Interaction Mechanisms of Insensitive Explosive FOX-7 and Graphene Oxides from Ab Initio Calculations


Abstract
:1. Introduction
2. Computational Methods and Models
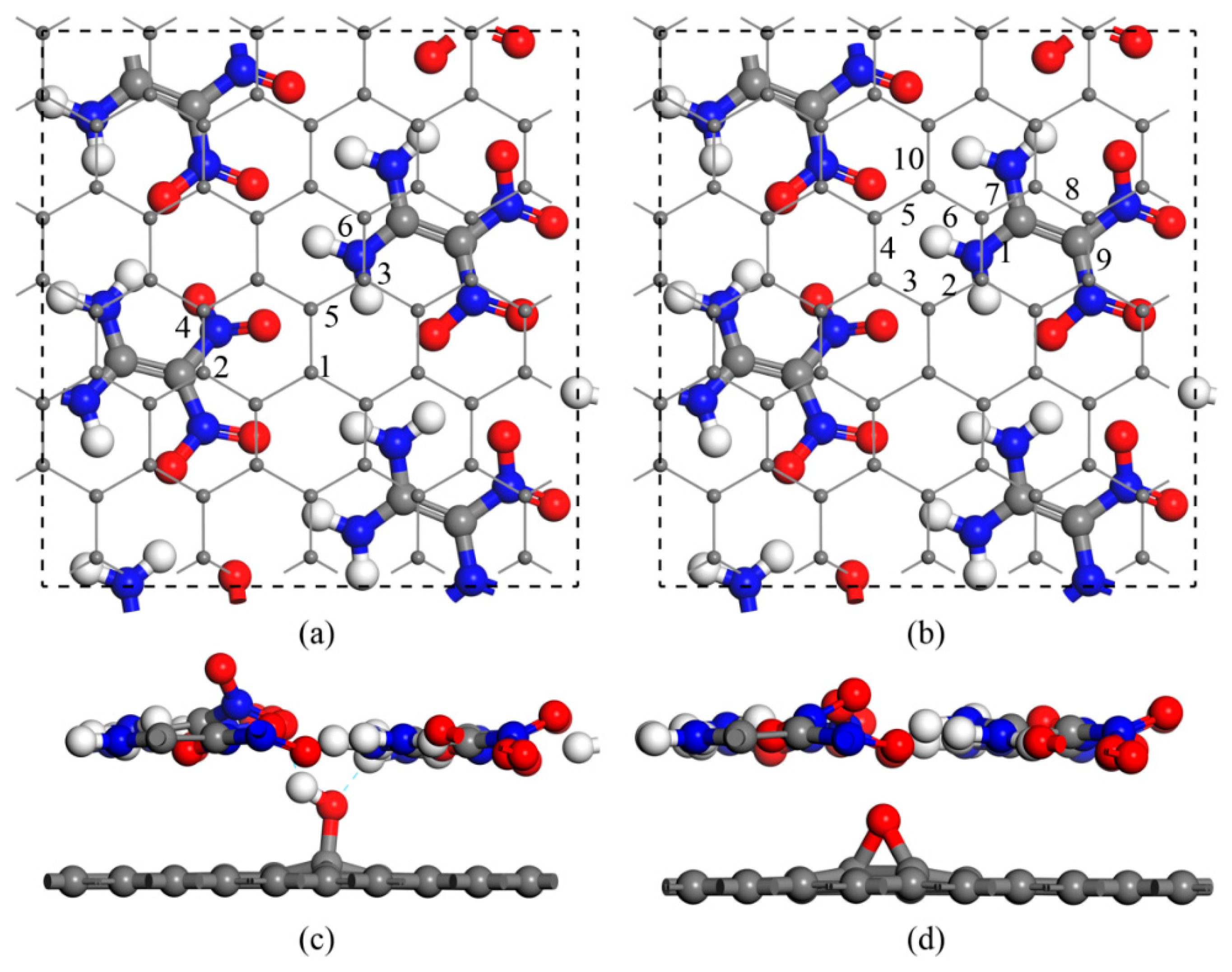
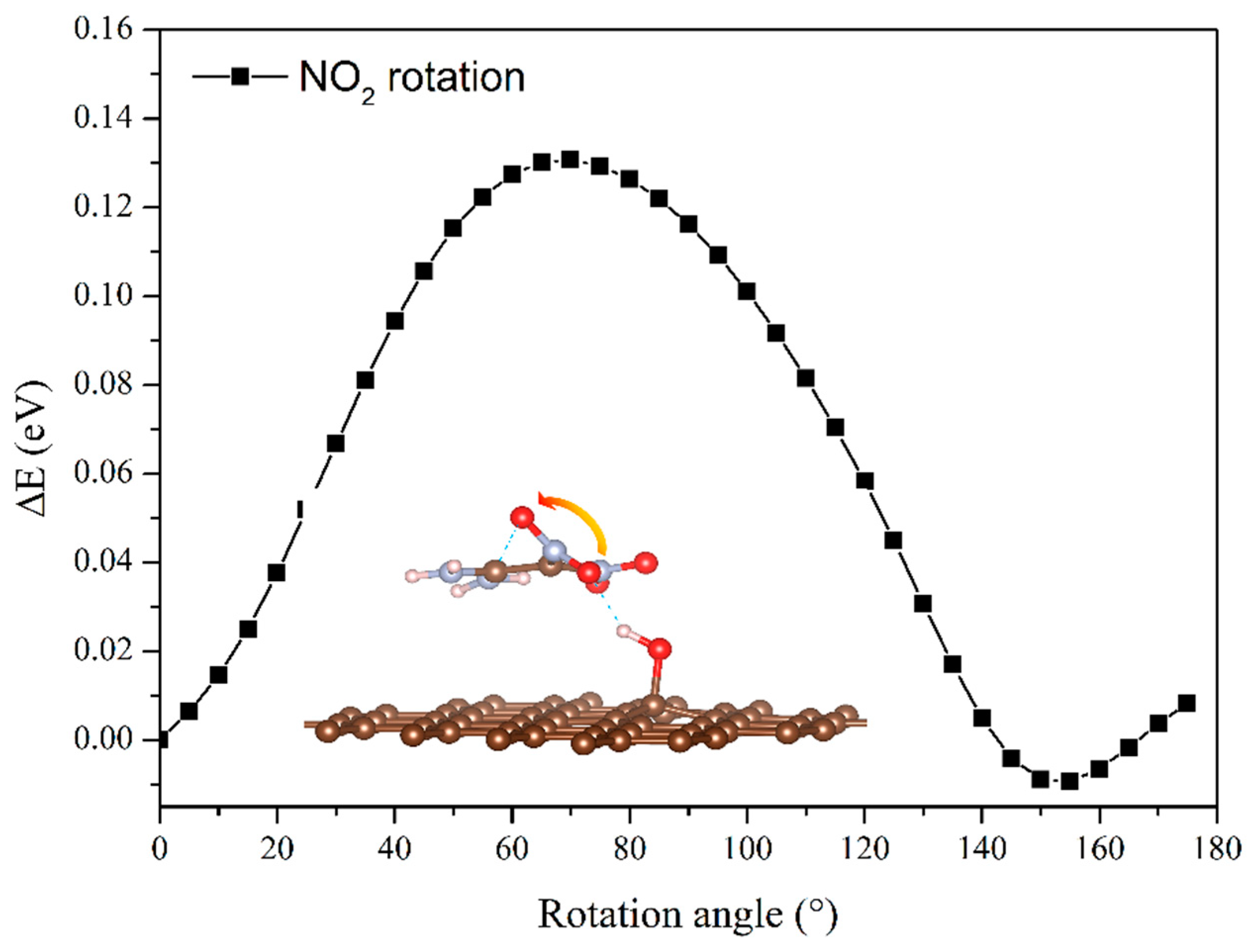
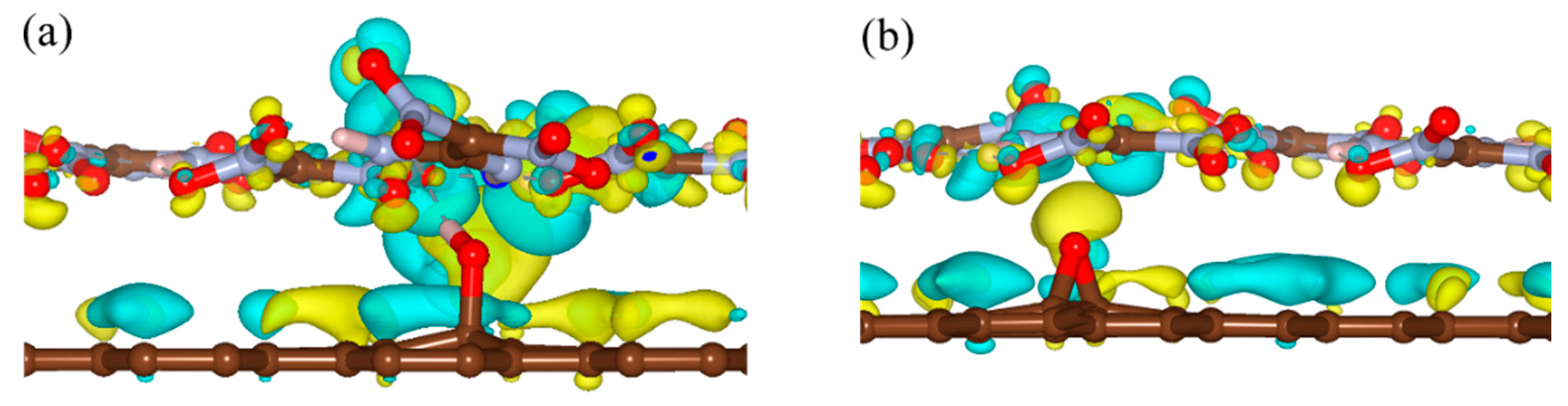
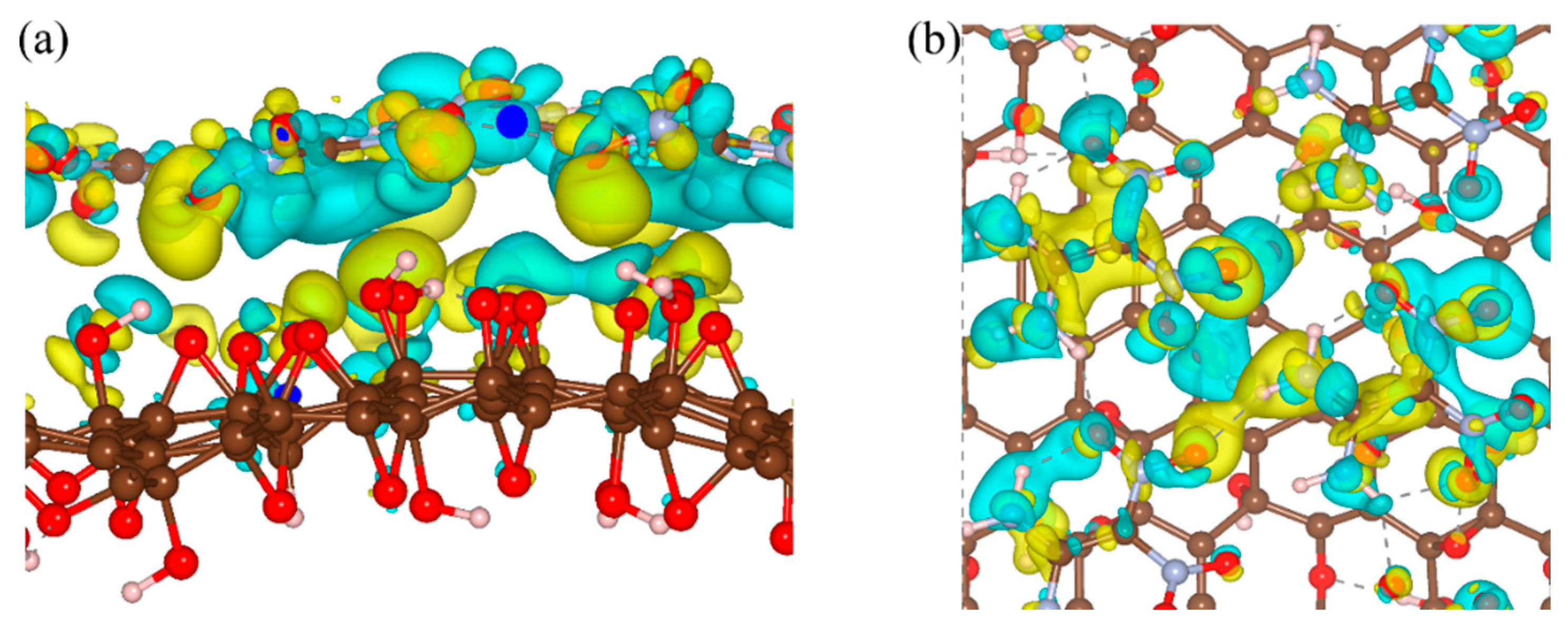
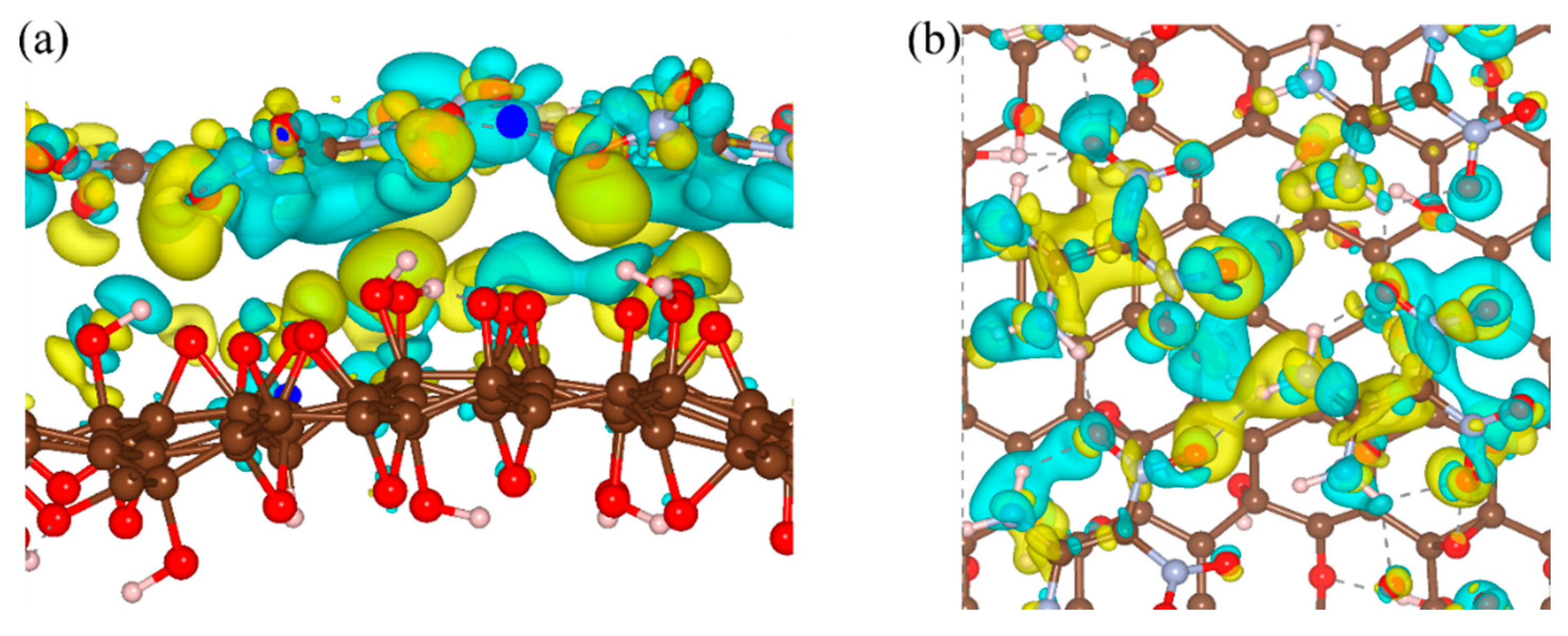
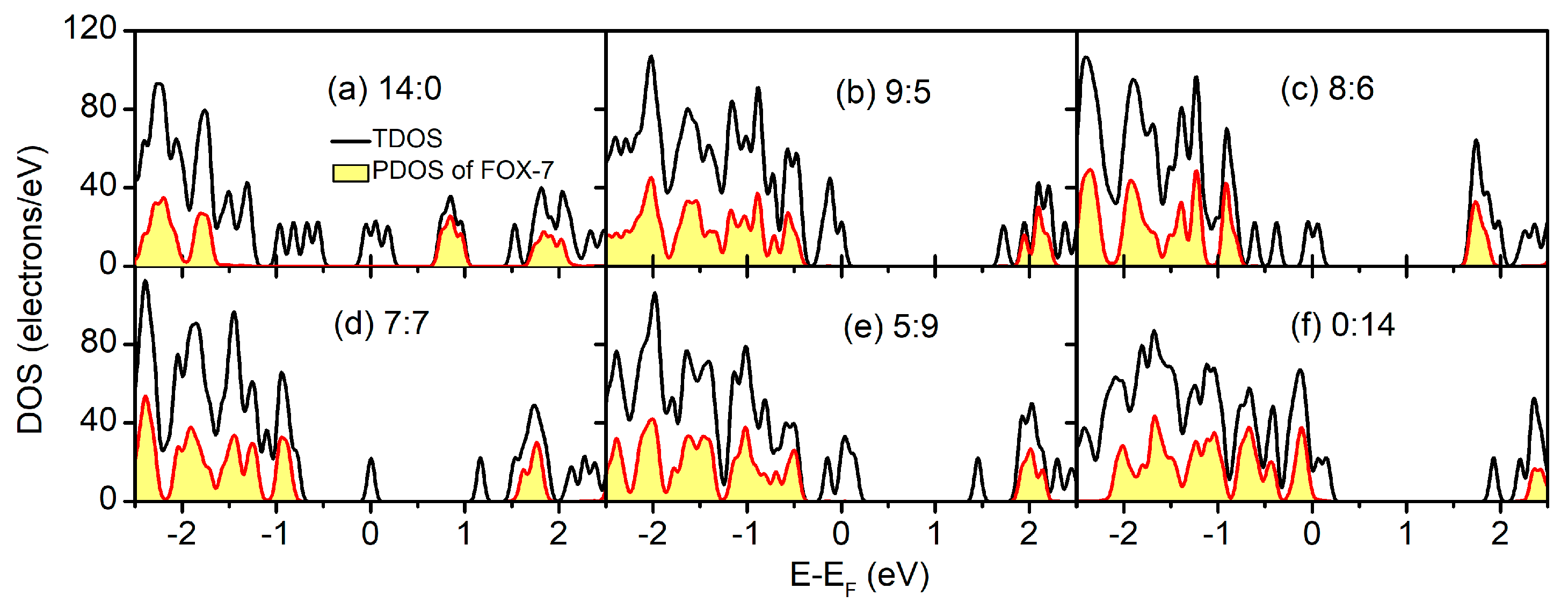
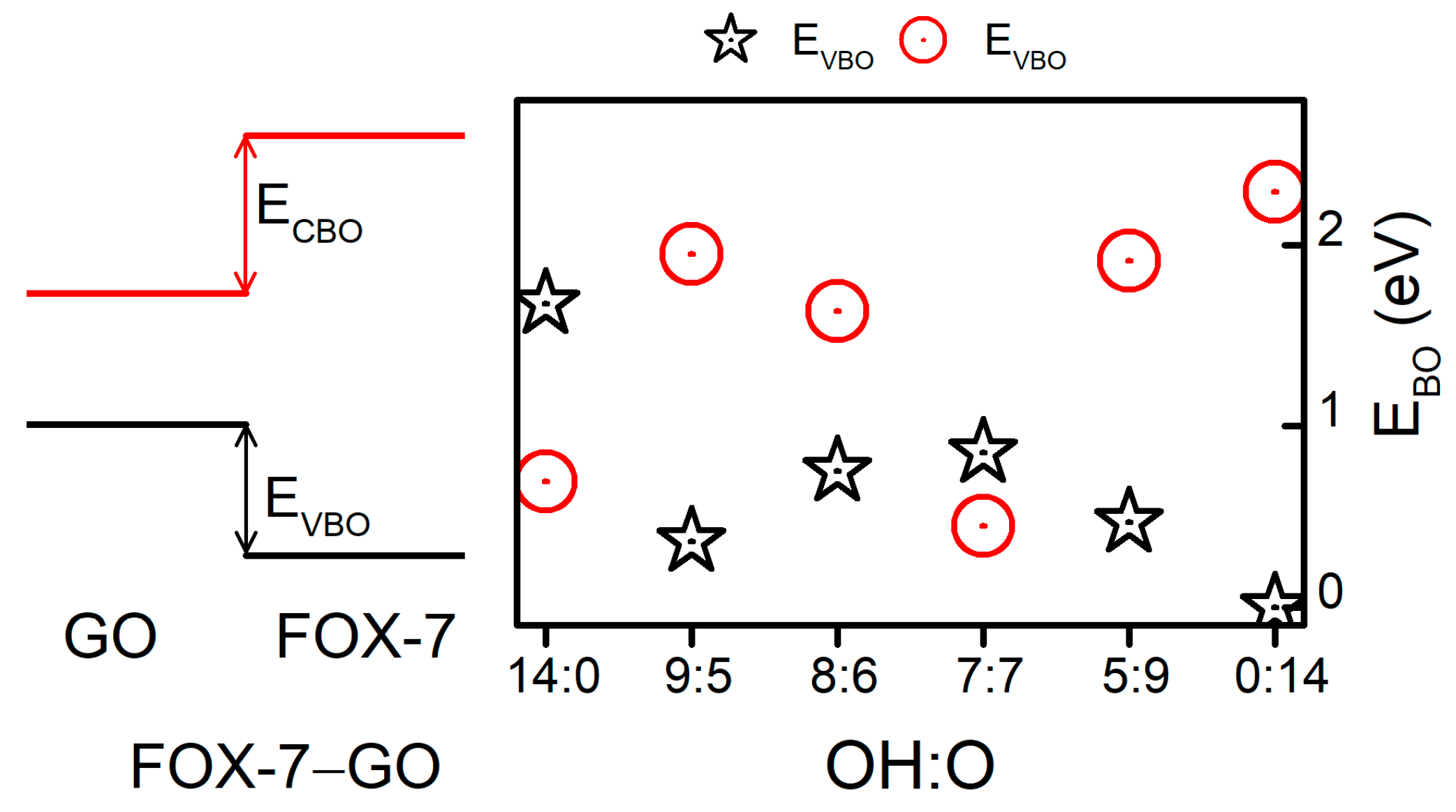
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sikder, A.K.; Maddala, G.; Agrawal, J.P.; Singh, H. Important aspects of behaviour of organic energetic compounds: A review. J. Hazard. Mater. 2001, 84, 1–26. [Google Scholar] [CrossRef]
- Badgujar, D.M.; Talawar, M.B.; Asthana, S.N.; Mahulikar, P.P. Advances in science and technology of modern energetic materials: An overview. J. Hazard. Mater. 2008, 151, 289–305. [Google Scholar] [CrossRef]
- van der Heijden, A.E.D.M.; Bouma, R.H.B. Crystallization and Characterization of RDX, HMX, and CL-20. Cryst. Growth Des. 2004, 4, 999–1007. [Google Scholar] [CrossRef]
- Jiao, F.B.; Xiong, Y.; Li, H.Z.; Zhang, C.Y. Alleviating the Energy & Safety Contradiction to Construct New Low Sensitive and Highly Energetic Materials through Crystal Engineering. CrystEngComm. 2018, 20, 1757–1768. [Google Scholar]
- Storm, C.B.; Stine, J.R.; Kramer, J.F. Sensitivity Relationships in Energetic Materials. In Chemistry and Physics of Energetic Materials; Springer: Dordrecht, The Netherlands, 1990; pp. 605–639. [Google Scholar] [Green Version]
- Mathieu, D. Sensitivity of Energetic Materials: Theoretical Relationships to Detonation Performance and Molecular Structure. Ind. Eng. Chem. Res. 2017, 56, 8191–8201. [Google Scholar] [CrossRef]
- Cowey, K.; Day, S.; Fryer, R. Examination of Wax-Coated RDX by Scanning Electron Microscopy and X-ray Photoelectron Spectroscopy. Propellants Explos. Pyrotech. 1985, 10, 61–64. [Google Scholar] [CrossRef]
- da Costa Mattos, E.; Moreira, E.D.; Diniz, M.F.; Dutra, R.C.L.; da Silva, G.; Iha, K.; Teipel, U. Characterization of Polymer-Coated RDX and HMX Particles. Propellants Explos. Pyrotech. 2008, 33, 44–50. [Google Scholar] [CrossRef]
- Ringbloom, V.D. Process for Coating Crystalline Explosives with Polyethylene Wax. U.S. Patent 4357185A, 2 November 1982. [Google Scholar]
- Jin, B.; Peng, R.F.; Chu, S.J.; Huang, Y.M.; Wang, R. Study of the Desensitizing Effect of Different [60] Fullerene Crystals on Cyclotetramethylenetetranitramine (HMX). Propellants Explos. Pyrotech. 2008, 33, 454–458. [Google Scholar] [CrossRef]
- Niu, C.H.; Jin, B.; Peng, R.F.; Shang, Y.; Liu, Q.Q. Preparation and characterization of insensitive HMX/rGO/G composites via in situ reduction of graphene oxide. RSC Adv. 2017, 7, 32275–32281. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Wang, J.; Shen, J.P.; Hua, C.; Yang, G.C. Preparation and Characterization of Insensitive HMX/Graphene Oxide Composites. Propellants Explos. Pyrotech. 2013, 38, 798–804. [Google Scholar] [CrossRef]
- Liu, T.; Geng, C.Z.; Zheng, B.H.; Li, S.B.; Luo, G. Encapsulation of Cyclotetramethylenetetranitramine (HMX) by Electrostatically Self-Assembled Graphene Oxide for Desensitization. Propellants Explos. Pyrotech. 2017, 42, 1057–1065. [Google Scholar] [CrossRef]
- Wang, J.F.; Chen, S.S.; Yao, Q.; Jin, S.H.; Zhao, S.W.; Yu, Z.F.; Li, J.X.; Shu, Q.H. Preparation, Characterization, Thermal Evaluation and Sensitivities of TKX-50/GO Composite. Propellants Explos. Pyrotech. 2017, 42, 1104–1110. [Google Scholar] [CrossRef]
- Zhang, X.; Hikal, W.M.; Zhang, Y.; Bhattacharia, S.K.; Li, L.; Panditrao, S.; Wang, S.R.; Weeks, B.L. Direct laser initiation and improved thermal stability of nitrocellulose/graphene oxide nanocomposites. Appl. Phys. Lett. 2013, 102, 141905. [Google Scholar] [CrossRef]
- Jiang, Y.; Deng, S.L.; Hong, S.; Zhao, J.H.; Huang, S.D.; Wu, C.; Gottfried, J.L.; Nomura, K.; Li, Y.; Tiwari, S.; et al. Energetic Performance of Optically Activated Aluminum/Graphene Oxide Composites. ACS Nano 2018, 12, 11366–11375. [Google Scholar] [CrossRef]
- Yan, Q.L.; Cohen, A.; Petrutik, N.; Shlomovich, A.; Burstein, L.; Pang, S.P.; Gozin, M. Highly insensitive and thermostable energetic coordination nanomaterials based on functionalized graphene oxides. J. Mater. Chem. A 2016, 4, 9941–9948. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.; Yang, Y.; Yan, Q.L.; Shlomovich, A.; Petrutik, N.; Burstein, L.; Pang, S.P.; Gozin, M. Highly Thermostable and Insensitive Energetic Hybrid Coordination Polymers Based on Graphene Oxide−Cu(II) Complex. Chem. Mater. 2016, 28, 6118–6126. [Google Scholar] [CrossRef]
- Smeu, M.; Zahid, F.; Ji, W.; Guo, H.; Jaidann, M.; Abou-Rachid, H. Energetic Molecules Encapsulated Inside Carbon Nanotubes and between Graphene Layers: DFT Calculations. J. Phys. Chem. C 2011, 115, 10985–10989. [Google Scholar] [CrossRef]
- Zhang, C.M.; Fu, X.L.; Li, J.Z.; Fan, X.Z.; Zhang, G.F. Desensitizing Effect of Graphene Oxide on Thermolysis Mechanisms of 4,4’-Azo-1,2,4-triazole Studied by Reactive Molecular Dynamics Simulations. J. Phys. Chem. A 2019, 123, 1285–1294. [Google Scholar] [CrossRef]
- Liu, L.M.; Car, R.; Selloni, A.; Dabbs, D.M.; Aksay, I.A.; Yetter, R.A. Enhanced Thermal Decomposition of Nitromethane on Functionalized Graphene Sheets: Ab Initio Molecular Dynamics Simulations. J. Am. Chem. Soc. 2012, 134, 19011–19016. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, J.C.; Jeon, W.C.; Cho, S.G.; Kwak, S.K. Explosion Study of Nitromethane Confined in Carbon Nanotube Nanocontainer via Reactive Molecular Dynamics. J. Phys. Chem. C 2017, 121, 6415–6423. [Google Scholar] [CrossRef]
- Trzciński, W.A.; Cudziło, S.; Chyłek, Z.; Szymańczyk, L. Detonation properties of 1,1-diamino-2,2-dinitroethene (DADNE). J. Hazard. Mater. 2008, 157, 605–612. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Lerf, A.; He, H.; Forster, M.; Klinowski, J. Structure of Graphite Oxide Revisited. J. Phys. Chem. B 1998, 102, 4477–4482. [Google Scholar] [CrossRef]
- Liu, L.Z.; Wang, L.; Gao, J.F.; Zhao, J.J.; Gao, X.F.; Chen, Z.F. Amorphous Structural Models for Graphene Oxides. Carbon 2012, 50, 1690–1698. [Google Scholar] [CrossRef]
- Evers, J.; Klapötke, T.M.; Mayer, P.; Oehlinger, G.; Welch, J. α- and β-FOX-7, Polymorphs of a High Energy Density Material, Studied by X-ray Single Crystal and Powder Investigations in the Temperature Range from 200 to 423 K. Inorg. Chem. 2006, 45, 4996–5007. [Google Scholar] [CrossRef]
- Zhao, J.J.; Liu, L.Z.; Li, F. Graphene Oxide: Physics and Applications; Springer: London, UK, 2015. [Google Scholar]
- Zhu, W.H.; Xiao, H.M. First-principles band gap criterion for impact sensitivity of energetic crystals: A review. Struct. Chem. 2010, 21, 657–665. [Google Scholar] [CrossRef]
- Zhao, J.J.; Liu, H. High-pressure behavior of crystalline FOX-7 by density functional theory calculations. Comput. Mater. Sci. 2008, 42, 698–703. [Google Scholar] [CrossRef]
- Appalakondaiah, S.; Vaitheeswaran, G.; Lebègue, S.J. Structural, vibrational, and quasiparticle band structure of 1,1-diamino-2,2-dinitroethelene from ab initio calculations. J. Chem. Phys. 2014, 140, 014105. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.F.; Xie, W.Y.; Zhao, J.J.; Zhang, S.B. Band alignment of two-dimensional lateral heterostructures. 2D Mater. 2017, 4, 015038. [Google Scholar] [CrossRef]
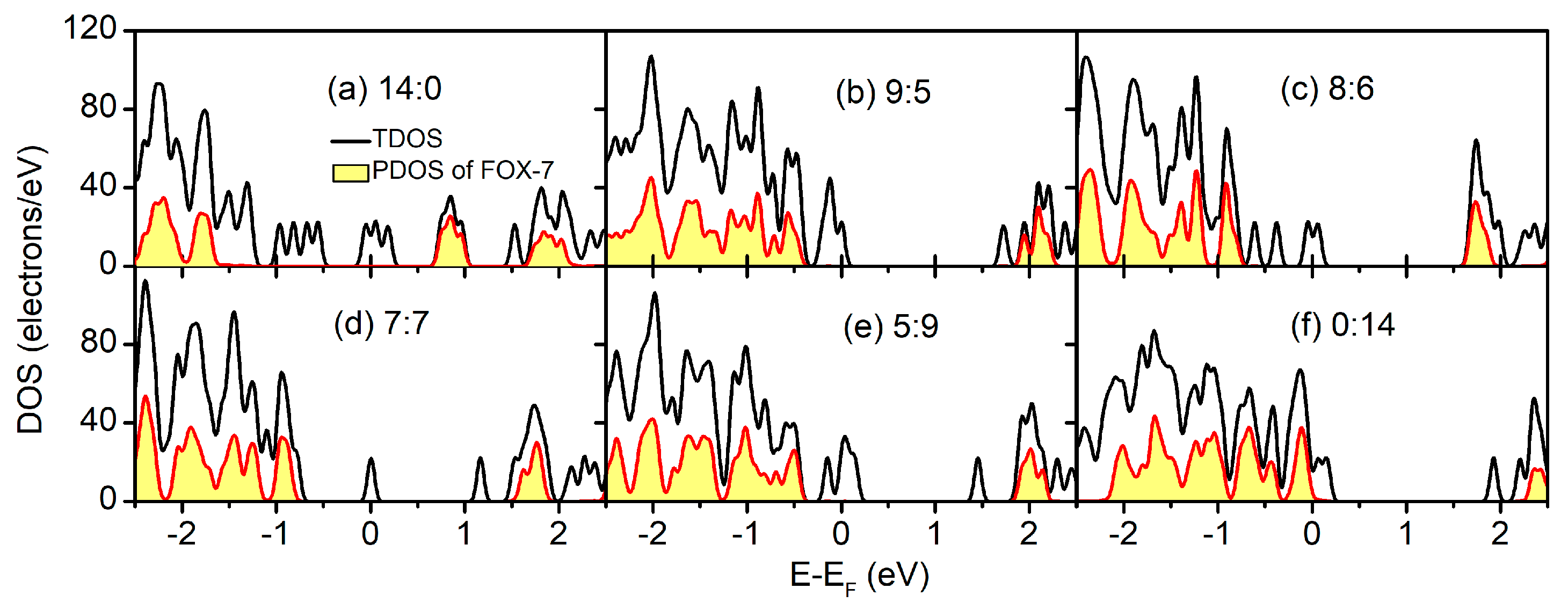
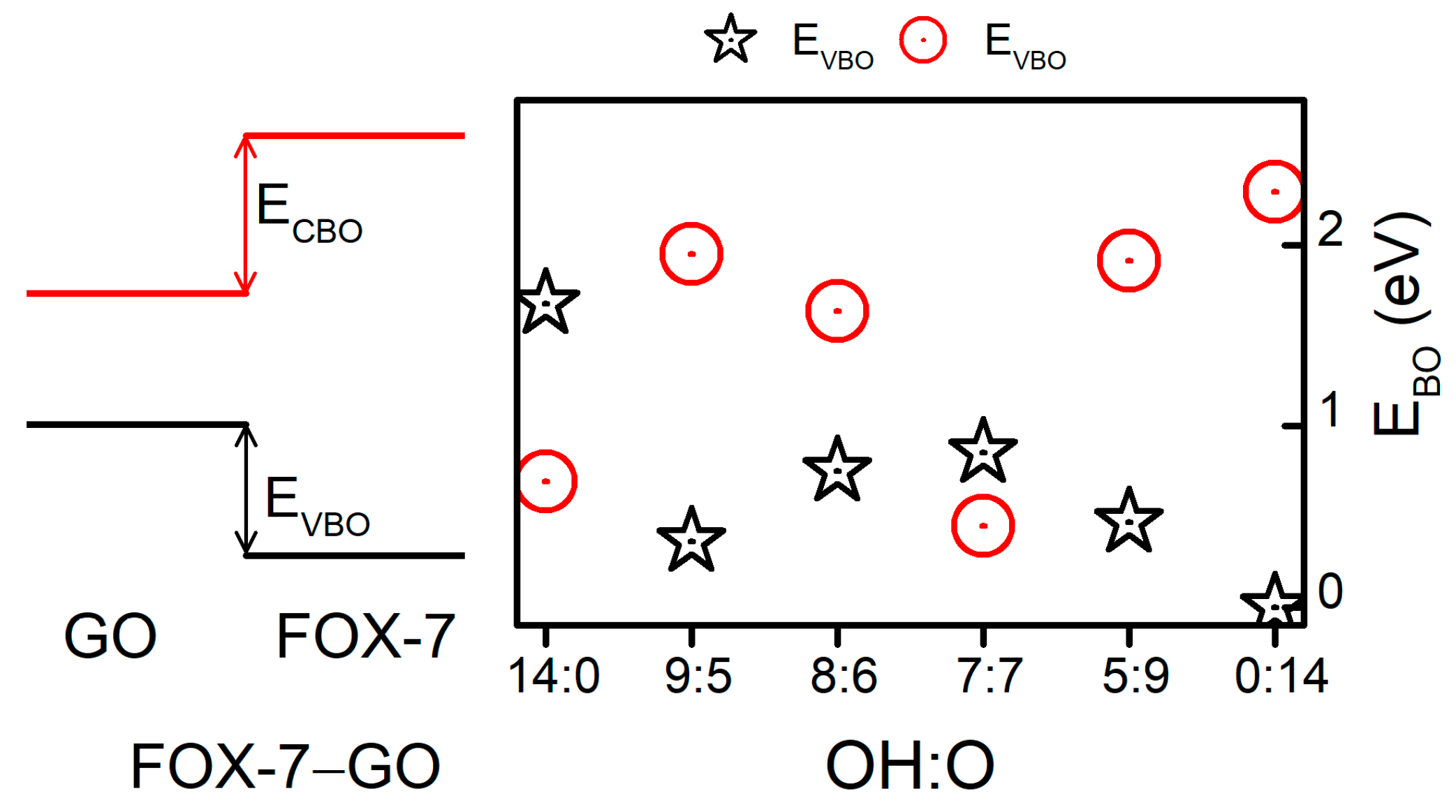

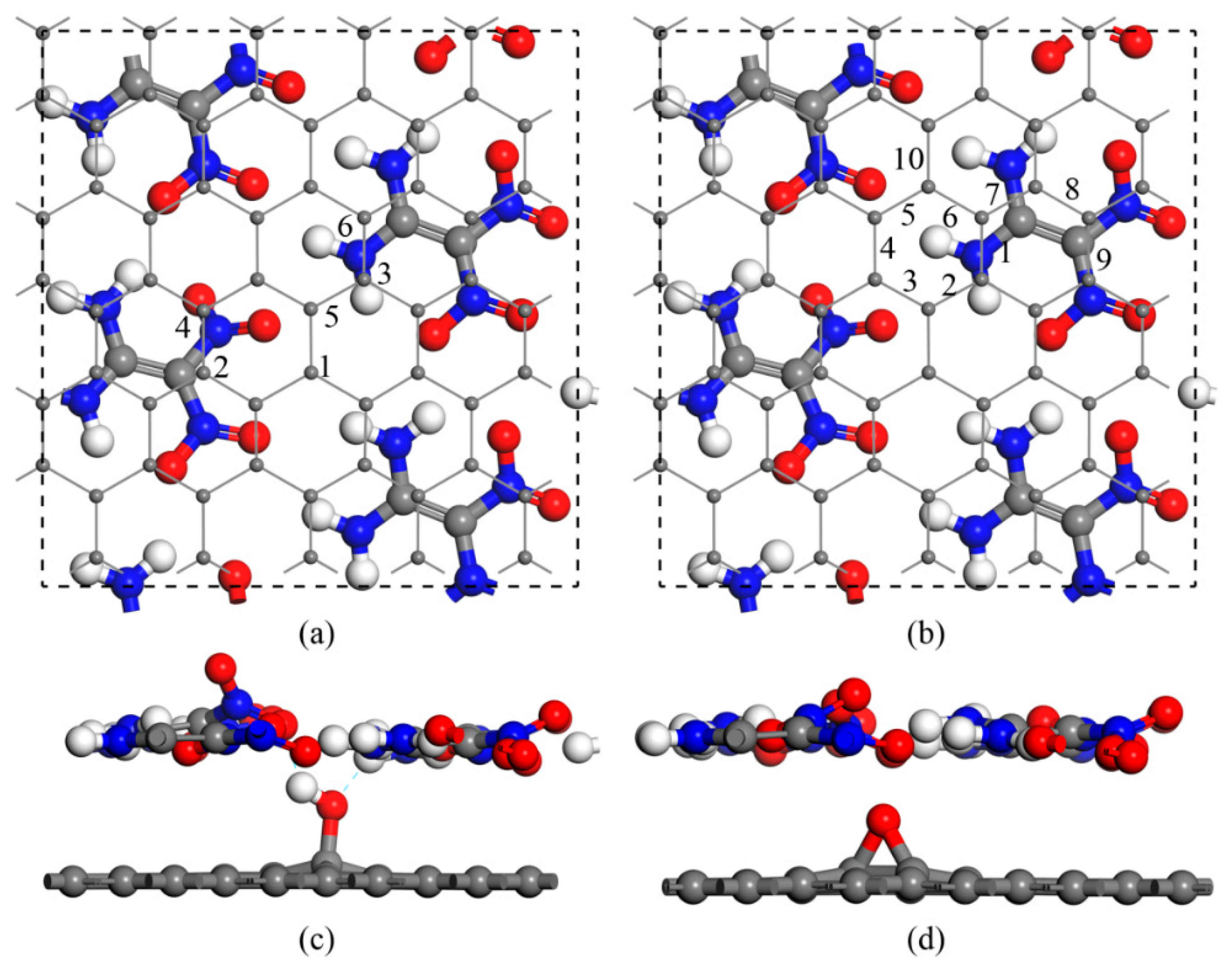{kind=link}
{kind=link}
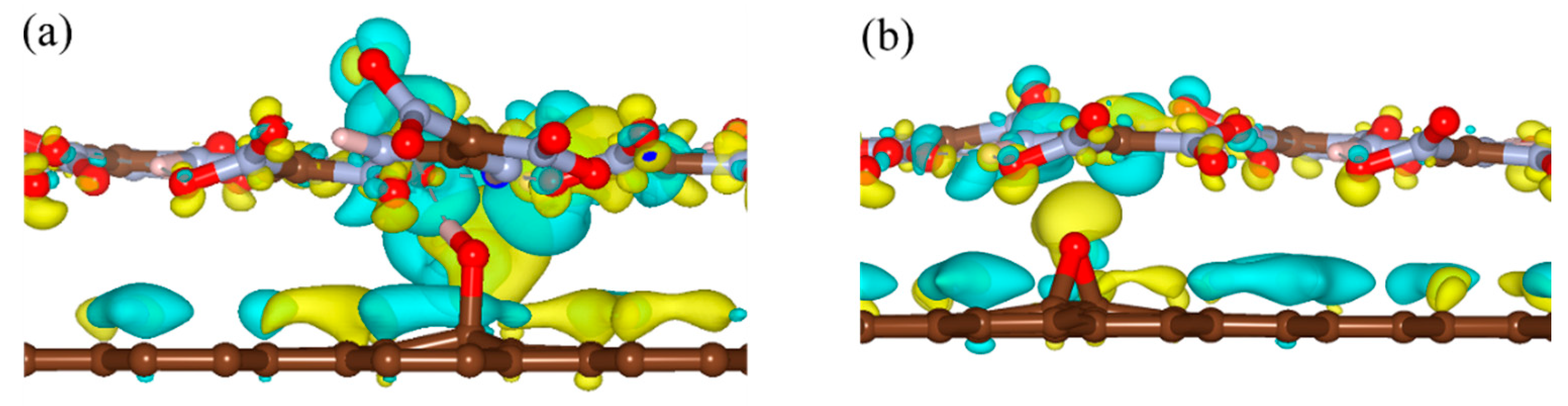{kind=link}
{kind=link}
{kind=link}
{kind=link}
| dHB/Å | CT/e | ΔEb/eV·nm−2 | ΔEgap/eV | ||
|---|---|---|---|---|---|
| FOX-7 | 2.490 | ||||
| FOX-7–G | - | −0.058 | −1.26 | 2.394 | |
| FOX-7–G–OH | site1 | 1.984/1.840 | −0.048 | −0.70 | 2.224 |
| site2 | 2.097 | −0.036 | −0.49 | 2.274 | |
| site3 | - | −0.040 | −0.50 | 2.161 | |
| site4 | 2.012 | −0.043 | −0.61 | 2.128 | |
| site5 | 1.979 | −0.044 | −0.63 | 2.363 | |
| site6 | - | −0.057 | −0.58 | 2.322 | |
| FOX-7–G–O | site1 | - | −0.043 | −1.05 | 2.404 |
| site2 | - | −0.051 | −1.09 | 2.327 | |
| site3 | - | −0.050 | −1.10 | 2.365 | |
| site4 | - | −0.059 | −1.11 | 2.320 | |
| site5 | - | −0.055 | −1.11 | 2.339 | |
| site6 | - | −0.050 | −1.07 | 2.295 | |
| site7 | - | −0.048 | −1.03 | 2.317 | |
| site8 | - | −0.047 | −1.04 | 2.322 | |
| site9 | - | −0.041 | −1.06 | 2.326 | |
| Site10 | - | −0.054 | −1.10 | 2.290 |
| OH:O | NHB | CT/e | ΔEb/eV⋅nm−2 | ΔEgap /eV | |
|---|---|---|---|---|---|
| FOX-7–G–OH | 14:0 | 4 | 0.061 | −0.96 | 2.444 |
| 9:5 | 2 | 0.045 | −0.75 | 2.403 | |
| 8:6 | 2 | 0.038 | −0.76 | 2.481 | |
| 7:7 | 2 | 0.042 | −0.86 | 2.459 | |
| 5:9 | 4 | 0.067 | −0.98 | 2.387 | |
| 1:13 | 1 | 0.062 | −0.83 | 2.317 | |
| 0:14 | 0 | 0.276 | −1.00 | 2.371 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, Y.; Sun, Y.; Zhao, J. Interaction Mechanisms of Insensitive Explosive FOX-7 and Graphene Oxides from Ab Initio Calculations. Nanomaterials 2019, 9, 1290. https://doi.org/10.3390/nano9091290
Su Y, Sun Y, Zhao J. Interaction Mechanisms of Insensitive Explosive FOX-7 and Graphene Oxides from Ab Initio Calculations. Nanomaterials. 2019; 9(9):1290. https://doi.org/10.3390/nano9091290
Chicago/Turabian StyleSu, Yan, Yuanze Sun, and Jijun Zhao. 2019. "Interaction Mechanisms of Insensitive Explosive FOX-7 and Graphene Oxides from Ab Initio Calculations" Nanomaterials 9, no. 9: 1290. https://doi.org/10.3390/nano9091290