Formation of 3-Dimensional Gold, Copper and Palladium Microelectrode Arrays for Enhanced Electrochemical Sensing Applications


Abstract
1. Introduction
2. Materials and Methods
2.1. Chemical Reagents
2.2. Electrochemical Experiments
2.2.1. Electrochemical Deposition of Metal Nanostructures
2.2.2. Electrochemical Sensing Experiments in Ionic Liquids
2.3. Electrode Imaging
3. Results and Discussion
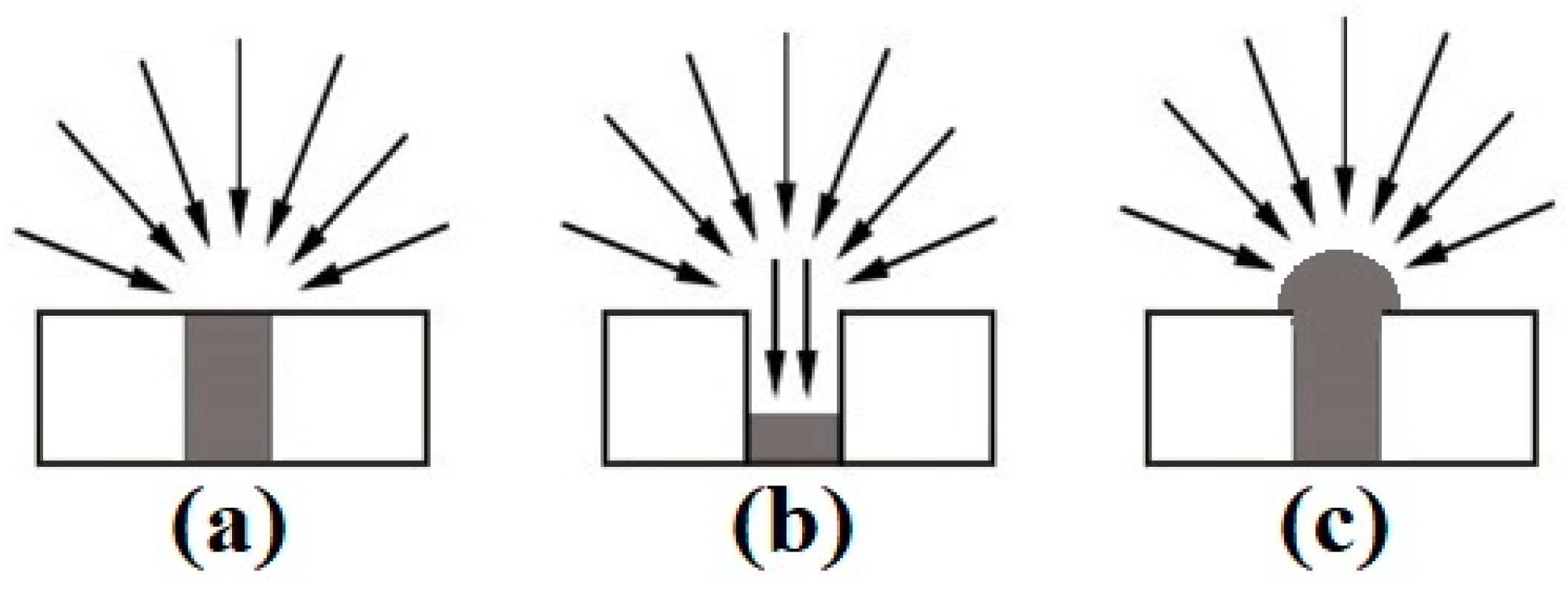
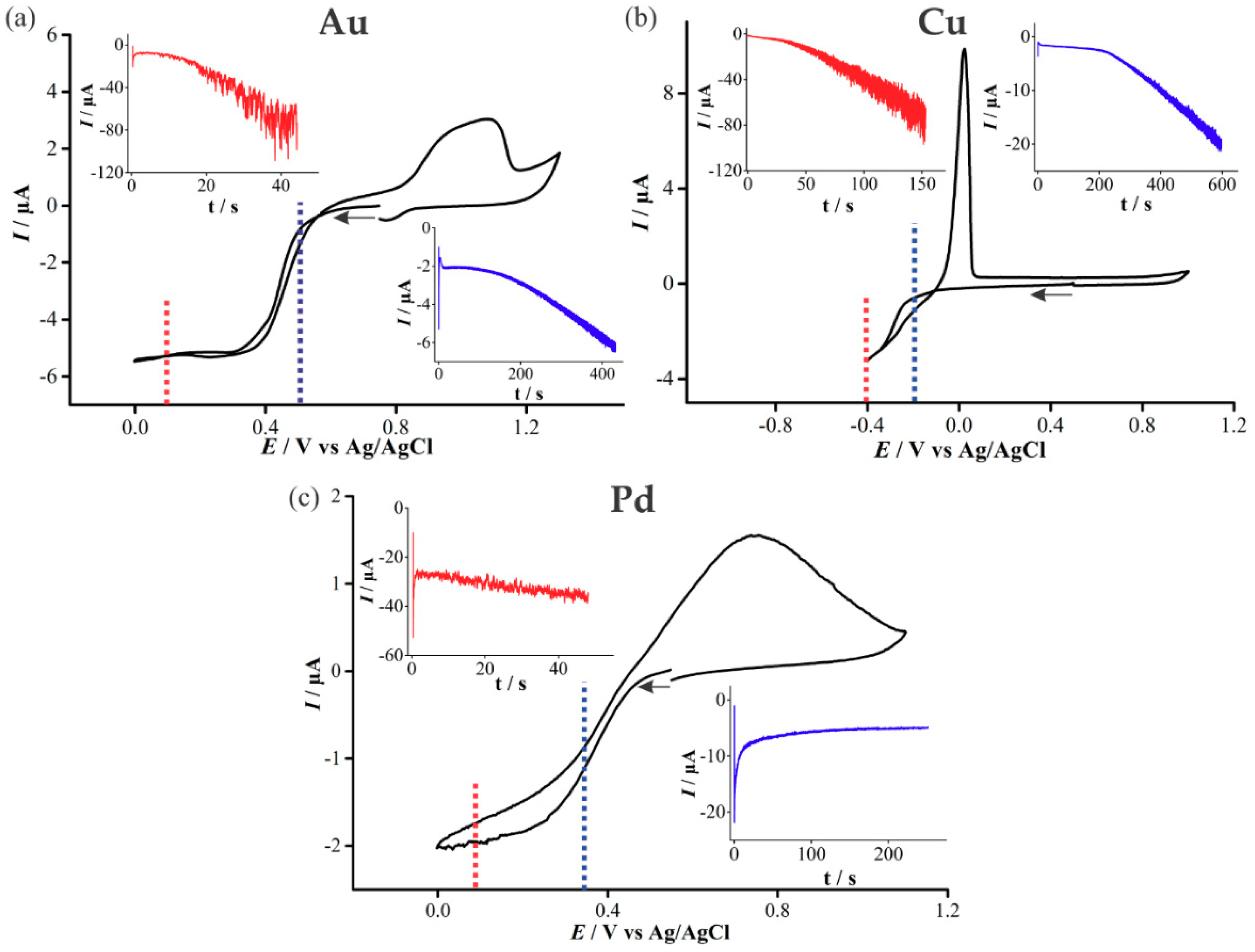
3.1. Electrochemical Deposition of 3D Nanostructured Microarrays
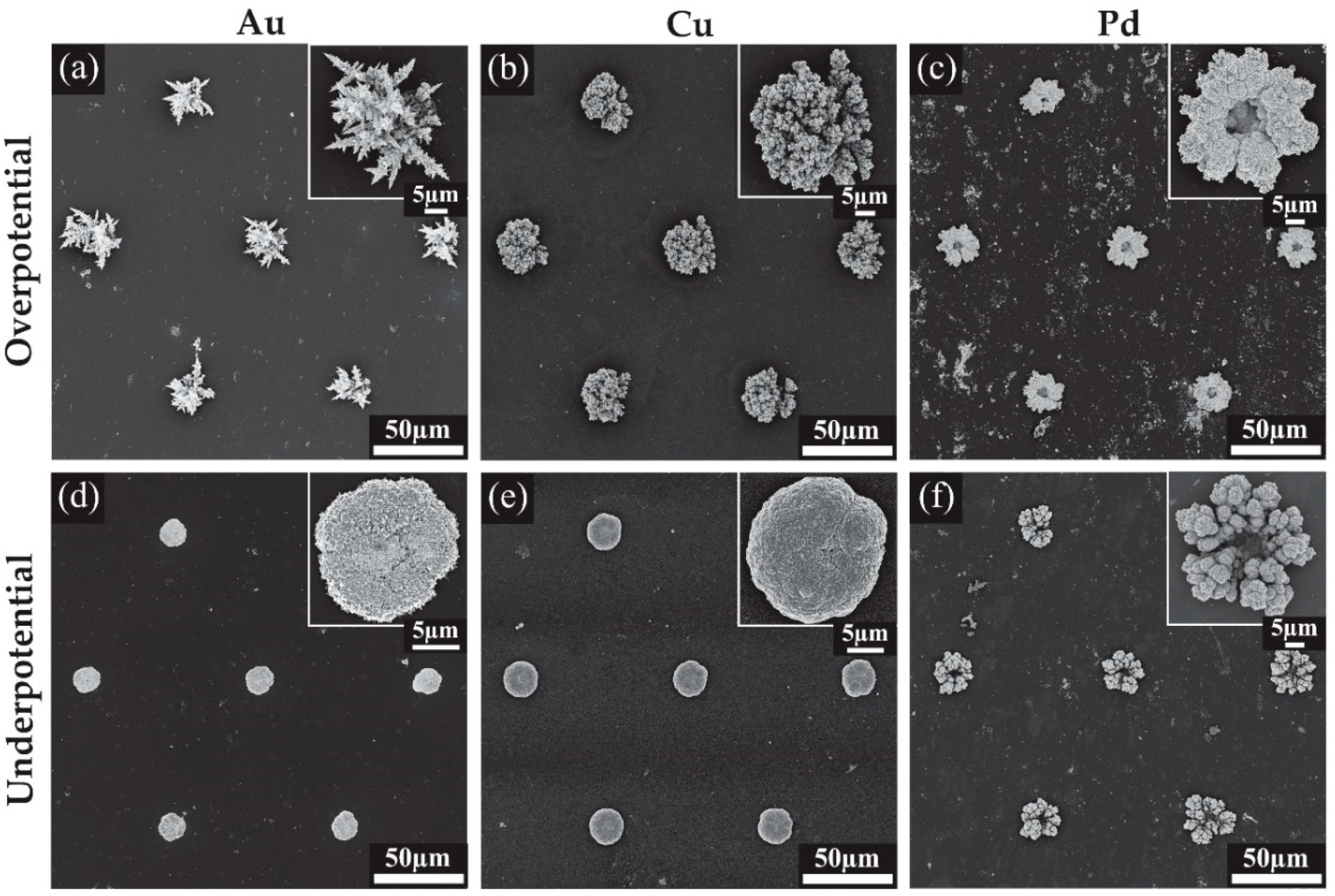
3.2. Characterisation of 3D Nanostructured Microarrays
3.3. Electroactive Surface Area Calculation
3.4. Electrochemical Sensing Experiments
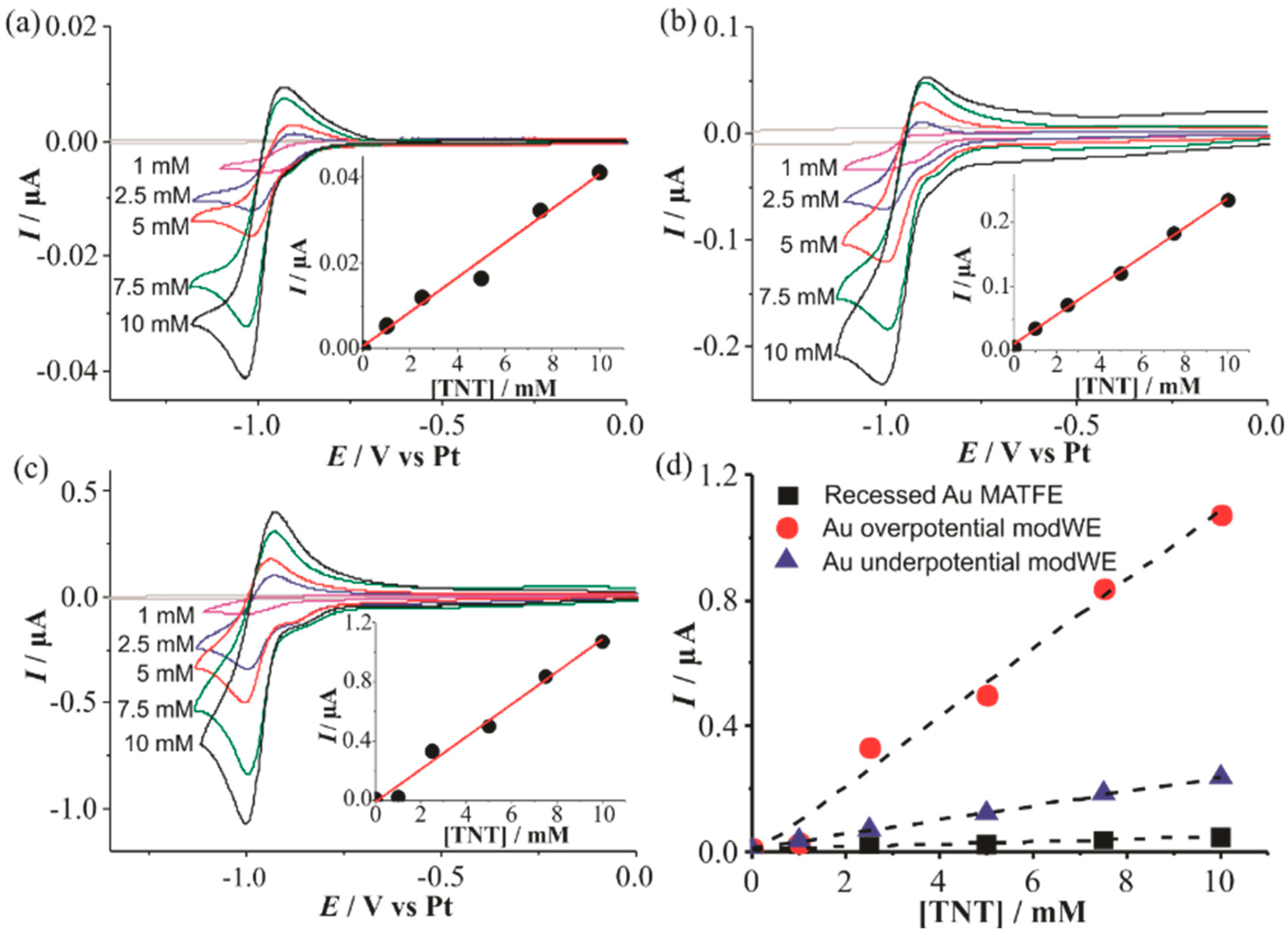
3.4.1. Electrochemical Detection of TNT on Au ModWEs
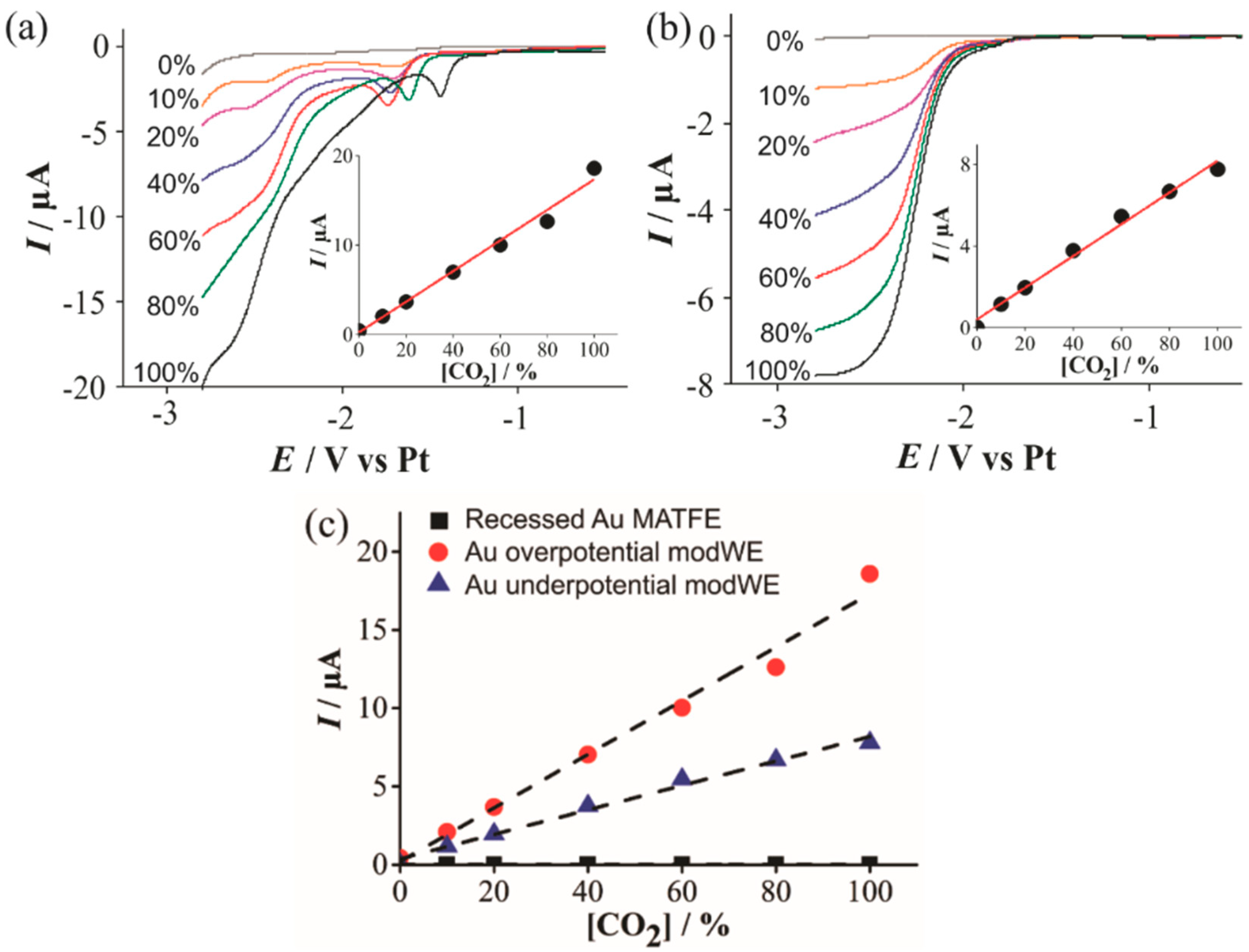
3.4.2. Electrochemical Detection of Carbon Dioxide on Cu ModWEs
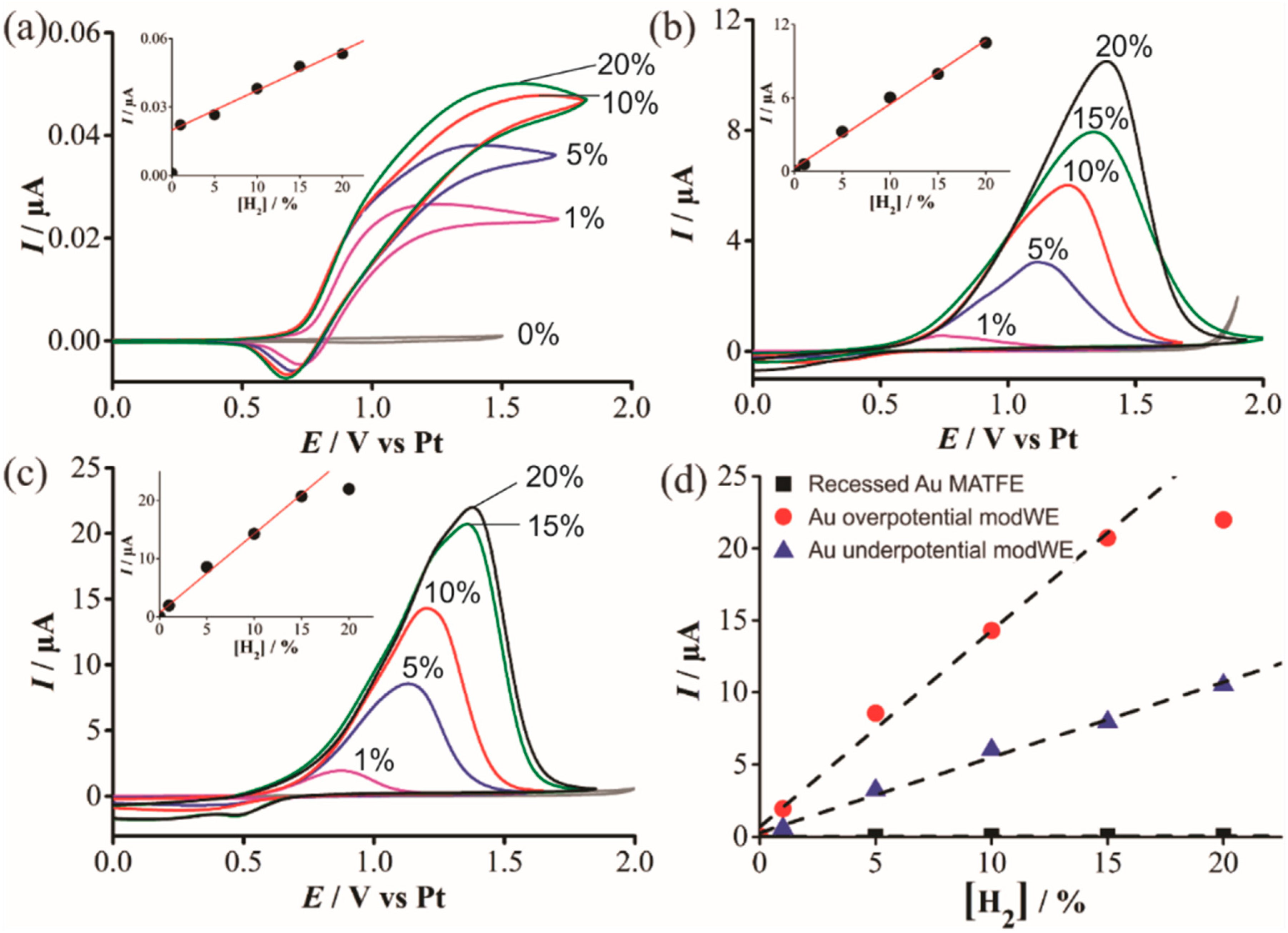
3.4.3. Electrochemical Detection of Hydrogen on Pd ModWEs
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xiong, L.; Compton, R.G. Amperometric Gas detection: A Review. Int. J. Electrochem. Sci. 2014, 9, 7152–7181. [Google Scholar]
- Stetter, J.; Penrose, W.; Yao, S. Sensors, Chemical Sensors, Electrochemical Sensors, and ECS. J. Electrochem. Soc. 2003, 150, S11–S16. [Google Scholar] [CrossRef]
- Stradiotto, N.R.; Yamanaka, H.; Zanoni, M.V.B. Electrochemical sensors: A powerful tool in analytical chemistry. J. Braz. Chem. Soc. 2003, 14, 159–173. [Google Scholar] [CrossRef]
- Huang, H.; Dasgupta, P.K. Electrochemical sensing of gases based on liquid collection interfaces. Electroanalysis 1997, 9, 585–591. [Google Scholar] [CrossRef]
- Renedo, O.D.; Alonso-Lomillo, M.A.; Martínez, M.J.A. Recent developments in the field of screen-printed electrodes and their related applications. Talanta 2007, 73, 202–219. [Google Scholar] [CrossRef] [PubMed]
- Kadara, R.O.; Jenkinson, N.; Banks, C.E. Characterisation of commercially available electrochemical sensing platforms. Sens. Act. B 2009, 138, 556–562. [Google Scholar] [CrossRef]
- MicruxTechnologies. Thin Film Electrochemical Sensors. Available online: https://www.micruxfluidic.com/en/electrochemical-solutions/thin-film-electrochemical-sensors/ (accessed on 15 August 2019).
- Lee, J.; Silvester, D. Low-cost microarray thin-film electrodes with ionic liquid gel-polymer electrolytes for miniaturised oxygen sensing. Analyst 2016, 141, 3705–3713. [Google Scholar] [CrossRef] [PubMed]
- Compton, R.G.; Banks, C.E. Understanding Voltammetry, 2nd ed.; World Scientific: Singapore, 2011. [Google Scholar]
- Arrigan, D.W.M. Nanoelectrodes, nanoelectrode arrays and their applications. Analyst 2004, 129, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Kadara, R.O.; Jenkinson, N.; Banks, C.E. Screen printed recessed microelectrode arrays. Sens. Act. B 2009, 142, 342–346. [Google Scholar] [CrossRef]
- Hussain, G.; Aldous, L.; Silvester, D.S. Preparation of Platinum-based ‘Cauliflower Microarrays’ and Their Enhanced Gas Sensing Abilities. Anal. Chim. Acta 2019, 1048, 12–21. [Google Scholar] [CrossRef]
- Hussain, G.; O’Mullane, A.P.; Silvester, D.S. Modification of Microelectrode Arrays with High Surface Area Dendritic Platinum 3D Structures: Enhanced Sensitivity for Oxygen Detection in Ionic Liquids. Nanomaterials 2018, 8, 735. [Google Scholar] [CrossRef] [PubMed]
- Hussain, G.; Silvester, D.S. Detection of sub-ppm Concentrations of Ammonia in an Ionic Liquid: Enhanced Current Density Using “Filled” Recessed Microarrays. Anal. Chem. 2016, 88, 12453–12460. [Google Scholar] [CrossRef] [PubMed]
- Plowman, B.; Bhargava, S.; Mullane, A.P. Electrochemical fabrication of metallic nanostructured electrodes for electroanalytical applications. Analyst 2011, 136, 5107–5119. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Du Plessis, G.; Arrigan, D.W.M.; Silvester, D.S. Towards improving the robustness of electrochemical gas sensors: Impact of PMMA addition on the sensing of oxygen in an ionic liquid. Anal. Methods 2015, 7, 7327–7335. [Google Scholar] [CrossRef]
- Bewick, A.; Fleischmann, M.; Thirsk, H.R. Kinetics of the electrocrystallization of thin films of calomel. Trans. Faraday Soc. 1962, 58, 2200–2216. [Google Scholar] [CrossRef]
- Cao, G.; Liu, Q.; Huang, Y.; Li, W.; Yao, S. Generation of gold nanostructures at the surface of platinum electrode by electrodeposition for ECL detection for CE. Electrophoresis 2010, 31, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- O’Mullane, A.P.; Ippolito, S.J.; Sabri, Y.M.; Bansal, V.; Bhargava, S.K. Premonolayer oxidation of nanostructured gold: An important factor influencing electrocatalytic activity. Langmuir 2009, 25, 3845–3852. [Google Scholar] [CrossRef] [PubMed]
- Suárez, M.F.; Marken, F.; Compton, R.G.; Bond, A.M.; Miao, W.; Raston, C.L. Evidence for Nucleation-Growth, Redistribution, and Dissolution Mechanisms during the Course of Redox Cycling Experiments on the C60/NBu4C60 Solid-State Redox System: Voltammetric, SEM, and in Situ AFM Studies. J. Phys. Chem. B 1999, 103, 5637–5644. [Google Scholar] [CrossRef]
- Fletcher, S.; Halliday, C.S.; Gates, D.; Westcott, M.; Lwin, T.; Nelson, G. The response of some nucleation/growth processes to triangular scans of potential. J. Electroanal. Chem. 1983, 159, 267–285. [Google Scholar] [CrossRef]
- Aguilar-Sánchez, M.; Palomar-Pardavé, M.; Corona-Avendaño, S.; Romero-Romo, M.; Ramírez-Silva, M.T.; Scharifker, B.R.; Mostany, J.; Rodríguez-Torres, I. Analysis of the copper electrodeposition current transients in nitrates media. ECS Trans. 2009, 20, 357–364. [Google Scholar]
- Grujicic, D.; Pesic, B. Electrodeposition of copper: The nucleation mechanisms. Electrochim. Acta 2002, 47, 2901–2912. [Google Scholar] [CrossRef]
- Gu, S.; Wang, X.; Wei, Y.; Fang, B. Mechanism for nucleation and growth of electrochemical deposition of palladium(II) on a platinum electrode in hydrochloric acid solution. Sci. China Chem. 2014, 57, 755–762. [Google Scholar] [CrossRef]
- Ren, B.; Jones, L.A.; Chan, M.; Oppedisano, D.K.; Qiu, D.; Ippolito, S.J.; Bhargava, S.K. The Effect of Electrodeposition Parameters and Morphology on the Performance of Au Nanostructures for the Detection of As (III). J. Electrochem. Soc. 2017, 164, H1121–H1128. [Google Scholar] [CrossRef]
- Lin, T.-H.; Lin, C.-W.; Liu, H.-H.; Sheu, J.-T.; Hung, W.-H. Potential-controlled electrodeposition of gold dendrites in the presence of cysteine. Chem. Commun. 2011, 47, 2044–2046. [Google Scholar] [CrossRef]
- Du, X.; Zhang, Z.; Miao, Z.; Ma, M.; Zhang, Y.; Zhang, C.; Wang, W.; Han, B.; Chen, Q. One step electrodeposition of dendritic gold nanostructures on β-lactoglobulin-functionalized reduced graphene oxide for glucose sensing. Talanta 2015, 144, 823–829. [Google Scholar] [CrossRef]
- Li, Y.S.; Sun, J.Z.; Bian, C.; Tong, J.H.; Dong, H.P.; Zhang, H.; Xia, S.H. Copper nano-clusters prepared by one-step electrodeposition and its application on nitrate sensing. AIP Adv. 2015, 5, 041312. [Google Scholar] [CrossRef]
- Li, Y.; Sun, J.; Bian, C.; Tong, J.; Xia, S. Electrodeposition of copper nano-clusters at a platinum microelectrode for trace nitrate determination. Procedia Eng. 2010, 5, 339–342. [Google Scholar] [CrossRef]
- Mercado, G.V.G.; González, C.J.; Oliva, M.I.; Brunetti, V.; Eimer, G.A. Morphology of Copper Deposits Obtained by Metallic Electrodeposition. Procedia Mater. Sci. 2015, 8, 635–640. [Google Scholar] [CrossRef][Green Version]
- Hepel, M.; Stobiecka, M. Interactions of adsorbed albumin with underpotentially deposited copper on gold piezoelectrodes. Bioelectrochemistry 2007, 70, 155–164. [Google Scholar] [CrossRef]
- Heydari, H.; Abdolmaleki, A.; Gholivand, M.B. Electrodeposition and characterization of palladium nanostructures on stainless steel and application as hydrogen sensor. CeN 2015, 37, 23. [Google Scholar] [CrossRef]
- Trasatti, S.; Petrii, O.A. Real surface area measurements in electrochemistry. J. Electroanal. Chem. 1992, 327, 353–376. [Google Scholar] [CrossRef]
- Hilmi, A.; Luong, J.H.T.; Nguyen, A.-L. Development of Electrokinetic Capillary Electrophoresis Equipped with Amperometric Detection for Analysis of Explosive Compounds. Anal. Chem. 1999, 71, 873–878. [Google Scholar] [CrossRef]
- Tanner, E.; Batchelor-Mcauley, C.; Compton, R.G. Carbon Dioxide Reduction in Room-Temperature Ionic Liquids: The Effect of the Choice of Electrode Material, Cation, and Anion. J. Phys. Chem. C 2016, 120, 26442–26447. [Google Scholar] [CrossRef]
- Antolini, E. Palladium in fuel cell catalysis. Energy Environ. Sci. 2009, 2, 915–931. [Google Scholar] [CrossRef]
- Gupta, R.; Guin, S.K.; Aggarwal, S.K. Electrocrystallization of palladium (Pd) nanoparticles on platinum (Pt) electrode and its application for electro-oxidation of formic acid and methanol. Electrochim. Acta 2014, 116, 314–320. [Google Scholar] [CrossRef]
- Kang, C.; Lee, J.; Silvester, D.S. Electroreduction of 2,4,6-Trinitrotoluene in Room Temperature Ionic Liquids: Evidence of an EC2 Mechanism. J. Phys. Chem. C 2016, 120, 10997–11005. [Google Scholar] [CrossRef]
- Rees, N.V.; Compton, R.G. Electrochemical CO2 sequestration in ionic liquids; a perspective. Energy Environ. Sci. 2011, 4, 403–408. [Google Scholar] [CrossRef]
- Silvester, D.; Ward, K.; Aldous, L.; Hardacre, C.; Compton, R. The electrochemical oxidation of hydrogen at activated platinum electrodes in room temperature ionic liquids as solvents. J. Electroanal. Chem. 2008, 618, 53–60. [Google Scholar] [CrossRef]
- Silvester, D.S.; Aldous, L.; Hardacre, C.; Compton, R.G. An Electrochemical Study of the Oxidation of Hydrogen at Platinum Electrodes in Several Room Temperature Ionic Liquids. J. Phys. Chem. B 2007, 111, 5000–5007. [Google Scholar] [CrossRef]
- Streeter, I.; Wildgoose, G.G.; Shao, L.; Compton, R.G. Cyclic voltammetry on electrode surfaces covered with porous layers: An analysis of electron transfer kinetics at single-walled carbon nanotube modified electrodes. Sens. Act. B 2008, 133, 462–466. [Google Scholar] [CrossRef]
- Hussain, G.; Sofianos, M.V.; Lee, J.; Gibson, C.; Buckley, C.E.; Silvester, D.S. Macroporous platinum electrodes for hydrogen oxidation in ionic liquids. Electrochem. Commun. 2018, 86, 43–47. [Google Scholar] [CrossRef]
- Tang, Y.; Lin, L.; Kumar, A.; Guo, M.; Sevilla, M.; Zeng, X. Hydrogen Electrooxidation in Ionic Liquids Catalyzed by the NTf2 Radical. J. Phys. Chem. C 2017, 121, 5161–5167. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Modification | Deposition Potential/V | Charge Limit/mC | Average Deposition Time/s | QH/µC | ESA/mm2 | |
|---|---|---|---|---|---|---|
| Au | Unmodified | - | - | - | 3.55 | 0.909 |
| Overpotential | 0.1 | −1.5 | 55 | 4.33 | 4.10 | |
| Underpotential | 0.5 | −1.5 | 450 | 1.87 | 2.57 | |
| Cu | Overpotential | −0.4 | −4.5 | 150 | - | - |
| Underpotential | −0.2 | −4.5 | 600 | - | - | |
| Pd | Unmodified | - | - | - | 2.15 | 1.02 |
| Overpotential | 0.1 | −1.5 | 50 | 2.17 | 5.17 | |
| Underpotential | 0.35 | −1.5 | 250 | 1.96 | 4.66 |
| Au Modification | Ip (10 mM TNT)/A | J/Am−2 | Sensitivity/AM−1 |
|---|---|---|---|
| Unmodified Au | −4.84 × 10−8 | −5.32 × 10−2 | 3.89 × 10−6 |
| Overpotential | −1.07 × 10−6 | −2.61 × 10−1 | 1.10 × 10−4 |
| Underpotential | −2.35 × 10−7 | −9.14 × 10−2 | 2.26 × 10−5 |
| Cu Modification | Ip (100% CO2)/A | Sensitivity/A%vol.−1 |
|---|---|---|
| Overpotential | −1.86 × 10−5 | 1.54 × 10−7 |
| Underpotential | −7.79 × 10−6 | 8.35 × 10−8 |
| Pd Modification | Ip (15% H2)/A | J/Am−2 | Sensitivity/A%vol.−1 |
|---|---|---|---|
| Unmodified | 4.17 × 10−8 | 4.07 × 10−2 | 1.05 × 10−9 |
| Overpotential | 2.07 × 10−5 | 4.01 | 1.36 × 10−6 |
| Underpotential | 7.94 × 10−6 | 1.71 | 5.22 × 10−7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hay, C.E.; Lee, J.; Silvester, D.S. Formation of 3-Dimensional Gold, Copper and Palladium Microelectrode Arrays for Enhanced Electrochemical Sensing Applications. Nanomaterials 2019, 9, 1170. https://doi.org/10.3390/nano9081170
Hay CE, Lee J, Silvester DS. Formation of 3-Dimensional Gold, Copper and Palladium Microelectrode Arrays for Enhanced Electrochemical Sensing Applications. Nanomaterials. 2019; 9(8):1170. https://doi.org/10.3390/nano9081170
Chicago/Turabian StyleHay, Catherine E., Junqiao Lee, and Debbie S. Silvester. 2019. "Formation of 3-Dimensional Gold, Copper and Palladium Microelectrode Arrays for Enhanced Electrochemical Sensing Applications" Nanomaterials 9, no. 8: 1170. https://doi.org/10.3390/nano9081170
APA StyleHay, C. E., Lee, J., & Silvester, D. S. (2019). Formation of 3-Dimensional Gold, Copper and Palladium Microelectrode Arrays for Enhanced Electrochemical Sensing Applications. Nanomaterials, 9(8), 1170. https://doi.org/10.3390/nano9081170