The Kinetic Behaviors of H Impurities in the Li/Ta Bilayer: Application for the Accelerator-Based BNCT


 ,
,
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Bulk Properties and Surface Energy
3.2. The Interface Model Geometry
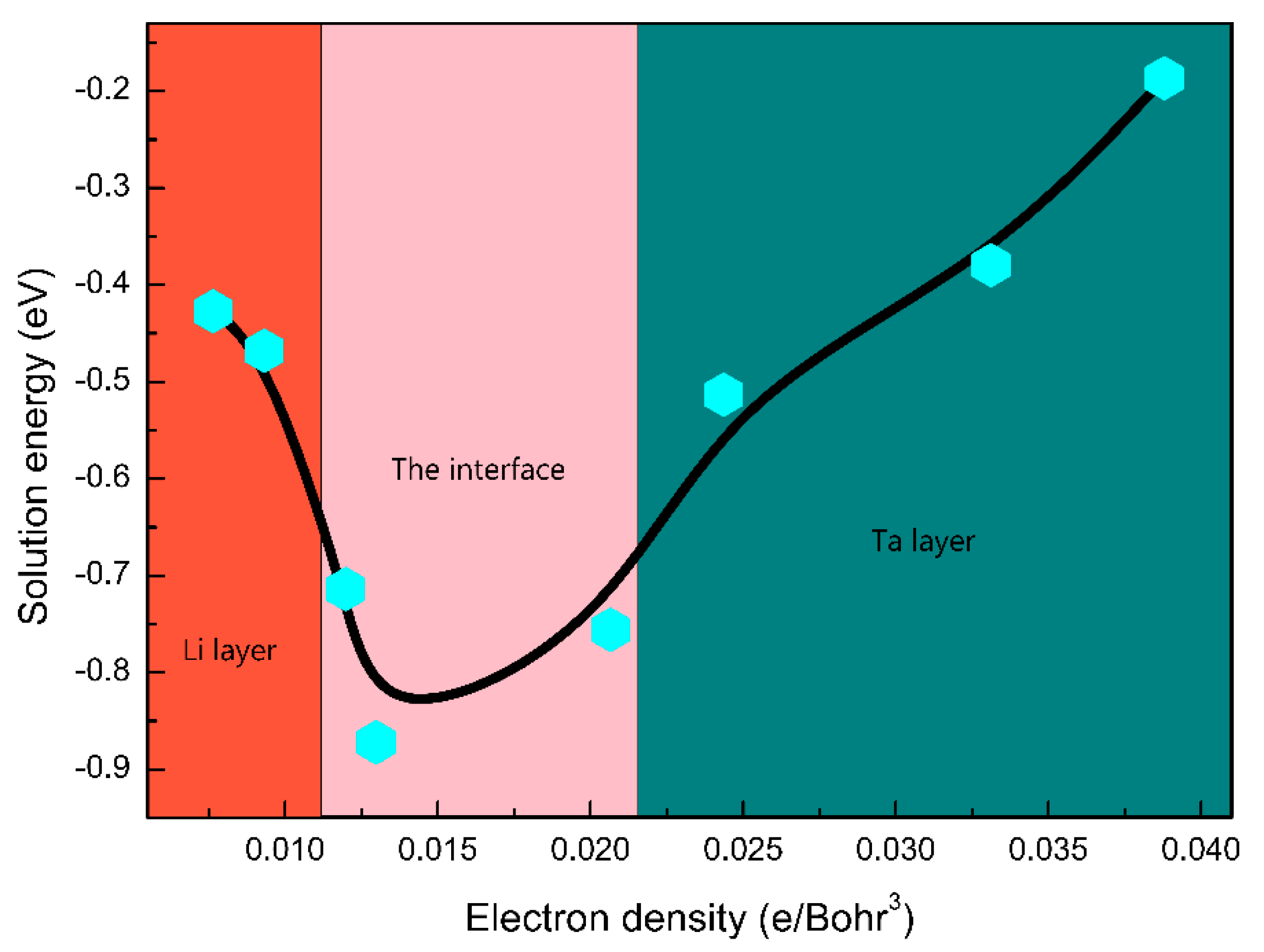
3.3. The H Solution Energy and Diffusion Mechanism
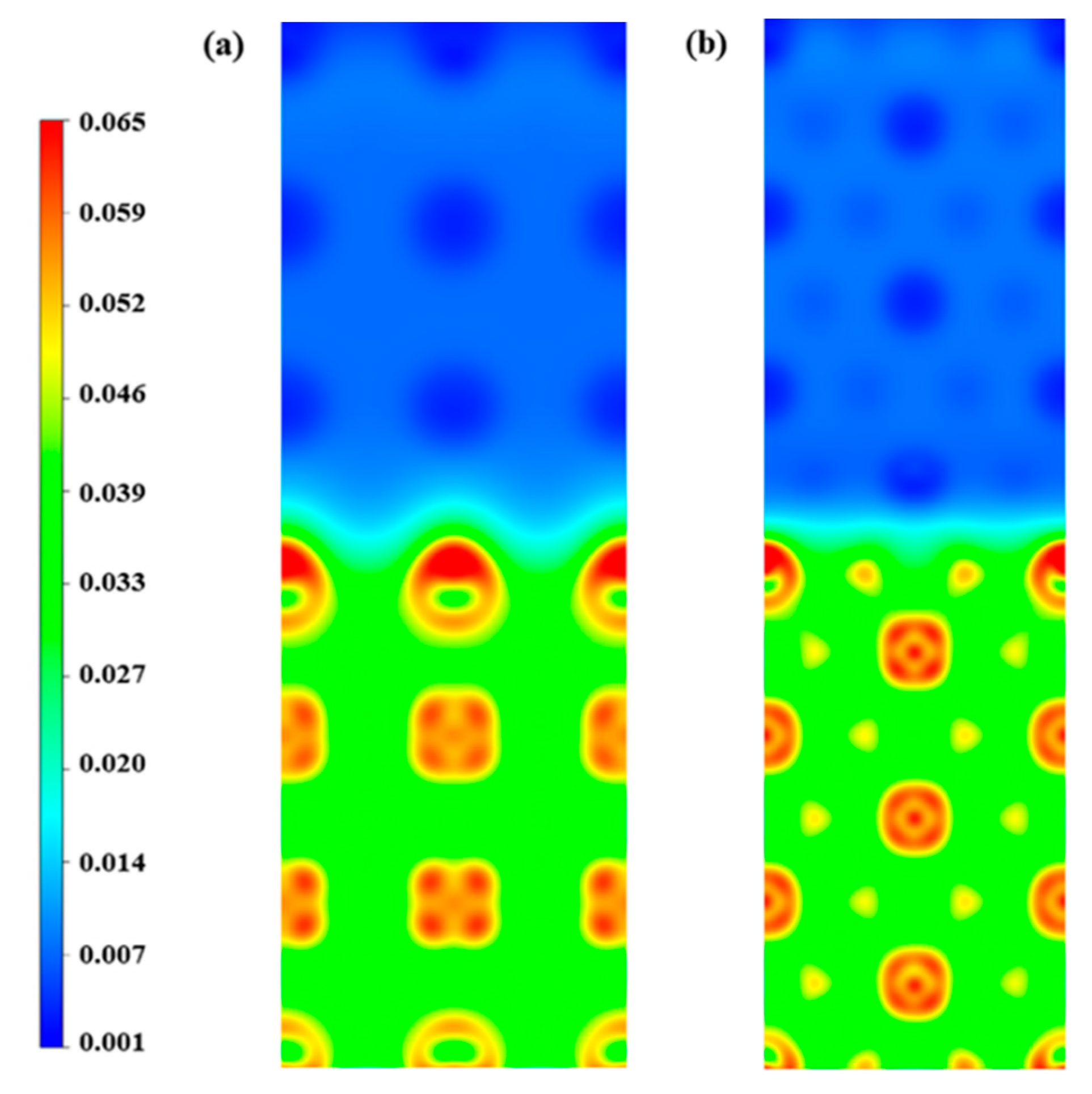
3.4. The H Bubbles Formation in the Li/Ta Bilayer
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. 6-Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Gothelf, A.; Mir, L.M.; Gehl, J. Electrochemotherapy: Results of cancer treatment using enhanced delivery of bleomycin by electroporation. Cancer Treat. Rev. 2003, 29, 371–387. [Google Scholar] [CrossRef]
- Maruyama, K.; Ishida, O.; Kasaoka, S.; Takizawa, T.; Utoguchi, N.; Shinohara, A.; Chiba, M.; Kobayashi, H.; Eriguchi, M.; Yanagie, H. Intracellular targeting of sodium mercaptoundecahydrododecaborate (BSH) to solid tumors by transferrin-PEG liposomes, for boron neutron-capture therapy (BNCT). J. Control. Release 2004, 98, 195–207. [Google Scholar] [CrossRef]
- Forton, E.; Stichelbaut, F.; Cambriani, A.; Kleeven, W.; Ahlback, J.; Jongen, Y. Overview of the IBA accelerator-based BNCT system. Appl. Radiat. Isot. 2009, 67, 262–265. [Google Scholar] [CrossRef]
- Kumada, H.; Kurihara, T.; Yoshioka, M.; Kobayashi, H.; Matsumoto, H.; Sugano, T.; Sakurai, H.; Sakae, T.; Matsumura, A. Development of beryllium-based neutron target system with three-layer structure for accelerator-based neutron source for boron neutron capture therapy. Appl. Radiat. Isot. 2015, 106, 78–83. [Google Scholar] [CrossRef]
- Barth, R.F.; Soloway, A.H. Boron neutron capture therapy of brain tumors—Current status and future prospects. J. Neurooncol. 1997, 33, 3–7. [Google Scholar] [CrossRef]
- Bleuel, D.L. Determination and Production of an Optimal Neutron Energy Spectrum for Boron Neutron Capture Therapy; University of California: Berkeley, CA, USA, 2003. [Google Scholar]
- Bayanov, B.; Belov, V.; Taskaev, S. Neutron producing target for accelerator based neutron capture therapy. J. Phys. Conf. Ser. 2006, 41, 460–465. [Google Scholar] [CrossRef]
- Tian, Y.S.; Hu, Z.L.; Tong, J.F.; Chen, J.Y.; Peng, X.Y.; Liang, T.J. Design of beam shaping assembly based on 3.5 MeV radio-frequency quadrupole proton accelerator for boron neutron capture therapy. Acta Phys. Sin. 2018, 67, 1–8. [Google Scholar]
- Guan, X.; Luo, Z.; Fu, S. Study of Beam Dynamics on High Current 3.5 MeV RFQ for China ADS. Chin. J. Nucl. Sci. Eng. 2003, 23, 73–78. [Google Scholar]
- Xie, D.; Wang, Z.; Sun, J.; Li, J.; Ma, E.; Shan, Z. In situ study of the initiation of hydrogen bubbles at the aluminium metal / oxide interface. Nat. Mater. 2015, 14, 899–904. [Google Scholar] [CrossRef]
- Barnoush, A.; Vehoff, H. Recent developments in the study of hydrogen embrittlement: Hydrogen effect on dislocation nucleation. Acta Mater. 2010, 58, 5274–5285. [Google Scholar] [CrossRef]
- Gavriljuk, V.G.; Shivanyuk, V.N.; Foct, J. Diagnostic experimental results on the hydrogen embrittlement of austenitic steels. Acta Mater. 2003, 51, 1293–1305. [Google Scholar] [CrossRef]
- Sugawara, H.; Kasatov, D.; Sokolova, E.; Miyazawa, T.; Taskaev, S.; Shchudlo, I.; Koshkarev, A.; Bykov, T.; Gromilov, S.; Makarov, A.; et al. In Situ Observations of Blistering of a Metal Irradiated with 2-MeV Protons. Metals 2017, 7, 558. [Google Scholar]
- Demkowicz, M.J.; Hoagland, R.G. Structure of Kurdjumov-Sachs interfaces in simulations of a copper-niobium bilayer. J. Nucl. Mater. 2008, 372, 45–52. [Google Scholar] [CrossRef]
- Demkowicz, M.J.; Hoagland, R.G.; Hirth, J.P. Interface Structure and Radiation Damage Resistance in Cu-Nb Multilayer Nanocomposites. Phys. Rev. Lett. 2008, 100, 136102. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Zhang, P.; Zhang, X.; Wang, R.H.; Liu, G.; Zhang, G.J.; Sun, J. Mechanical properties of fcc / fcc Cu / Nb nanostructured multilayers. Mater. Sci. Eng. A 2012, 545, 118–122. [Google Scholar] [CrossRef]
- González, C.; Iglesias, R. Energetic analysis of He and monovacancies in Cu/W metallic interfaces. Mater. Des. 2016, 91, 171–179. [Google Scholar] [CrossRef]
- He, W.H.; Gao, X.; Wang, D.; Gao, N.; Cui, M.H.; Pang, L.L.; Wang, Z.G. First-principles investigation of grain boundary morphology effects on helium solutions in tungsten. Comput. Mater. Sci. 2018, 148, 224–230. [Google Scholar] [CrossRef]
- Demkowicz, M.J.; Misra, A.; Han, W.Z.; Fu, E.G.; Wang, Y.Q. Effect of grain boundary character on sink efficiency. Acta Mater. 2012, 60, 6341–6351. [Google Scholar]
- Han, W.; Demkowicz, M.J.; Mara, N.A.; Fu, E.; Sinha, S.; Rollett, A.D.; Wang, Y.; Carpenter, J.S.; Beyerlein, I.J.; Misra, A. Design of Radiation Tolerant Materials Via Interface Engineering. Adv. Mater. 2013, 25, 6975–6979. [Google Scholar] [CrossRef]
- Di Stefano, D.; Mrovec, M.; Elsässer, C. First-principles investigation of hydrogen trapping and diffusion at grain boundaries in nickel. Acta Mater. 2015, 98, 306–312. [Google Scholar] [CrossRef]
- Spišák, D.; Hafner, J. Diffusion of Fe atoms on W surfaces and Fe/W films and along surface steps. Phys. Rev. B 2004, 70, 195426. [Google Scholar] [CrossRef]
- Li, N.; Fu, E.G.; Wang, H.; Carter, J.J.; Shao, L.; Maloy, S.A.; Misra, A.; Zhang, X. He ion irradiation damage in Fe/W nanolayer films. J. Nucl. Mater. 2009, 389, 233–238. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamcis for liquid metals. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple John. Phys Rev Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- He, C.; Cheng, M.; Zhang, M.; Zhang, W.X. Interfacial Stability and Electronic Properties of Ag/M (M = Ni, Cu, W, and Pd) and Cu/Cr Interfaces. J. Phys. Chem. C 2018, 122, 17928–17935. [Google Scholar] [CrossRef]
- Kiejna, A. Surface atomic structure and energetics of tantalum. Surf. Sci. 2005, 598, 276–284. [Google Scholar] [CrossRef]
- Liu, Y.L.; Zhang, Y.; Zhou, H.B.; Lu, G.H.; Liu, F.; Luo, G.N. Vacancy trapping mechanism for hydrogen bubble formation in metal. Phys. Rev. B 2009, 79, 172103. [Google Scholar] [CrossRef]
- Sun, L.; Jin, S.; Li, X.C.; Zhang, Y.; Lu, G.H. Hydrogen behaviors in molybdenum and tungsten and a generic vacancy trapping mechanism for H bubble formation. J. Nucl. Mater. 2013, 434, 395–401. [Google Scholar] [CrossRef]
- Seko, A.; Takahashi, A.; Tanaka, I. First-principles interatomic potentials for ten elemental metals via compressed sensing. Phys. Rev. B 2015, 92, 054113. [Google Scholar] [CrossRef]
- Zhang, P.; Ma, Z.; Wang, Y.; Zou, Y.; Lei, W. RSC Advances A fi rst principles study of the mechanical properties of Li–Sn alloys. RSC Adv. 2015, 5, 36022–36029. [Google Scholar] [CrossRef]
- Fajen, O. Neutron powder-diffraction studies of lithium, sodium, and potassium metal. Phys. Rev. B 1989, 40, 86–97. [Google Scholar]
- Koči, L.; Ma, Y.; Oganov, A.R.; Souvatzis, P.; Ahuja, R. Elasticity of the superconducting metals V, Nb, Ta, Mo, and W at high pressure. Phys. Rev. B 2008, 77, 214101. [Google Scholar] [CrossRef]
- Purja Pun, G.P.; Darling, K.A.; Kecskes, L.J.; Mishin, Y. Angular-dependent interatomic potential for the Cu-Ta system and its application to structural stability of nano-crystalline alloys. Acta Mater. 2015, 100, 377–391. [Google Scholar] [CrossRef]
- Liu, Z.; Qi, Y.; Lin, Y.X.; Chen, L.; Lu, P.; Chen, L.Q. Interfacial Study on Solid Electrolyte Interphase at Li Metal Anode: Implication for Li Dendrite Growth. J. Electrochem. Soc. 2016, 163, 592–598. [Google Scholar] [CrossRef]
- Bauer, E.; Poppa, H. The surface energy of metals. Thin Solid Films 1972, 12, 167–185. [Google Scholar] [CrossRef]
- Tyson, W.R.; Miller, W.A. Surface free energies of solid metals: Estimation from liquid surface tension measurements. Surf. Sci. 1977, 62, 267–276. [Google Scholar] [CrossRef]
- Flood, E.A.; Benson, G.C. Surface energy and work function of elemental metals. Can. J. Chem. 2006, 46, 1297–1316. [Google Scholar] [CrossRef]
- Trinkaus, H.; Singh, B.N. Helium accumulation in metals during irradiation—Where do we stand? J. Nucl. Mater. 2003, 323, 229–242. [Google Scholar] [CrossRef]
- Fu, C.C.; Willaime, F. Ab initio study of helium in α-Fe: Dissolution, migration, and clustering with vacancies. Phys. Rev. B 2005, 72, 064117. [Google Scholar] [CrossRef]
- Huang, G.Y.; Juslin, N.; Wirth, B.D. First-principles study of vacancy, interstitial, noble gas atom interstitial and vacancy clusters in bcc-W. Comput. Mater. Sci. 2016, 123, 121–130. [Google Scholar] [CrossRef]
- Blomqvist, A.; Pálsson, G.K.; Araújo, C.M.; Ahuja, R.; Hjörvarsson, B. Significance of self-trapping on hydrogen diffusion. Phys. Rev. Lett. 2010, 105, 185901. [Google Scholar] [CrossRef]
- Mooij, L.; Huang, W.; Droulias, S.A.; Johansson, R.; Hartmann, O.; Xin, X.; Palonen, H.; Scheicher, R.H.; Wolff, M.; Hjörvarsson, B. Influence of site occupancy on diffusion of hydrogen in vanadium. Phys. Rev. B 2017, 95, 064301. [Google Scholar] [CrossRef]
- You, Y.W.; Kong, X.S.; Wu, X.B.; Xu, Y.C.; Fang, Q.F.; Chen, J.L.; Luo, G.N.; Liu, C.S.; Pan, B.C.; Wang, Z. Dissolving, trapping and detrapping mechanisms of hydrogen in bcc and fcc transition metals. AIP Adv. 2013, 3, 012118. [Google Scholar] [CrossRef]
- Johansson, R.; Ahuja, R.; Eriksson, O.; Hjörvarsson, B.; Scheicher, R.H. Effect of uniaxial strain on the site occupancy of hydrogen in vanadium from density-functional calculations. Sci. Rep. 2015, 5, 10301. [Google Scholar] [CrossRef]
- Review, P. Atoms embedded in an electron gas: Immersion energies. Phys. Rev. B 1981, 24, 3037–3047. [Google Scholar]
- Nørskov, J.K.; Besenbacher, F. Theory of hydrogen interaction with metals. J. Less-Common Met. 1987, 130, 475–490. [Google Scholar] [CrossRef]
- Jónsson, H.; Mills, G.; Jacobsen, K.W. CHAPTER 16 Nudged elastic band method for nding minimum en- ergy paths of transitions. In Classical and Quantum Dynamics in Condensed Phase Simulations; World Scientific: Singapore, 1997. [Google Scholar]
- Wicke, E.; Nernst, G.H.; Jones, H.; Hydrides, P.S.; Pub-, I.; Poeschel, E.; Brodowsky, H.; Galstaun, L.S.; Wilkinson, M.K.; Shull, C.G.; et al. Phase Transitions of Hydrogen in Metals due to Elastic Interaction. 1964. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a (Å) | Bulk Modulus (Mbar) | References (Å and Mbar) | |
|---|---|---|---|
| Li | 3.439 | 1.373 | 3.439 a; 3.436, 1.406 b; 3.479 c |
| Ta | 3.308 | 1.995 | 3.311, 1.96 d; 3.309, 2.11 e; 3.304, 1.947 f |
| (100) | (110) | (111) | |
|---|---|---|---|
| Li | 0.456 | 0.500 | 0.522 |
| Ref. | 0.48 a, 0.522 b | 0.51a, 0.556 b,0.522 c | 0.56 a, 0.590 b |
| Ta | 2.486 | 2.303 | 2.749 |
| Ref. | 2.32 d | 2.31, 2.90 e | 2.71 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Chen, H.; Tong, J.; He, W.; Li, X.; Liang, T.; Li, Y.; Yin, W. The Kinetic Behaviors of H Impurities in the Li/Ta Bilayer: Application for the Accelerator-Based BNCT. Nanomaterials 2019, 9, 1107. https://doi.org/10.3390/nano9081107
Liu X, Chen H, Tong J, He W, Li X, Liang T, Li Y, Yin W. The Kinetic Behaviors of H Impurities in the Li/Ta Bilayer: Application for the Accelerator-Based BNCT. Nanomaterials. 2019; 9(8):1107. https://doi.org/10.3390/nano9081107
Chicago/Turabian StyleLiu, Xiao, Huaican Chen, Jianfei Tong, Wenhao He, Xujing Li, Tianjiao Liang, Yuhong Li, and Wen Yin. 2019. "The Kinetic Behaviors of H Impurities in the Li/Ta Bilayer: Application for the Accelerator-Based BNCT" Nanomaterials 9, no. 8: 1107. https://doi.org/10.3390/nano9081107
APA StyleLiu, X., Chen, H., Tong, J., He, W., Li, X., Liang, T., Li, Y., & Yin, W. (2019). The Kinetic Behaviors of H Impurities in the Li/Ta Bilayer: Application for the Accelerator-Based BNCT. Nanomaterials, 9(8), 1107. https://doi.org/10.3390/nano9081107