3.1. Framework of Kinetic Measurements
L-lysine-α-oxidase from
Trichoderma viride (LO) was co-immobilised with an inert protein, bovine serum albumin, by crosslinking with glutaraldehyde. This valuable co-crosslinking immobilisation approach [
17] allowed us to obtain an immobilised enzyme layer with high biocomponent activity and good stability properties by simply casting a small amount of the proper co-crosslinking enzyme solution onto the electrode surface [
18]. Of course, the influence of the inert protein and crosslinker concentrations have strong impact on the efficiency of LO immobilisation and its catalytic properties and have been already studied and optimised elsewhere [
12].
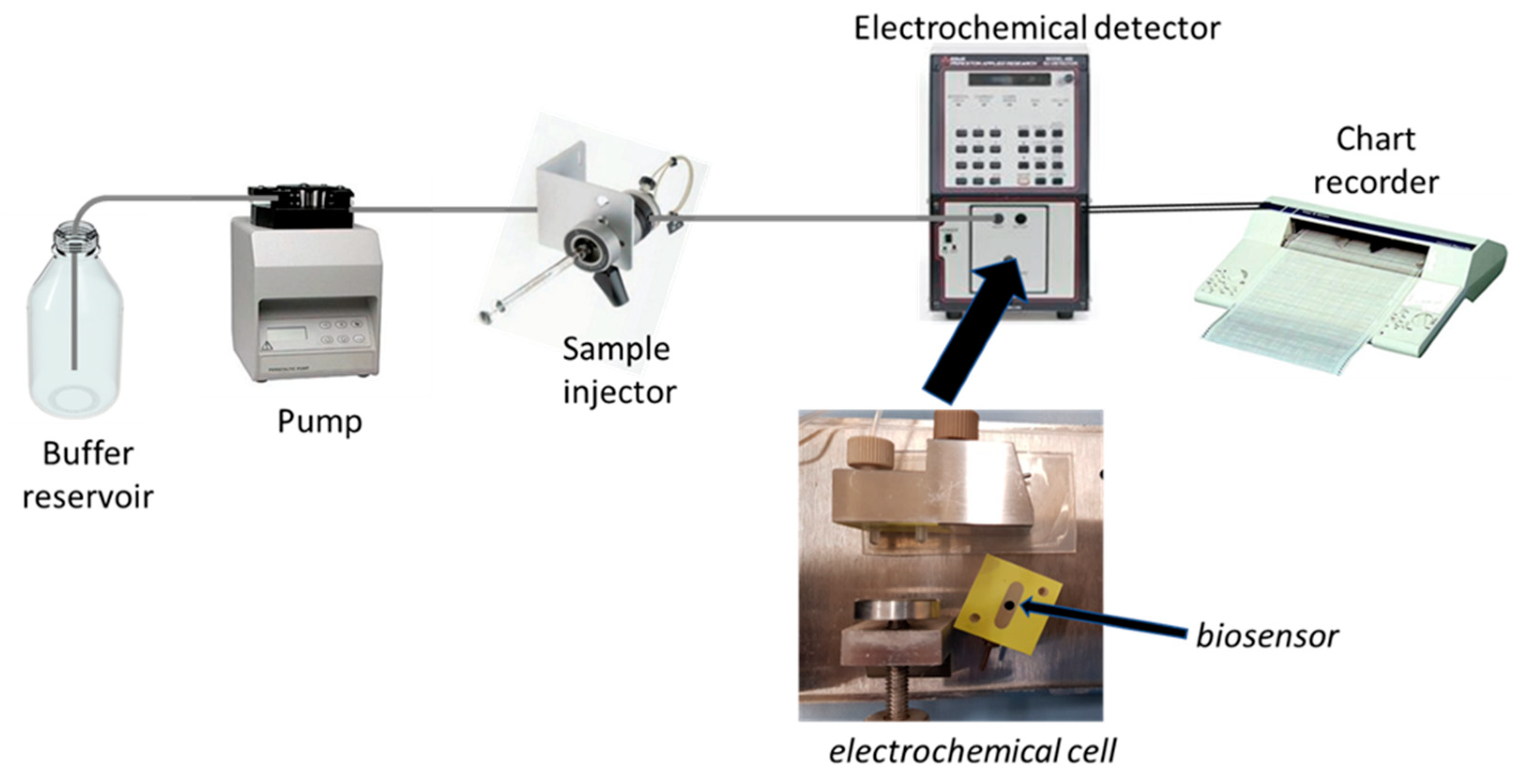
Accordingly, this procedure permitted us to realise an amperometric enzyme electrode showing high sensitivity and fast response time, with an enzyme layer so stable under stirring or flowing solutions to permit a flow injection analysis of L-lysine (Lys) sample, and thus, a kinetic study of the immobilised enzyme (see
Figure 1 for a schematic diagram of the flow injection setup used in the kinetic measurements). In fact, the flow injection system permitted here to study the kinetics of the immobilised enzyme in a simple straightforward way as demonstrated by
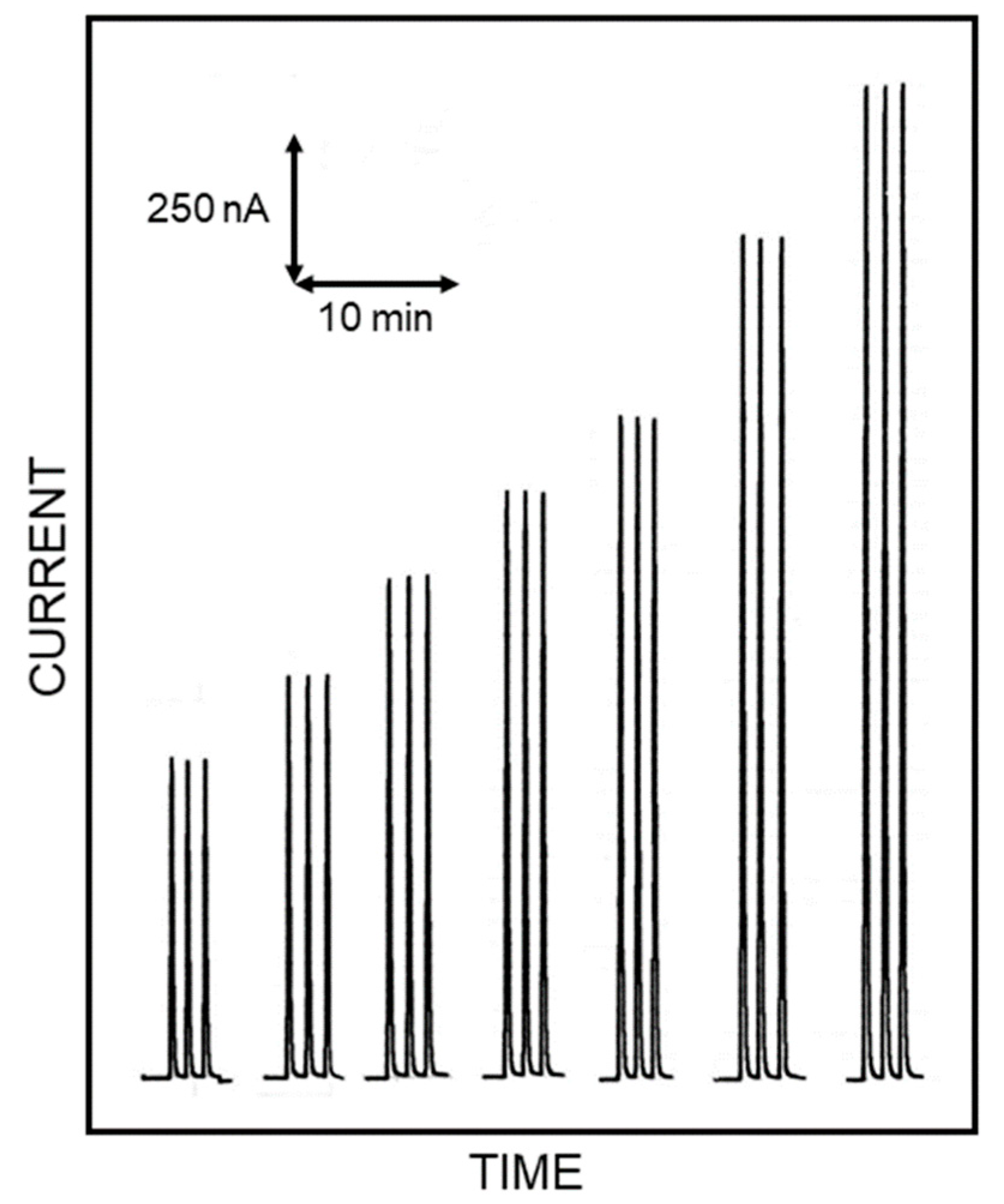
Figure 2, showing flow injection responses due to repeated injections of standard Lys solutions, acquired in optimal conditions for this study. Indeed, the responses were practically identical for each Lys level demonstrating the high repeatability of the present approach (within-a-day coefficients of variation for replicate (
n = 5) Lys injections were 0.92% and 1.35% at 4 mM and 0.2 mM Lys levels, respectively); the sensor-to-sensor repeatability was also tested by fabricating three different biosensors on different days. In the worst case scenario, a response deviation of 4.6% (at 1.25 mM Lys level) was observed, demonstrating the good repeatability of biosensor production. Moreover, the amperometric enzyme electrode here used showed a linear range of almost three decades [
13], permitting a limit of detection (at a signal to noise ratio of 3) as low as 1 µM in batch experiments [
12], whereas in flow injection analysis it was 4 µM [
13] (corresponding to a limit of quantification of 13 µM). Furthermore, fast responses were also observed due to the low response time of the biosensor here used [
12], which allowed a high sample throughput (less than 1 sample min
−1) and permitted us a complete kinetic study in a few minutes, thus minimising any undesired side effects due to, e.g., long-term variations of enzyme activity or substrate permeability in membrane. Not less important, the flow injection setup showed a rapid start-up time (typically less than minutes), no drift in signal measurements while allowing very low sample sizes (typically 20 µL) and low carrier consumption.
In this respect,
Scheme S1 in the
Supplementary Materials represents a figurative illustration of the amperometric enzyme electrode used in the kinetic measurements, pointing out the enzyme catalysis and the relevant mass flows involved (see ref. [
19] for a further deepening about). Here, Lys (
S) reacts with the enzyme LO (
E) immobilised in the enzyme membrane producing hydrogen peroxide (
P), which is easily detected (by electrooxidation) at the electrode surface and generates a current that is proportional to the amount of hydrogen peroxide produced by the enzyme. Naturally, this permitted us to follow the kinetic of LO in a simple way while the coupled flow injection setup allowed for easy and fast substrate sampling. With respect to the classic kinetic in solution, however, here, the enzyme kinetic is coupled (from left to right in
Scheme S1) to the electron transfer kinetics, diffusion of the substrate/product in membrane and to the mass transfer of the substrate supplying solution (for sake of simplicity, any partitioning or electrostatic effects coming from enzyme membrane have been here considered); of course, these mass transfers involve a substrate concentration gradient both in membrane and in solution which is roughly shown by shading in
Scheme S1. Finally, the effect of dioxygen on the LO enzymatic reaction was here neglected, since it has been demonstrated that its influence is reduced when dioxygen is supplied by the electrolyte solution and its diffusion is limited through the enzyme membrane [
20] as in the present case. Nevertheless, in the present study, air-saturated solutions were used both for carrier stream and standards to further limit any dioxygen dependence. Indeed, the highly within- and between-days repeatability (see above) indirectly confirmed the dioxygen independence.
The electrooxidation of hydrogen peroxide at the electrode surface is very fast, and thus never limiting; furthermore, it is always proportional to the hydrogen peroxide concentration, whatever its concentration or pH, thus, nonlinear behaviours due to electron transfer kinetics are unlikely. Accordingly, no build-up of
P in membrane is expected, and quickly after the substrate supply, a steady-state condition can be assumed, being limited by enzyme kinetics and/or substrate mass transfer. The “external” substrate mass transfer (i.e., that coming out from the substrate supplying solution) can be easily increased by increasing the flow rate in the flow injection measurements but reaches a limiting value due to the membrane thickness, since no convective flow is possible within the membrane [
21]; hence, in these studies, flow rate is an optimal tool to switch between an external diffusive control through solution (low flow rates) to a diffusive, limiting control through the membrane (high flow rates). Conversely, the “internal” substrate mass transfer (i.e., substrate diffusion in the enzyme entrapping membrane) is fixed depending on the substrate permeability of the used membrane, the latter of which can be limiting for fast enzyme kinetics or, vice versa, limited by enzyme kinetics for poor enzyme activity; since pH usually modulates the activity of the enzymes, changing the pH on the enzyme kinetic studies is another helpful tool to switch between enzyme or limiting diffusion kinetics. Please note that even under substrate mass transfer limiting conditions, the kinetic measurements reflect the catalytic enzyme behaviour since these limiting conditions simply involve an apparent dilution of substrate concentration.
3.2. pH Dependence of the Allosteric Behaviour of the Enzyme
The influence of pH on the activity of the LO enzyme as immobilised in the present study has been already reported elsewhere [
12,
13]. Briefly, the activity of the immobilised enzyme (studied in a pH range of 5–9.5 and measured as the sensitivity to the Lys response) increased the pH, reaching a maximum and nearly levelling off from pH 7.5. This behaviour agrees with that already reported for the native enzyme in solution [
2], indicating that the entrapped membrane and the ammonia enzymatic production and hydrogen peroxide electrooxidation in the membrane do not significantly influence the ionisation processes of both substrate and enzyme in membrane; furthermore, pre-incubation studies ruled out any potential changes in enzyme stability in that pH range [
12]. Remarkably, the dependence of the apparent Michaelis-Menten constant
K’
M with pH behaved similarly but in a specular fashion, i.e.,
K’
M decreased with pH till about pH 7 (from about 3.5 mM down to nearly 1.7 mM, while a 0.04 mM value was reported for the native enzyme [
2]), which then remained constant up to pH 9.5 [
12]. Lys shows its isoelectric point (p
I) at pH 9.7, while its p
Ka values are 2.2, 8.9 and 10.3; accordingly, the formation of a proper Lys ionic form to explain the observed enzyme activity increase in the pH range 5–7.5 should be ruled out, since the diprotic Lys form is already the main Lys specie in that pH range (see
Scheme S2 in
Supplementary Materials). On the other hand, both the pH behaviours previously described are in agreement with competitive inhibition kinetics [
22] by H
+ ions. After increasing the pH, the enzyme molecules are converted from the inactive, enzyme-inhibitor (E-H
+) complex, dead-end form to the full affinity form (E); thus, enzyme activity increases and
K’
M decreases. LO has its p
I at pH 4.35 [
2] so that the active site of LO loses a proton to bind Lys and catalyse its oxidation; due to the pH range in which the enzyme activity increase is observed, the deprotonation of the side chain of an histidine residue may be hypothesised here (p
Ka values 1.77, 6.10 and 9.18), considering that about 24 histidine residues have been found in LO [
2] and that histidine plays an active role in the mechanism of many enzyme reactions [
23]. Indeed, a histidine residue has been invoked in L-amino acid oxidase mechanism, acting as a base to catalyse proton removal from L-leucine [
24,
25].
As pointed out elsewhere [
15,
16], kinetics studies of LO from
Trichoderma viride and from
Trichoderma cf.
aureoviride Rifai VKM F-4268D showed a notable allosteric behaviour of these enzymes; in any case, those studies were performed at fixed pH values and the influence of pH on the cooperative behaviour of the enzyme was never explored. Accordingly, the pH influence on the enzyme kinetics was here studied using a Britton-Robinson universal buffer (i.e., acetate/phosphate/borate) at fixed ionic strength (0.1 M), to avoid any change in the ionic composition of the supporting electrolyte. As can be seen from
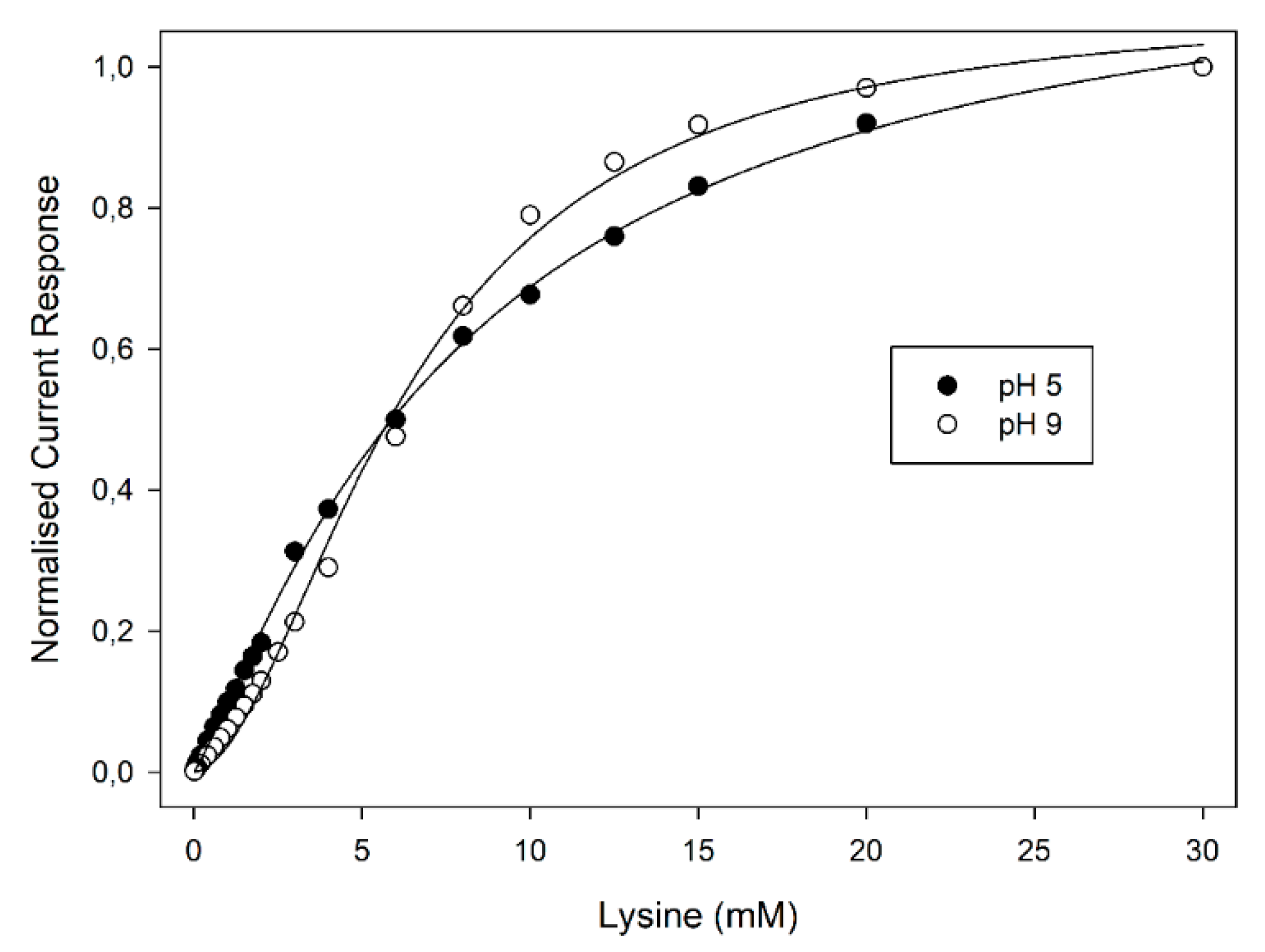
Figure 3, reporting the normalised current responses of the amperometric enzyme electrode vs. Lys concentration at two pH values for the sake of clarity, the plots display the well-known kinetic behaviour expected for enzyme catalysis (i.e., linear and saturated responses at low and high substrate concentrations, respectively). A critical inspection of
Figure 3 showed a significant deviation from the expected hyperbolic behaviour (i.e., Michaelis-Menten model) to a sigmoidal-like plot as the pH increased; furthermore, these deviations were particularly evident starting from about pH 7 and reached their maximal deviation at about pH 9. The sigmoidal behaviour of LO kinetics of course confirmed the allosteric behaviour of this enzyme. Indeed, as pointed out in the Introduction section, LO is a dimeric enzyme consisting of two identical subunits each one containing a FAD unit [
2,
4,
5,
6,
7,
8,
9,
10], thus, cooperative binding could arise; nevertheless, a pH dependence of the cooperation was never reported before and accordingly a deeper study was performed. Please note that the pH dependence on the allosteric behaviour of enzymes is not surprising, since it was already described in 1904 by Christian Bohr while studying the oxygen binding affinity of haemoglobin, a striking pH effect well-known as the Bohr effect (see for example ref. [
26] for some historical nods and the relevant modelling).
The modelling of an allosteric enzyme is rather intricate. Frequently, as a first approach, the Hill equation is used for the kinetics of such enzymes [
22]. In this case, it can be proven that for an allosteric enzyme with
n equivalent subunits (2 for the present enzyme), the velocity equation
v is:
where [
s] is the substrate concentration,
Vmax the maximum velocity and
K0.5 the substrate concentration at
Vmax/2 (which reduces to
KM, the Michaelis-Menten constant, when
n = 1). Please note that the Hill approach is strictly valid for high cooperativity [
22]: yet, where cooperativity is not so high, enzyme kinetics can be even described by Hill-type equations but the Hill coefficient,
n, corresponding to the number of substrate binding sites per enzyme molecule (2 in the present case) misses its physiochemical sense becoming an apparent Hill coefficient,
napp, still describing the cooperative degree between the active sites of the enzyme, but assuming non-integer values, usually less or at least approaching the actual (integer) number of sites present in the enzyme [
22]. Using the Hill model as the first approach in fitting the current responses observed at the amperometric enzyme electrode as a function of Lys concentration gave the
napp values reported in
Table 1 for different pH values and several flow rates. As can be seen,
napp, and hence, the cooperative behaviour of LO, increased with pH increase, starting from about pH 7, and reaching its maximal (expected) value at about pH 9. Naturally, as already pointed out above, the enzyme kinetic is complicated by substrate mass transfer; thus, discrimination between enzyme and mass transfer limitations is essential.
The influence of mass transfer on to the response of the present amperometric enzyme electrode has been already reported elsewhere [
12,
13]. As those studies showed, increasing the flow rate permitted to maximise the “external” substrate mass transfer until reaching (starting from flow rates higher than 1 mL min
−1) a limiting value due to the (finite) thickness of the membrane entrapping the enzyme; furthermore, the same studies showed that changing the pH (e.g., changing the activity of the immobilised enzyme) permitted to change from enzyme to “internal” substrate mass transfer limitations at every flow rates. Accordingly, further kinetic studies were performed at different flow rates, and the relevant current responses observed at the amperometric enzyme electrode as a function of Lys concentration were fitted using the Hill model. As
Table 1 shows, the
napp observed at several flow rates but at a fixed pH values were all nearly the same, demonstrating that the cooperative behaviour observed for LO and its pH dependence was not due to mass transfer complications but simply due to enzyme kinetics. The observed non-dependence of
napp from the flow rate is not unexpected; indeed, mass transfer limitations usually involves variations in
K’
M and the apparent maximum velocity
V’max in immobilised enzyme systems, due to the dilutions effects raising from those limitations in membrane [
19]; on the contrary,
napp would denote the “equation form” used to describe the enzyme kinetics, i.e., the kinetic mechanisms involved in enzyme catalysis, which are of course independent on substrate supply.
3.3. Application of Monod-Wyman-Changeux model
As is well known, the Hill velocity equation can be linearised by converting it on its logarithmic form [
22], the well-known Hill plot:
As pointed out in many studies, the Hill plot is not a straight line for many allosteric enzymes [
26]; apart from demonstrating the simplicity (and weakness) of the Hill model, those deviations from the expected linearity show that, usually, cooperativity binding in an enzyme is not fixed but depends on the saturation of the enzyme, i.e., from substrate concentration, that is, the
n parameter is substrate concentration dependent.
The first kinetics studies evidencing the allosteric behaviour of LO [
15,
16] used the Hill equation to demonstrate the departure from the classical Michaelis-Menten approach and calculate the relevant Michaelis-Menten constant. Even in the present study, the Hill equation was preliminary used to show the dependence of the cooperativity on pH; a closer inspection of data compared to those reported in
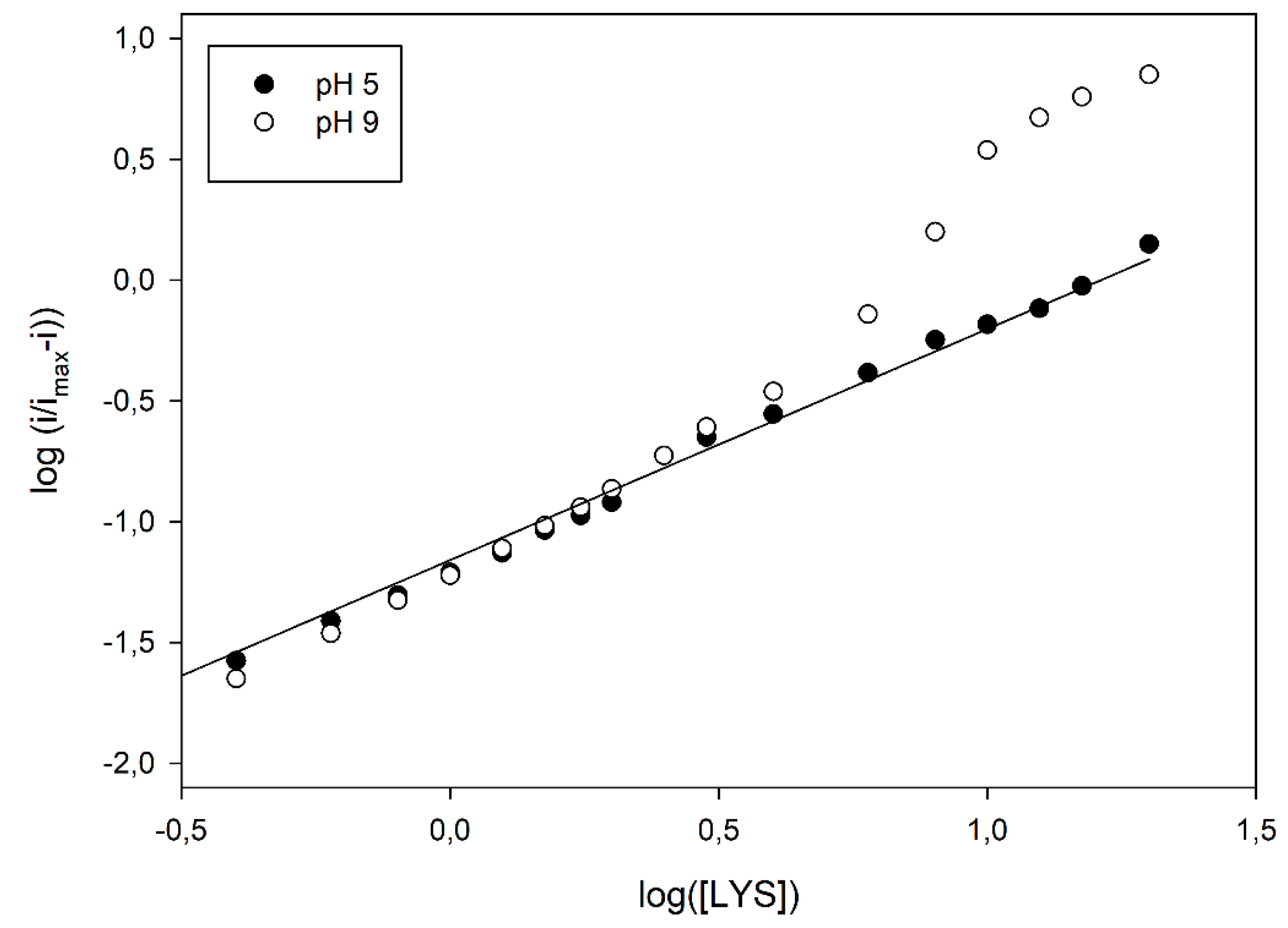
Figure 3 already evidenced a departure from the simple sigmoidal fitting. Indeed, conversion of kinetic data in the relevant logarithmic Hill plots displayed a significant nonlinear behaviour, at higher pH values, as shown in
Figure 4; similar deviations were also observed at different flow rates (see
Figure S1 in
Supplementary Materials), demonstrating once again that the enzyme kinetics and the relevant departures from the Michaelis-Menten model were not artefacts originating from mass transfer limitations. In particular, as shown in
Figure 4, a linear Hill plot was observed at pH 5, of which the slope (0.96 ± 0.02, correlation coefficient better than 0.998, three replicates, 17 data points fitted) agreed with the Michaelis-Menten model, as already pointed out in
Table 1; on the contrary, striking deviations from linearity were evident from about pH 7, which reached their maximal deviations at about pH 9, where a skewed sigmoidal plot was observed.
From the initial, phenomenological application of Hill equation, several different approaches and models were developed to describe the cooperativity in enzyme kinetics, in an attempt to offer a biochemical vision of the underlying mechanism (see ref. [
26] for a review of the relevant approaches developed). In particular, one of the most recent and best approaches describing cooperativity was proposed by Monod et al. [
27,
28], known as the Monod-Wyman-Changeux (MWC) model. Briefly, in this approach, the allosteric enzyme is modelled as two (or more) interconvertible conformational states, namely the tense (
T) and relaxed (
R) states, coexisting in a thermal equilibrium and differing in their affinity for the substrate. The binding of ligand molecules stabilises the higher affinity state, controls the ratio between the two states and induces a change of all enzyme subunits states at the same time, a phenomenon known as “concerted transition”. If the ligand dissociation constants for the
T and
R states are
and
, respectively, their ratio
c =
/
offers a suggestion of the difference of substrate affinities for the two states. Of course, if
c = 1, the affinities are the same and the MWC model simplifies to the Michaelis-Menten model; on the contrary, if
c is less than unity, the substrate shows much more affinity towards the relaxed
R state and, therefore, the equilibrium between the two states shifts towards the
R state after one substrate binding.
According to the MWC model, the skewed sigmoidal plots here observed at the higher pH values (as in
Figure 4) can be viewed as a progressive transition from the tense, low affinity
T state to the relaxed, high affinity
R state as the saturation of the enzyme, i.e., the substrate concentration, increases. In particular, by analysing the Hill plot at pH 9 (see
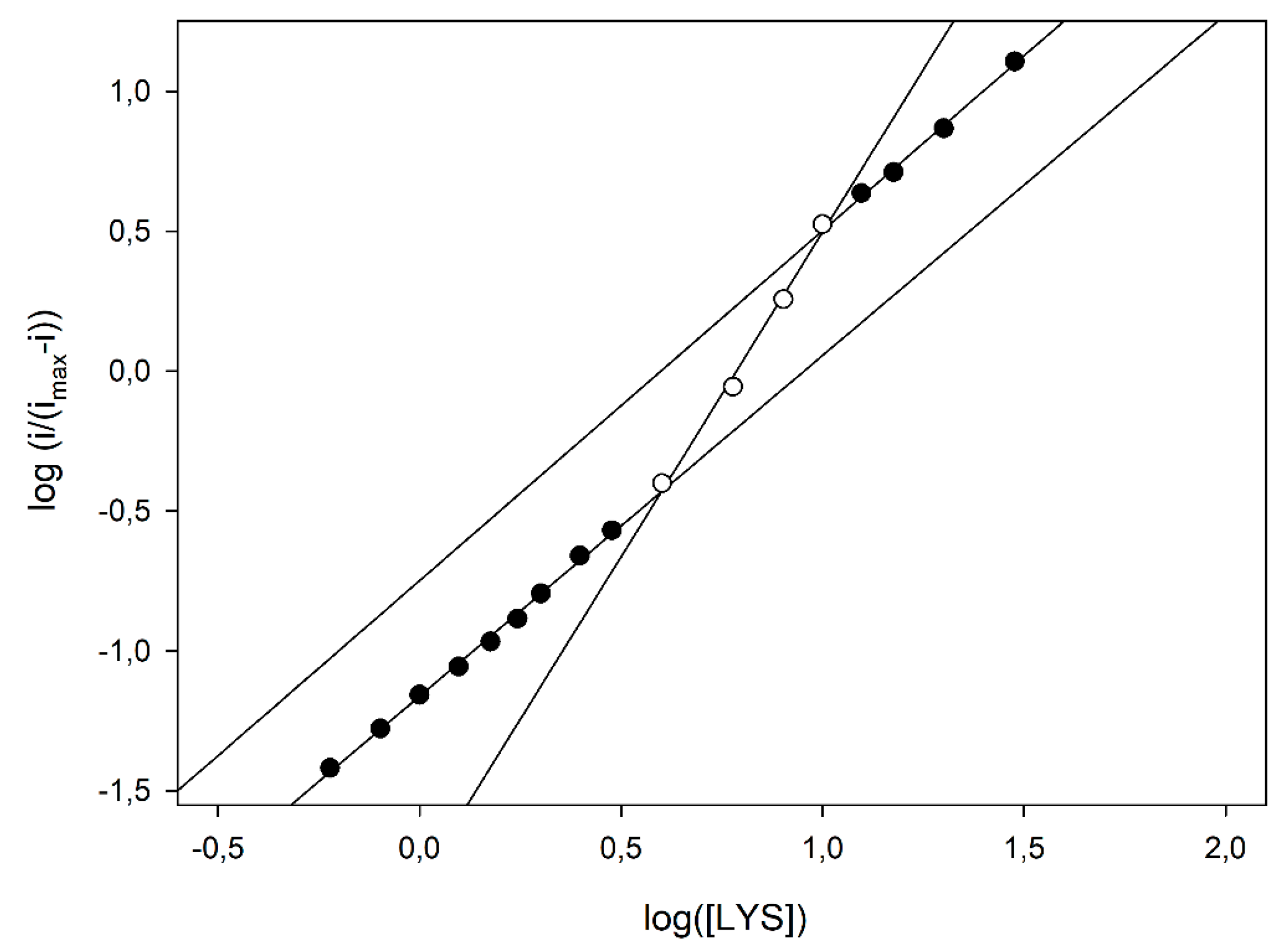
Figure 5), some kinetic data can be inferred using the MWC approach. In fact, the apparent Hill coefficient,
napp, can be estimated by the slope of the curve and, as Figure shows, it starts from an initial value of 1.20 ± 0.03 (left, low substrate concentrations, linear branch, three replicates, nine data points fitted), reaches its maximal value of 2.32 ± 0.13 at the inflexion point, i.e., at about half enzyme saturation (central linear branch, three replicates, four data points fitted) and decreases again to a low value of 1.25 ± 0.05 (right, high substrate concentrations, linear branch, three replicates, four data points fitted). Of course, this is a clear indication that, in the LO enzyme, cooperativity is Lys concentration dependent, since the maximum is at about half enzyme saturation and the minimum is at low and high saturations; accordingly, the
napp values listed in
Table 1, obtained by the simple Hill fitting, could represent a weighed mean value of cooperativity, since the Hill approach does not consider any substrate concentration dependence on cooperativity. Furthermore, the intercept of two asymptotes of the skewed sigmoidal plot (left and right linear branches) and the
y axis in
Figure 5 can permit to estimate the logs of
and
constants, respectively, and thus, the parameter
c. By means of a linear fitting (correlation coefficients better than 0.996), logs of
and
were −1.162 ± 0.006 (three replicates, nine data points fitted, equivalent to about 69 µM) and −1.82 ± 0.11 (three replicates, four data points fitted, equivalent to about 15 µM), respectively, and the ratio
c about 0.22, demonstrating that by increasing the LO saturation (i.e., the Lys concentration), the equilibrium between the two states shifted towards the relaxed, high affinity
R state.
According to this point of view, catalytic schemes of LO reactions, such as those pointed out for LO from
Trichoderma cf.
aureoviride Rifai VKM F-4268D [
16], can be hardly invoked in the present case, since they do not rely with any pH dependence of cooperativity, as is demonstrated in the present study. Since the MWC approach appears to be the best model describing the observed kinetics of LO from
Trichoderma viride, some considerations and hypotheses need here to be postulated to tentatively explain the presence of two interconvertible conformational states of LO (the tense
T and relaxed
R states), their difference in affinity for Lys and their dependence on pH. In this respect, many studies on the oxidation of amines by flavoproteins pointed out that the enzyme mechanism currently accepted involves the removal of a proton from the α-carbon of substrate by a base (the here supposed histidine residue in the present case) and the concerted hydride transfer from the neutral α-amino group of substrate to the FAD group of the oxidised enzyme [
25,
29]; naturally, if the α-amino group of substrate is in its protonated, acid form
, the relevant hydrogen transfer requires deprotonation, probably mediated by water molecules filling activity site of the enzyme, as they were found in the structure of a bacterial L-amino acid oxidase from
Rhodococcus opacus [
30]. Under this light, the enzyme–substrate complexes with the protonated or neutral α-amino group substrate might differ in their conformational states and affinity for the substrate, originating the cooperativity of LO. To corroborate this point of view and, more importantly, the pH dependence of cooperativity, the distribution diagram of Lys species with pH might help (see
Scheme S2 in
Supplementary Materials). As can be seen, in the pH range 7–9 (i.e., in the pH range where cooperativity was observed) the diprotic Lys form decreases with pH while the monoprotic form increases; if there is an affinity towards the latter, the neutral α-amino group form is higher with respect to the charged, diprotic form, the pH dependence of cooperativity can be explained in a first instance by the increase of this highly affinity form with pH.
3.4. Influence of LO Allostery and Its pH Dependence on Biosensor Performance
Allosteric enzymes appear quite interesting in principle since modulation by effectors or inhibitors on enzyme catalysis are clearly anticipated and can be strategically used for biosensor production [
31]. Even in cases where substrate and effector/inhibitor are the same molecule (as in the present case), allostery can be advantageous. Single-site binding enzymes (i.e., Michaelis-Menten kinetics) are characterised by a hyperbolic relationship where the dynamic range is fixed (
napp = 1); on the contrary, allosteric enzymes or a network of enzymes with different affinities toward the same substrate, due to their different, sigmoidal-like kinetics, can be useful used to extend (
napp < 1) or narrow (
napp > 1) the dynamic range of related biosensors [
32]. This should extend the usefulness of electrochemical biosensors in applications where the concentration of the analyte can vary over many orders of magnitude or, vice versa, where monitoring of the analyte within a narrow concentration window is required and high sensitivity and a steep relationship are preferred. As
Figure 3 shows, in the present case, LO allostery and its pH dependence can be advantageous for controlling the sensitivity and the dynamic range of LO-based biosensors, demonstrating that the working pH has a powerful influence on kinetics and hence on biosensor operation; at the same time, pH also controls the selectivity of Lys assay [
13], thus, care should be applied in pH optimisation.
Finally, it is worth of note that allostery could be also useful for generating sharp, all-or-none responses (
napp much more than 1), i.e., in enzyme “logical gate” applications where it is necessary e.g., to discriminate between physiological and pathological levels [
33]. Unfortunately, LO displays a maximum
napp = 2, thus, sharper responses are unlikely; in any case, coupling LO with another Lys enzyme displaying different affinity towards Lys (e.g., lysine 2-monooxygenase) could permit steeper input/output responses. This approach could be quite useful in the production of extracorporeal reactor devices to be used to remove pathological Lys levels.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}