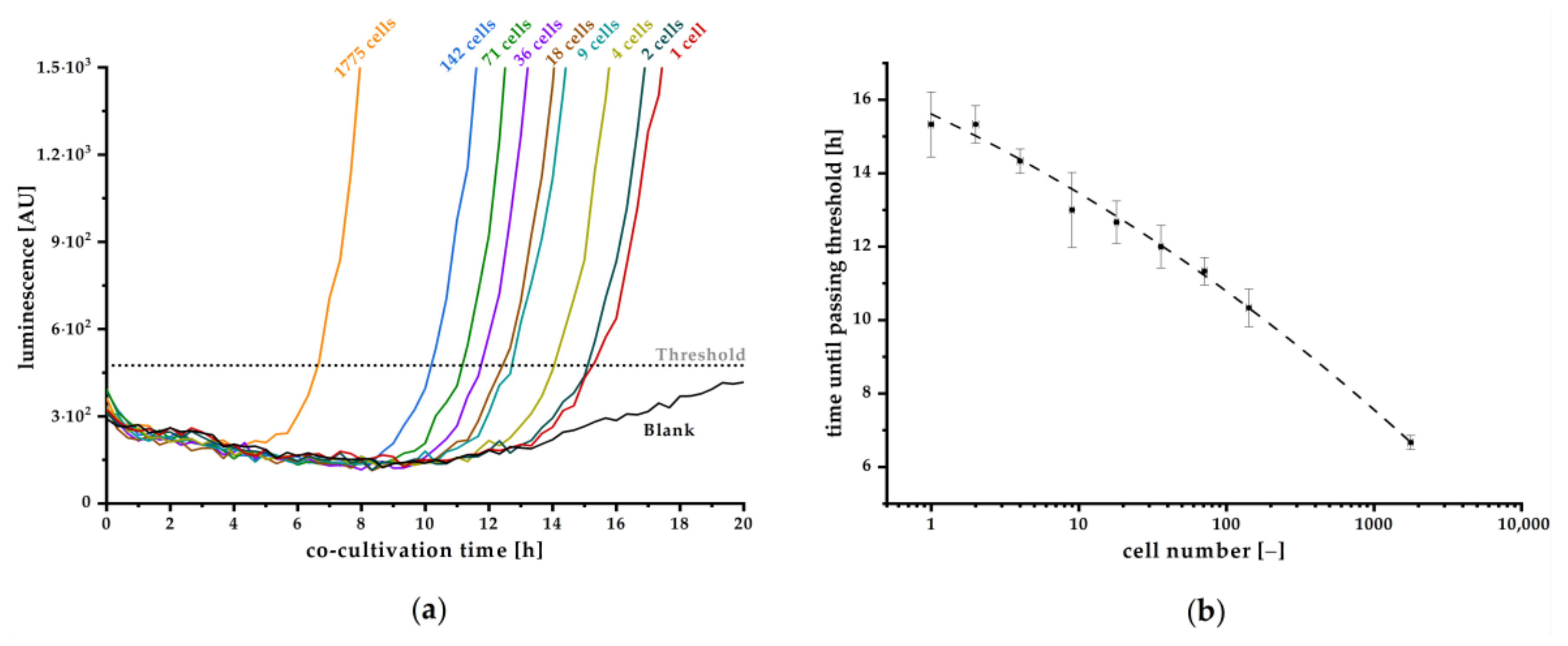
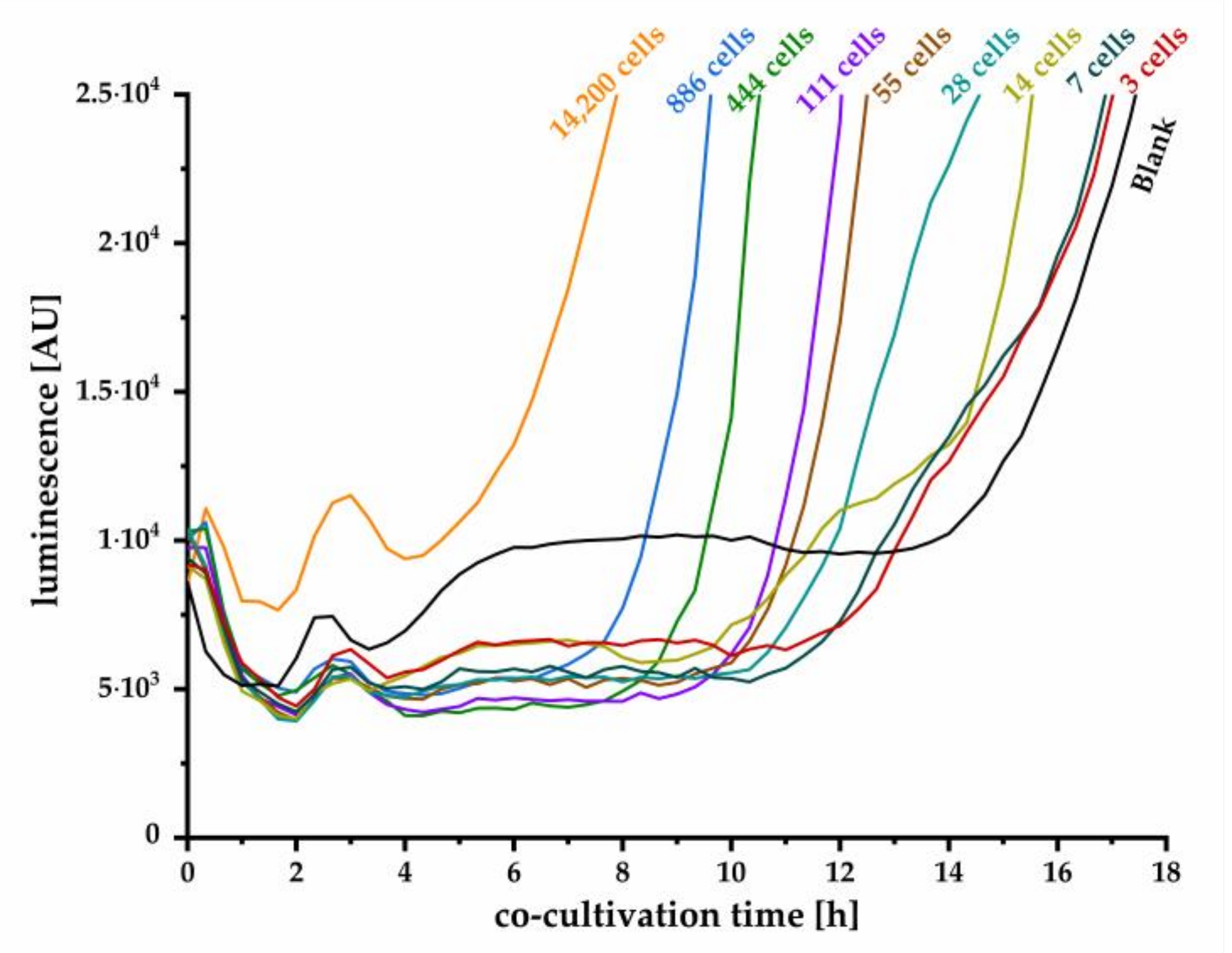
Y. enterocolitica for the first time by monitoring produced OHHL. We further demonstrated that even single cell detection and reliable quantification is possible. Although the detection of small cell number requires several hours of co-cultivation, the presented method is considerably faster than classic microbiological cultivation techniques. The assay set up was successfully optimized, resulting in a stable blank signal throughout relevant duration of cultivation, facilitating the determination of a threshold value that defines the minimal luminescence signal, confirming a positive detection. With this approach, we achieved high sensitivity for detection of one initial cell in a sample after 15.3 h of co-cultivation. Hence, monitoring the luminescence signal and the time to exceed threshold also enables quantitative estimation of the initial cell concentration on the basis of the calibration curve plotted in
Figure 6b. Here, the detection limit of one cell strongly depends on the reproducible low blank signal as realized in our improved experimental approach in combination with a reliable correlation factor between OD
600 and cells per milliliter. The quantitative detection range is limited by high cell numbers, where autoinducer concentration is already high enough to induce expression of the
lux operon. In this case, expression will start instantly, and strong luminescence signal increase can be observed within few minutes, as can be concluded from kinetics in
Figure 2. Our established method is more time-effective than the conventional microbiological detection methods and ensures the detection of living cells as the release of AHLs is an active process. For a specific detection of
Y. enterocolitica, further optimization has to be done, especially excluding luminescence signals induced by AIs from other bacteria. It is known that OHHL and similar AHLs that also might stimulate the
lux expression cassette in the pMA biosensor are produced by a huge number of
Enterobacteria. Therefore, a short pre-enrichment incubation step (e.g., 3 h) in CIN selection media might be beneficial to reduce the number of other bacteria and to mainly enrich
Y. enterocolitica. However, CIN medium is not 100% selective to
Y. enterocolitica, and therefore additional steps such as, e.g., an alkali treatment, could be necessary to increase selectivity [
46,
47]. Hence, further experiments have to be performed to reveal the specificity of our here presented assay when using real food samples. Nevertheless, our approach is well suited for fast an early detection of bacterial contamination and could be used in food security or veterinary applications. Further research will focus on the adaption of the luminescence cassette to specifically detect further AI molecules enabling fast detection of other pathogenic bacteria such as
Salmonella,
Campylobacter, or
Legionella. Exchanging the luminescence gene by fluorescence cassettes expressing different fluorophores might additionally enable multiplex detection of different bacteria in a single sample.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}