Improving the Thermal and Mechanical Properties of Poly(l-lactide) by Forming Nanocomposites with an in Situ Ring-Opening Intermediate of Poly(l-lactide) and Polyhedral Oligomeric Silsesquioxane


Abstract
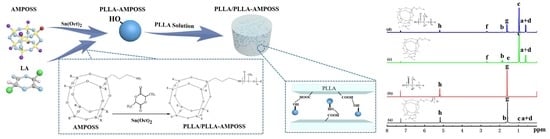
:
1. Introduction
2. Experimental
2.1. Materials
2.2. Sample Preparation
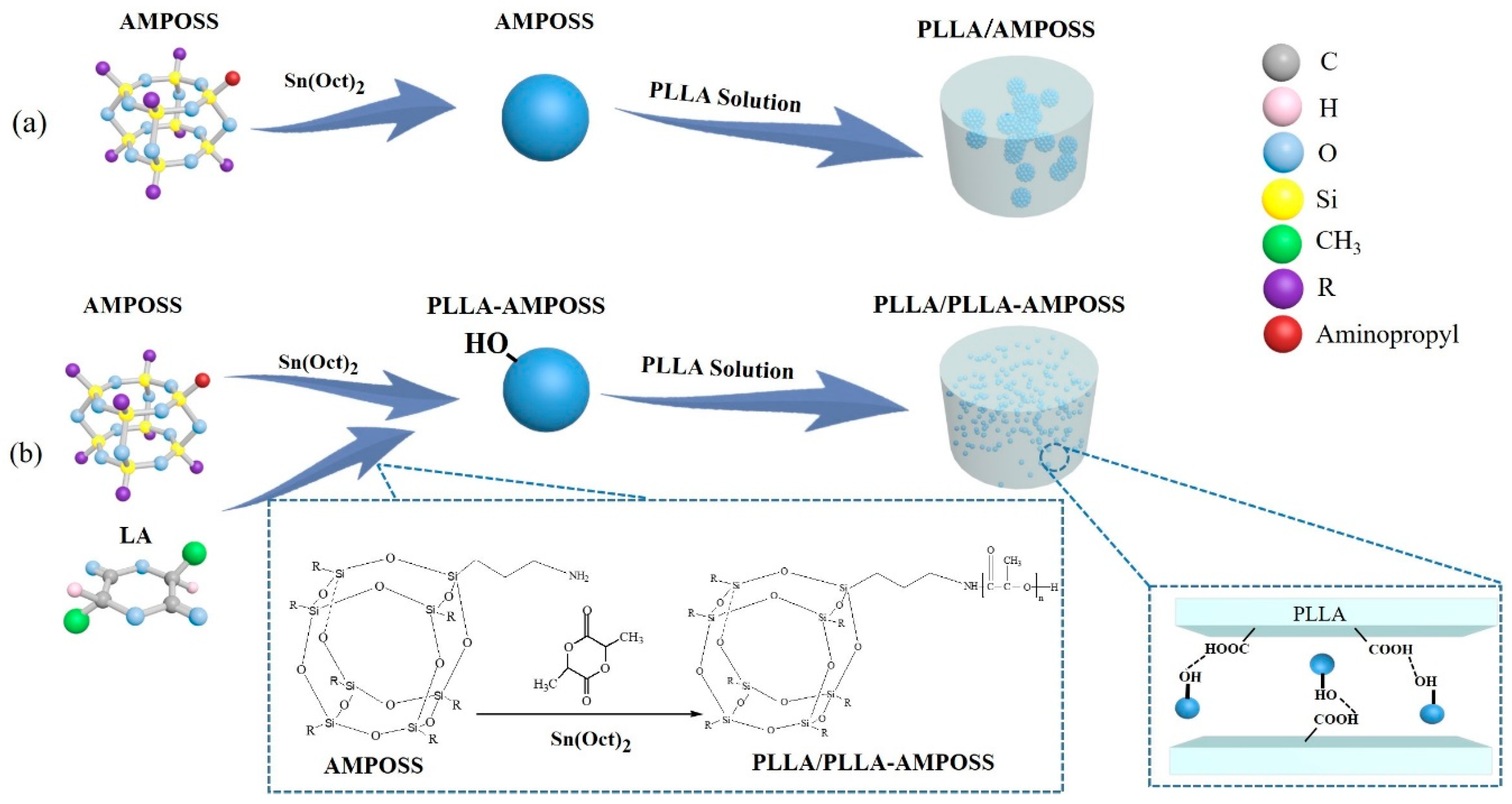
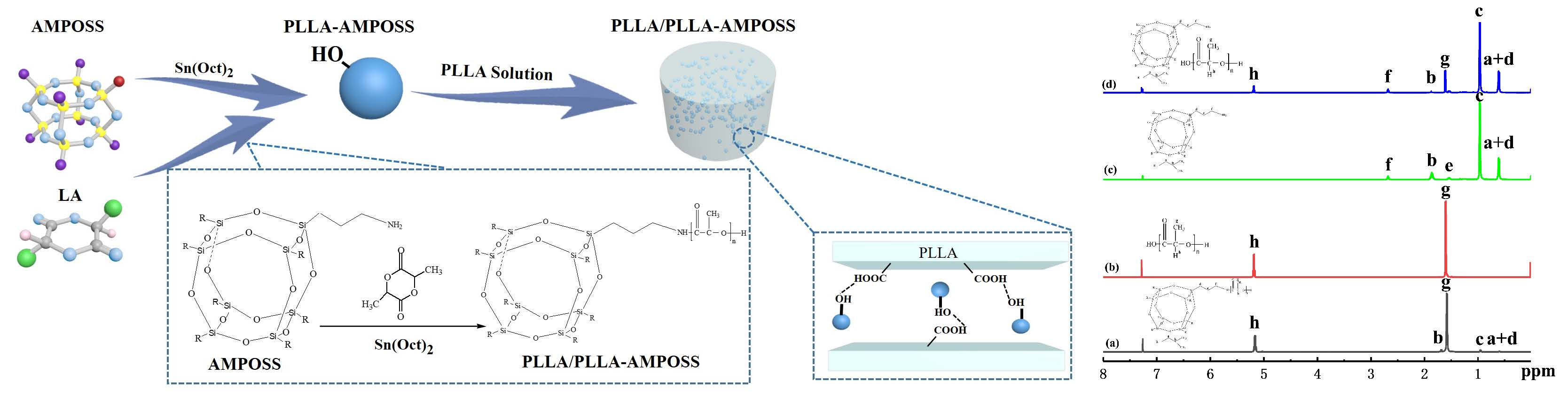
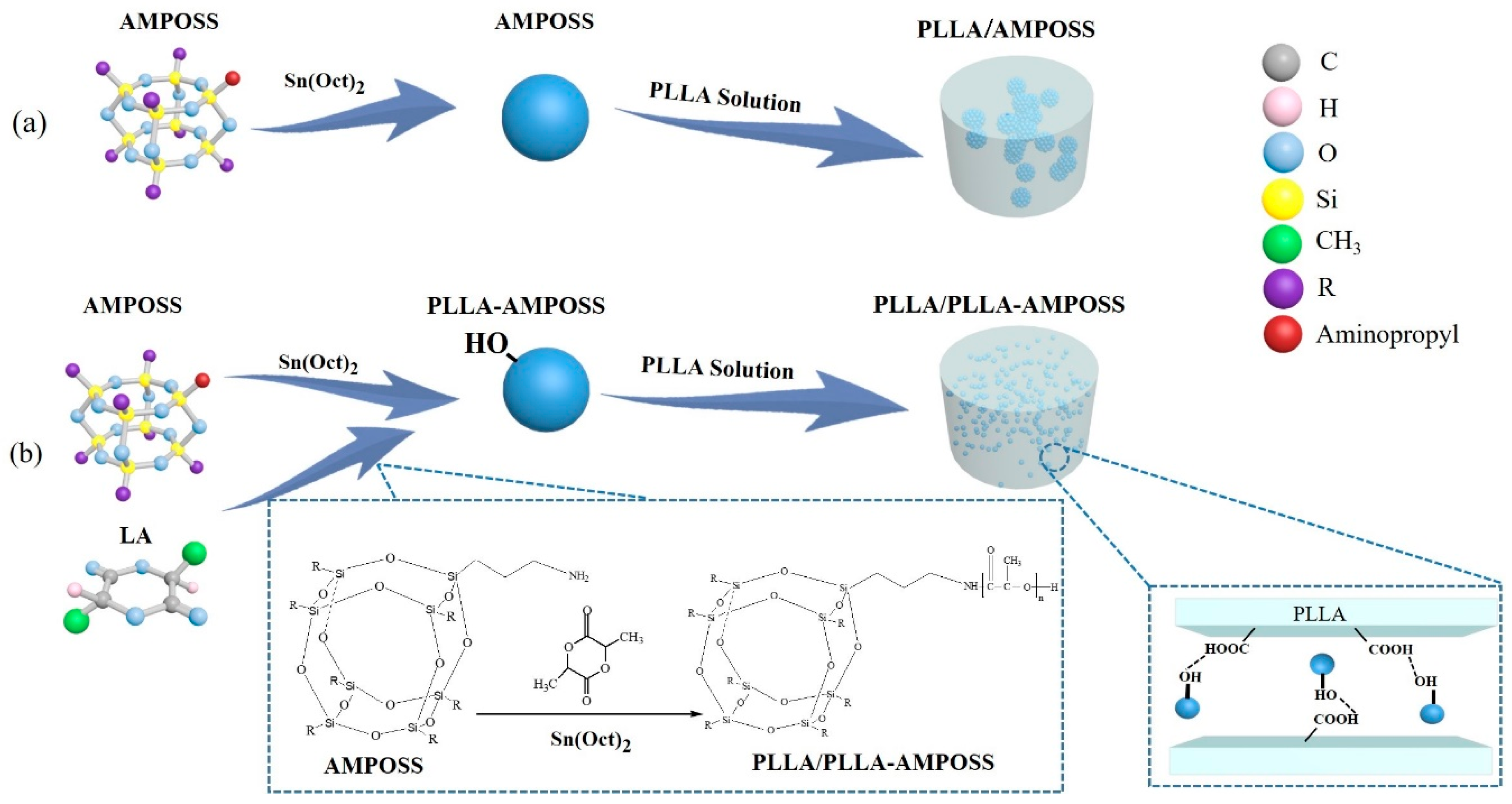
2.2.1. Synthesis of PLLA-AMPOSS Intermediates by Ring-Opening Polymerization
2.2.2. Fabrication of PLLA/PLLA-AMPOSS Nanocomposites
2.3. Sample Tests
3. Results and Discussion
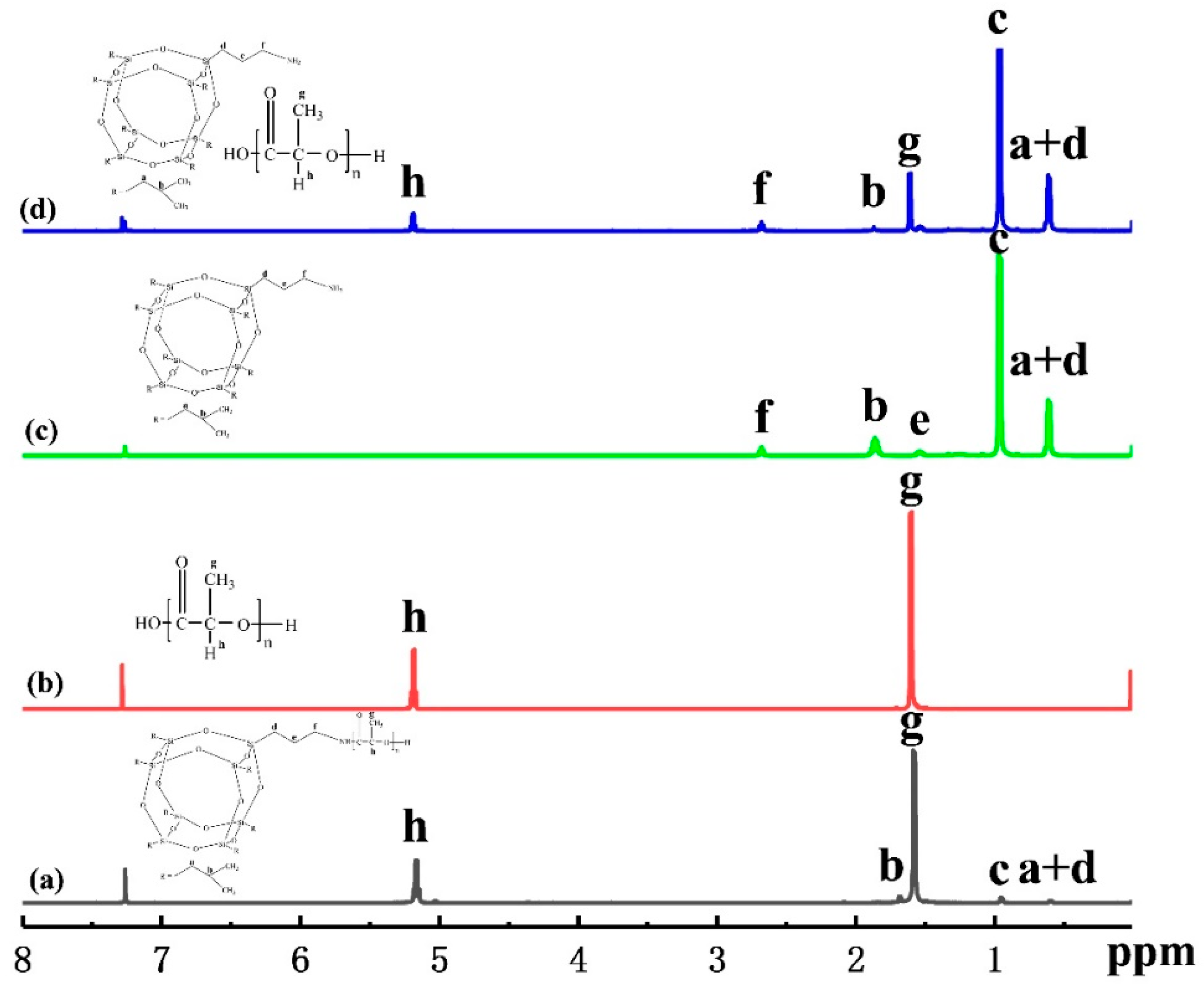
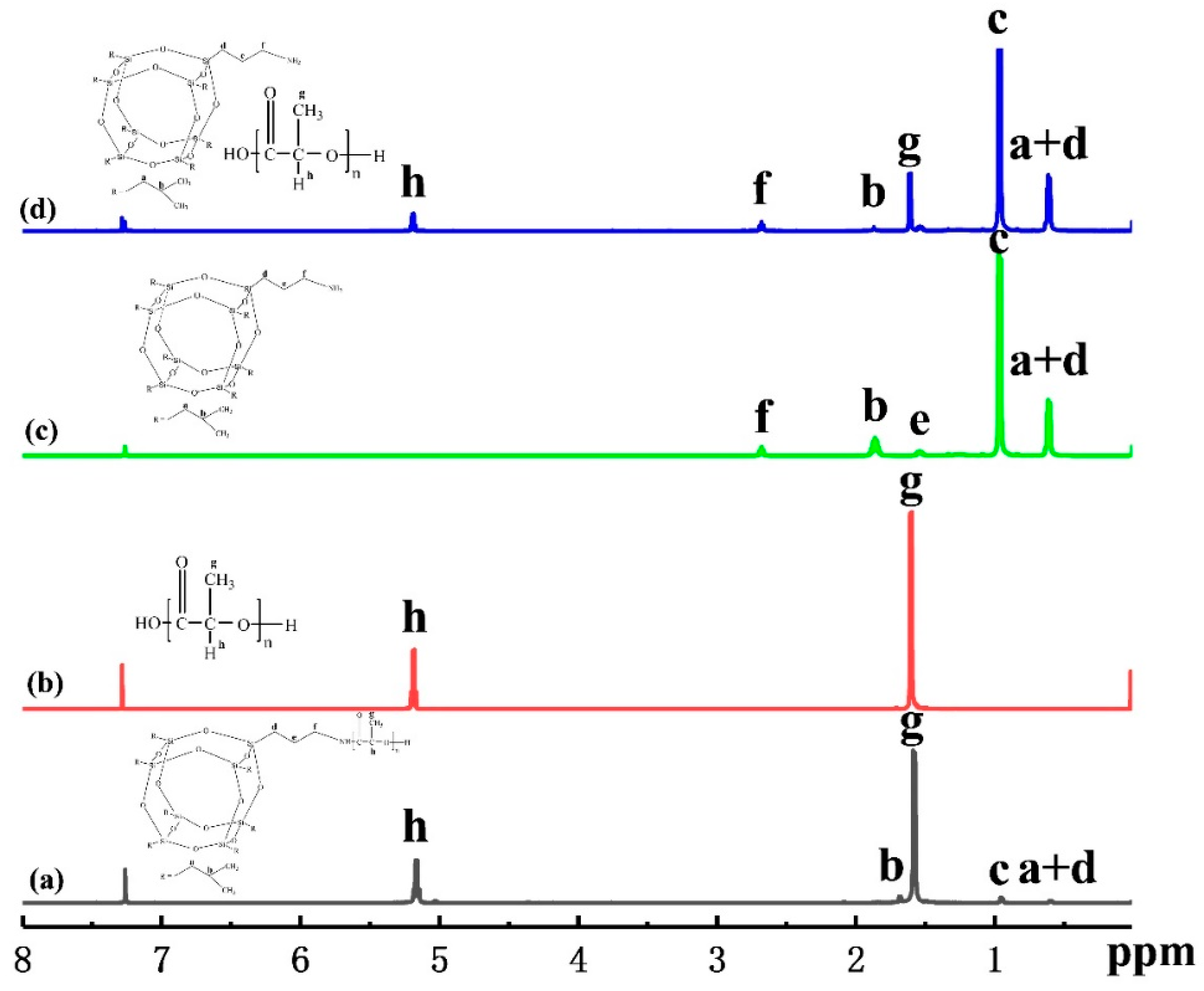
3.1. Characterization of PLLA-AMPOSS Intermediates
3.2. Structure and Performance of PLLA/PLLA-AMPOSS Composites
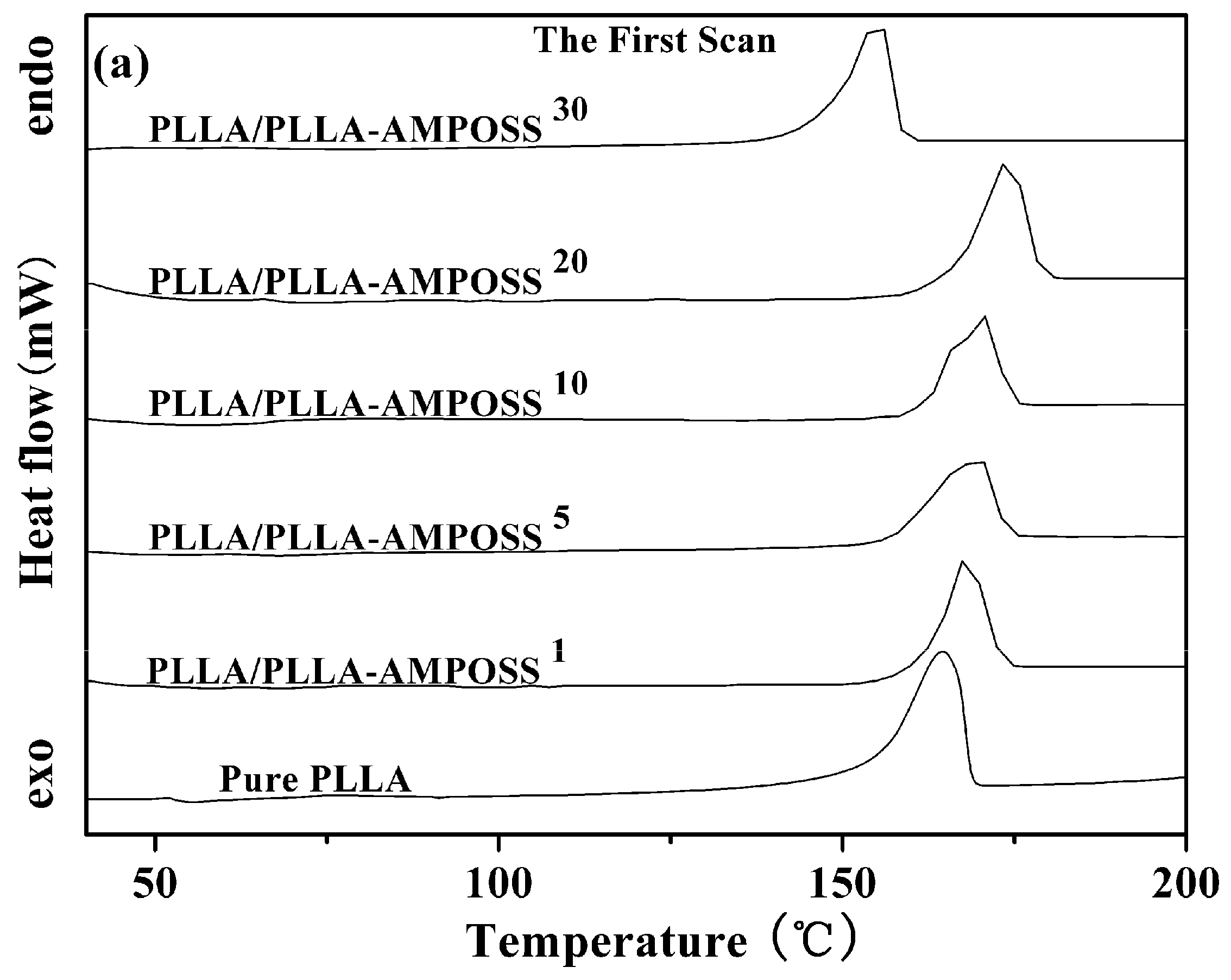
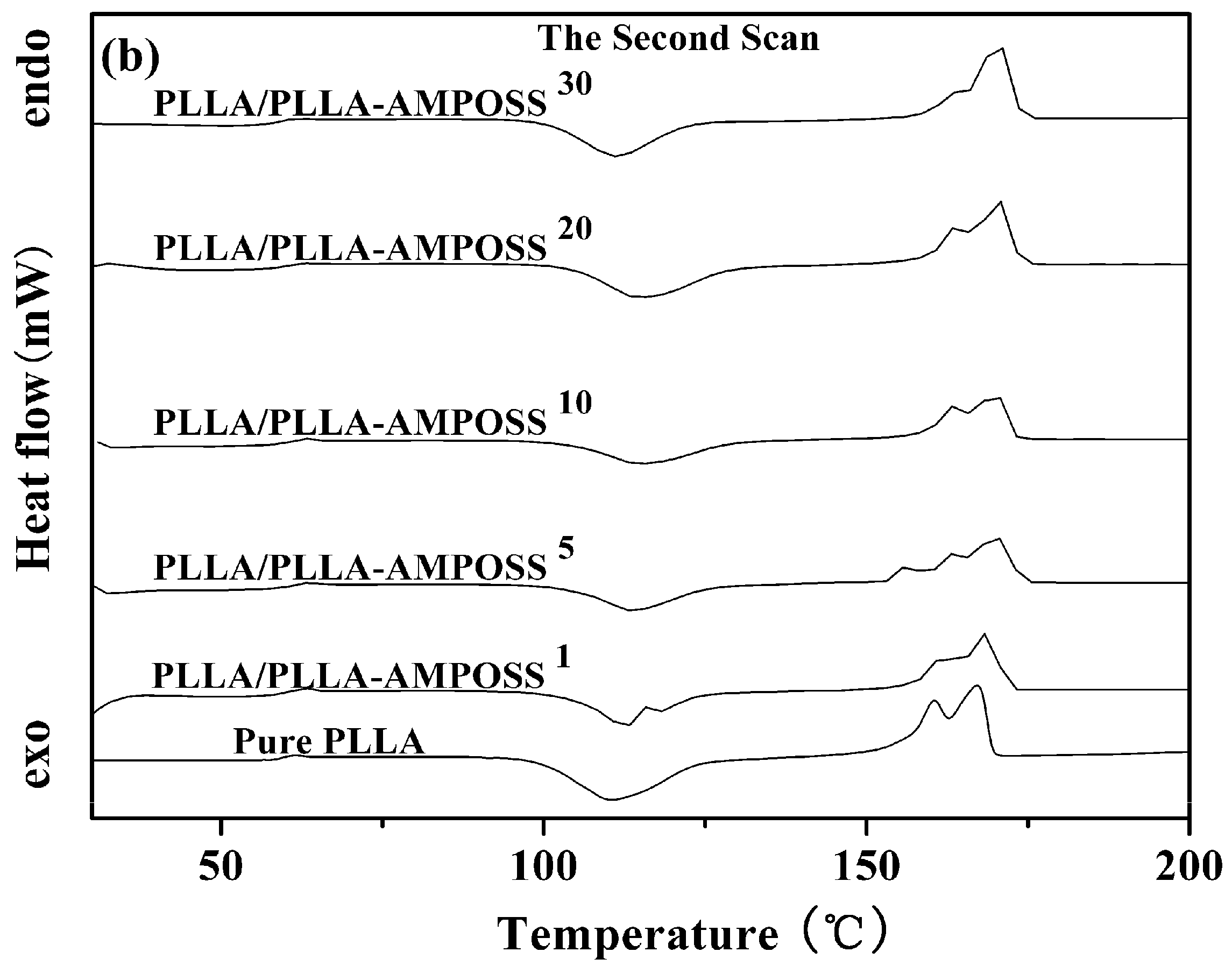
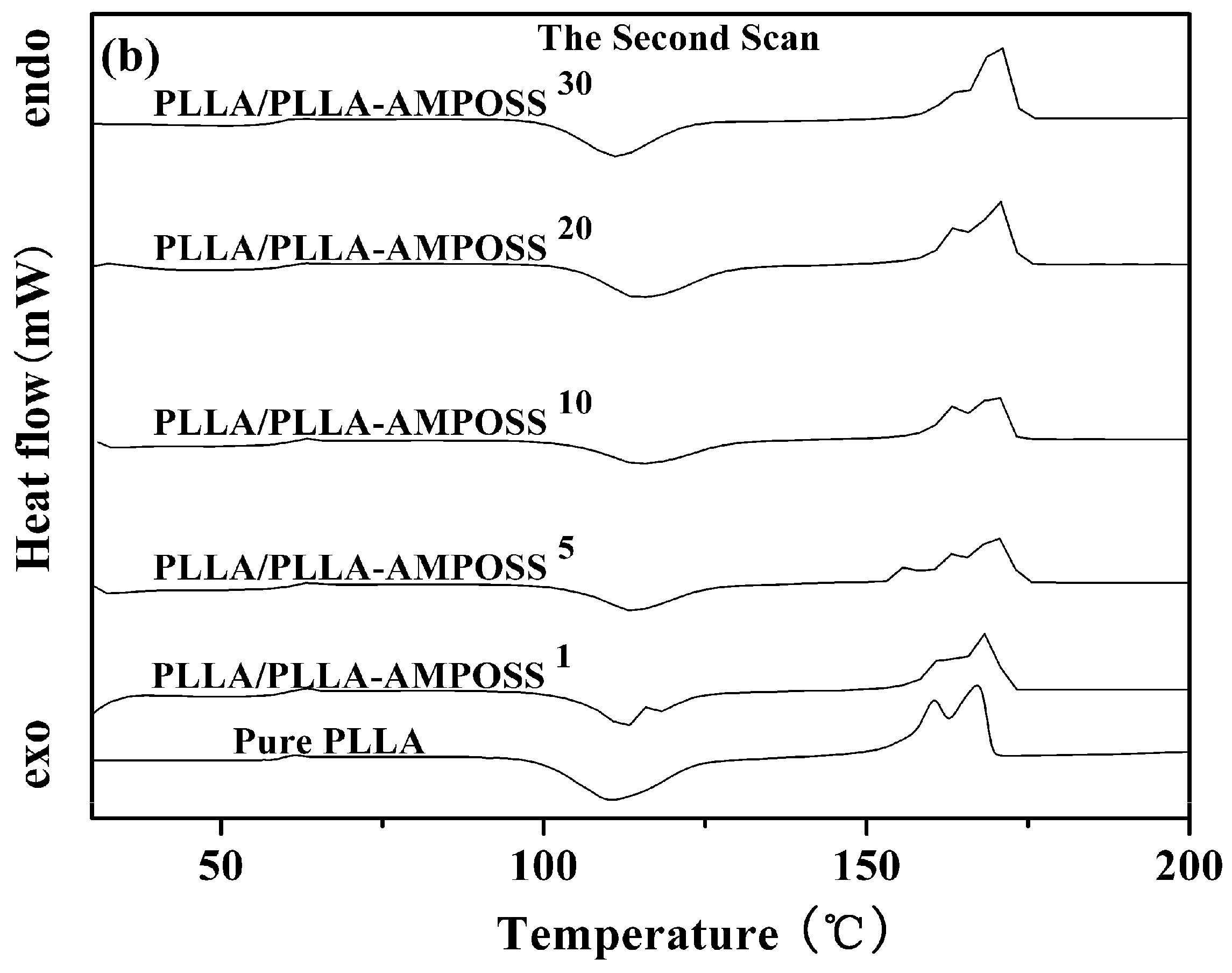
3.2.1. Thermal Properties of PLLA and PLLA/PLLA-AMPOSS Nanocomposites
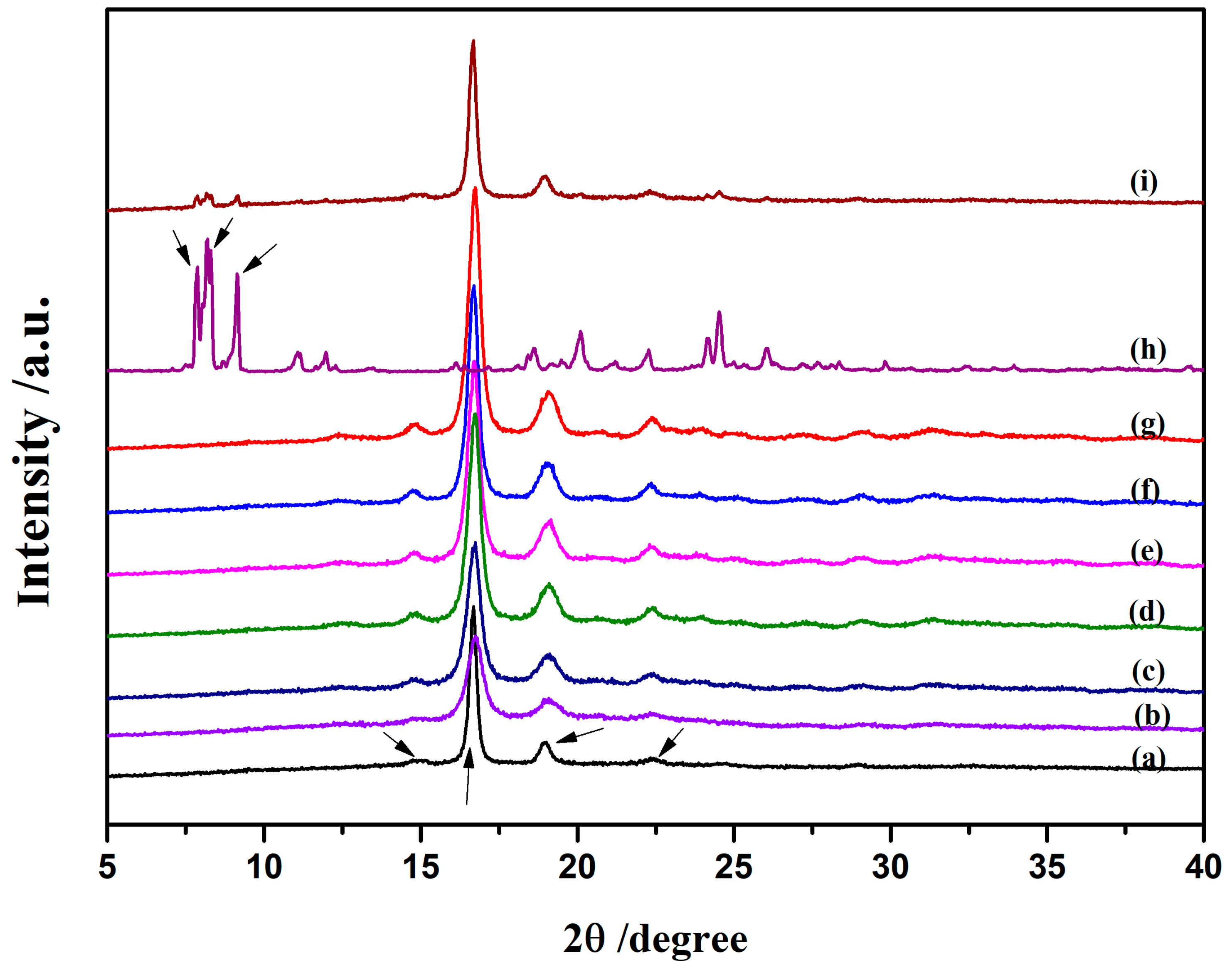
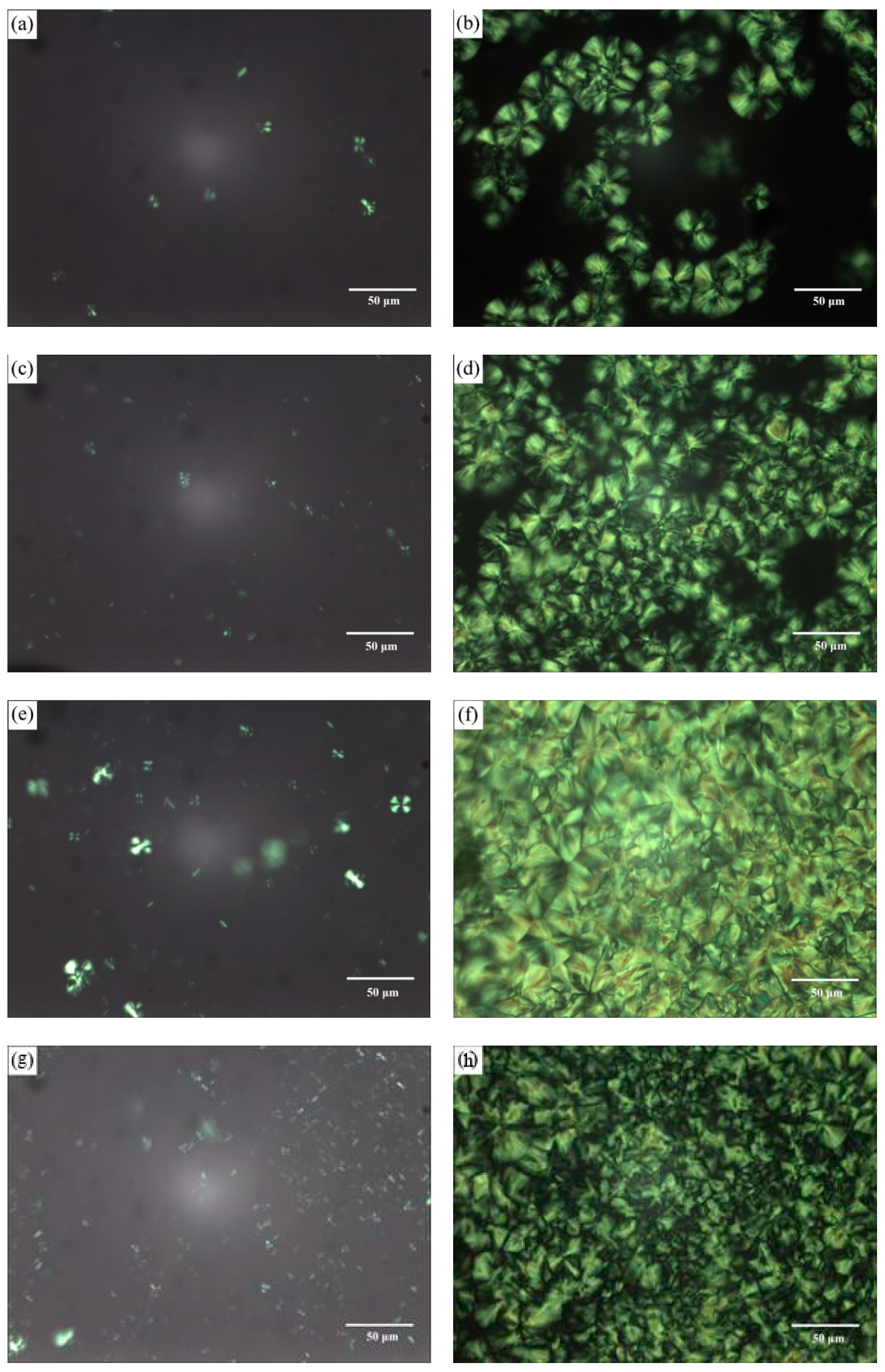
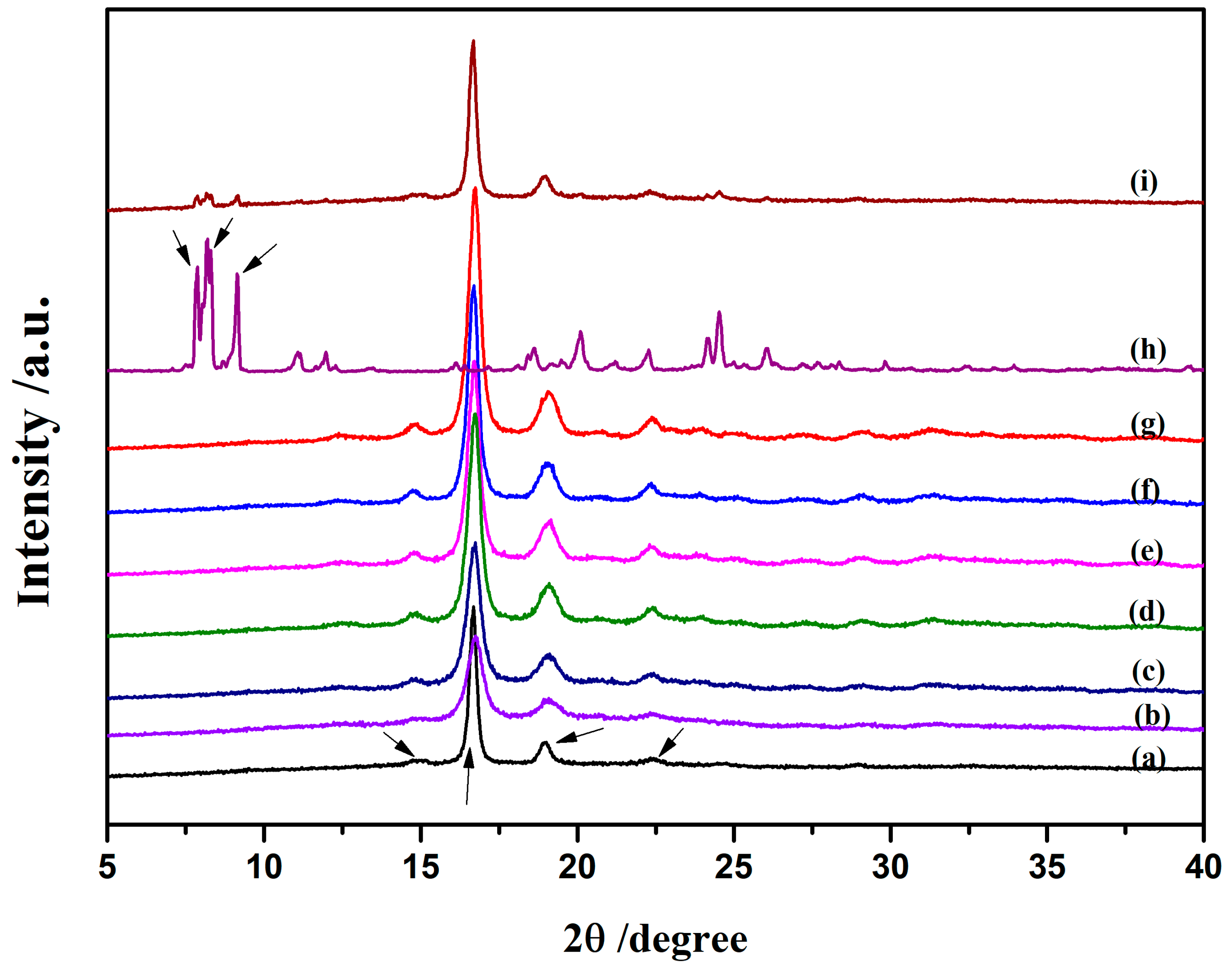
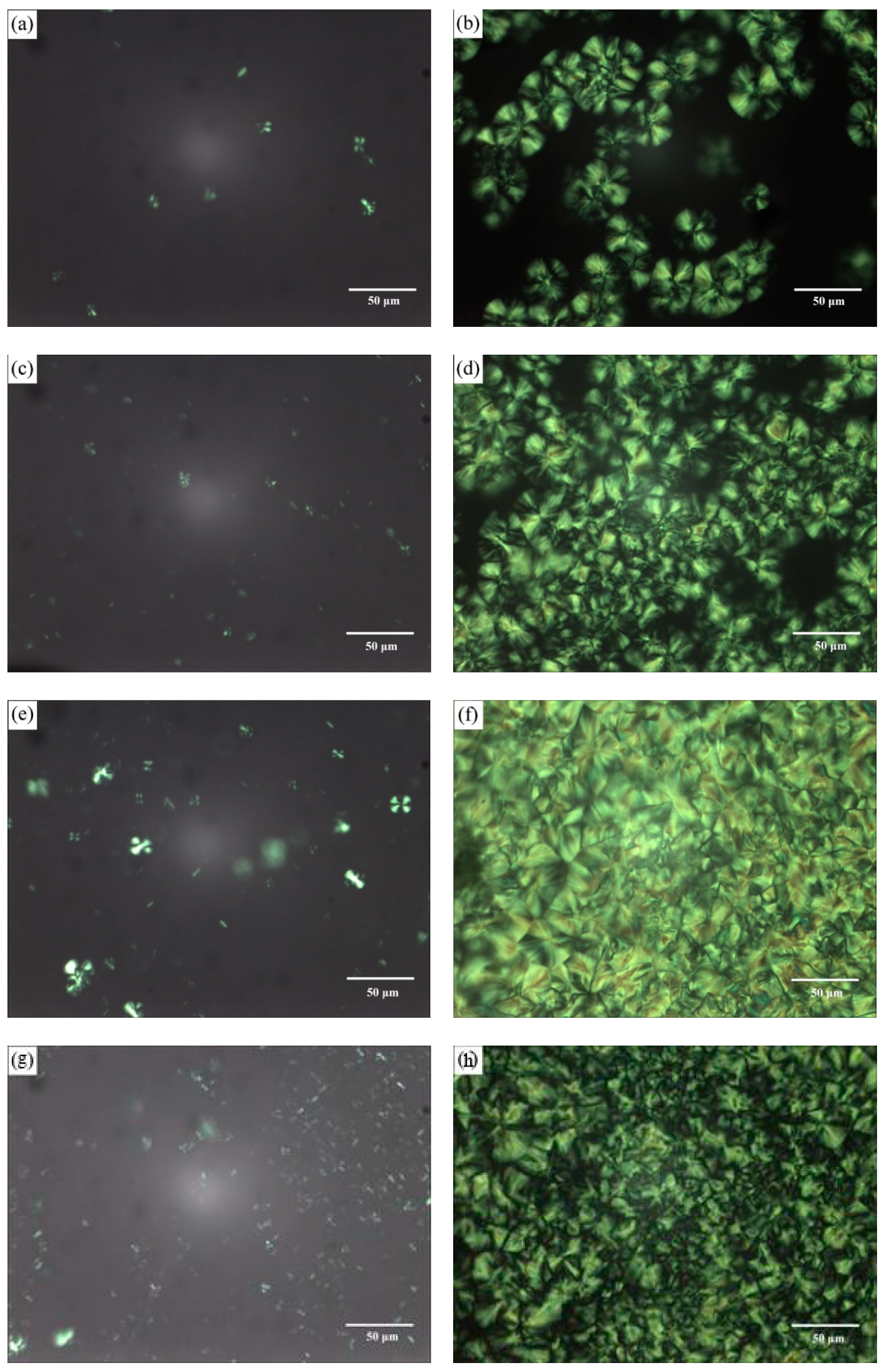
3.2.2. Morphology and Structure of Crystallization
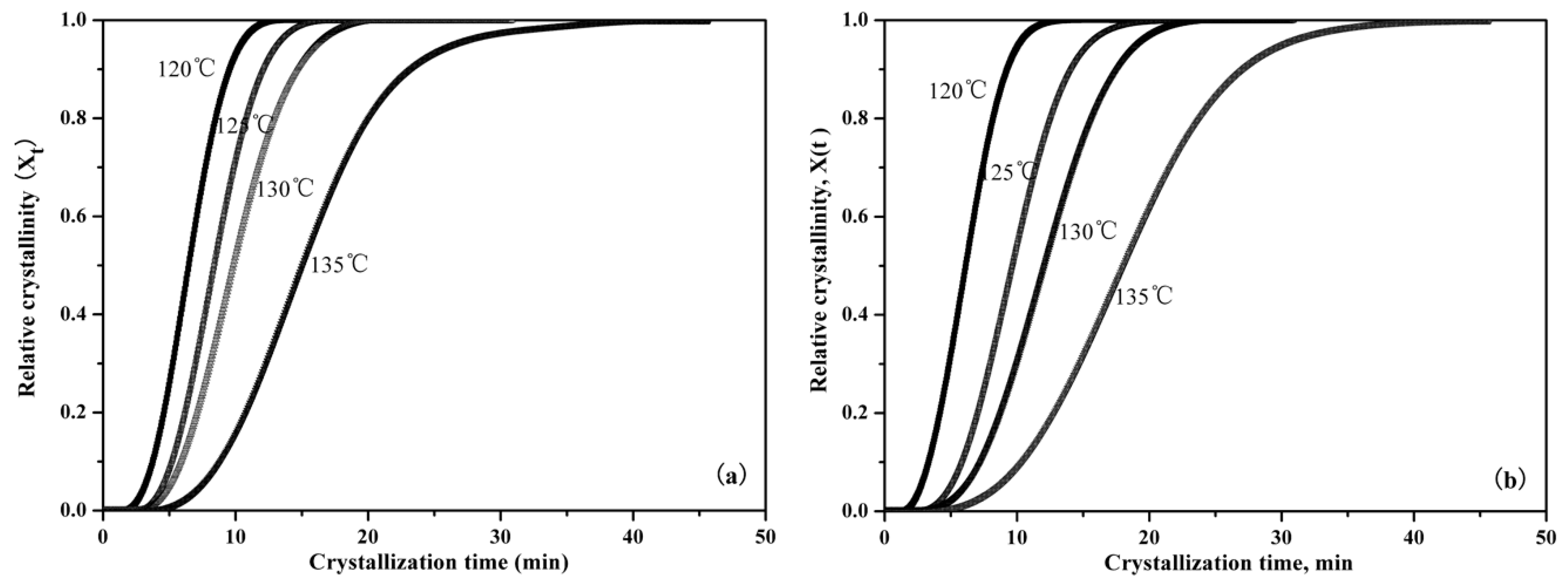
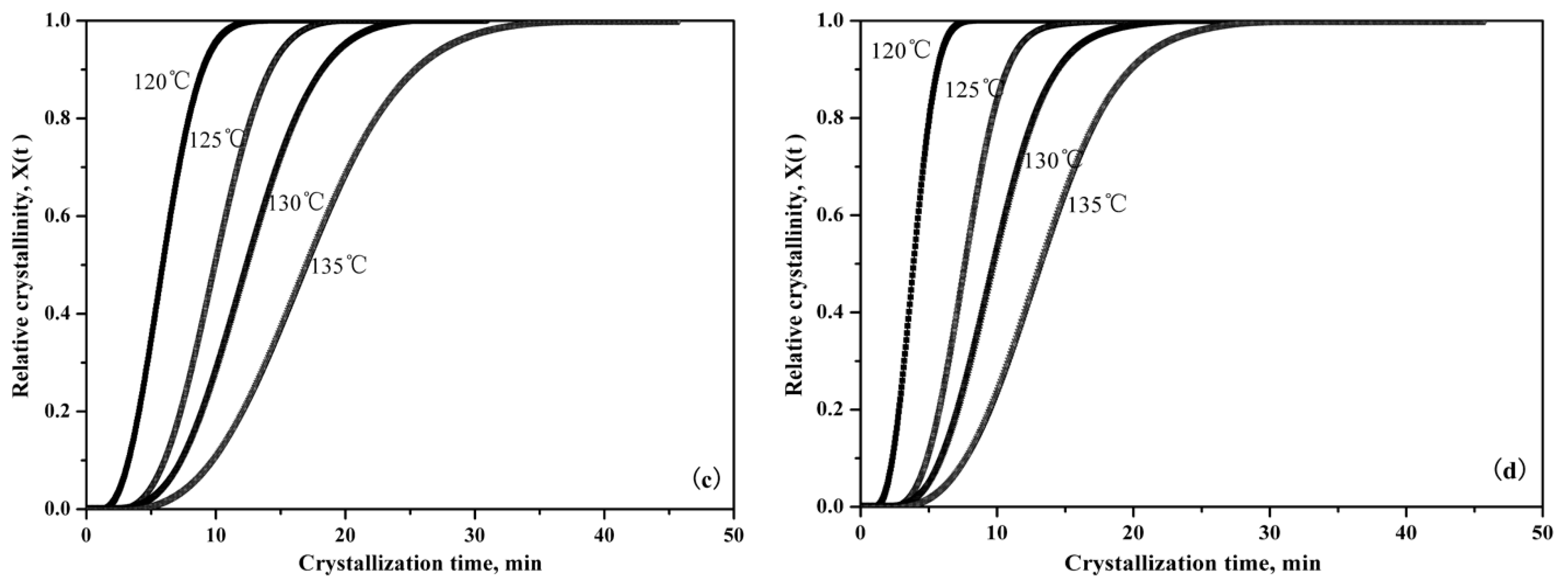
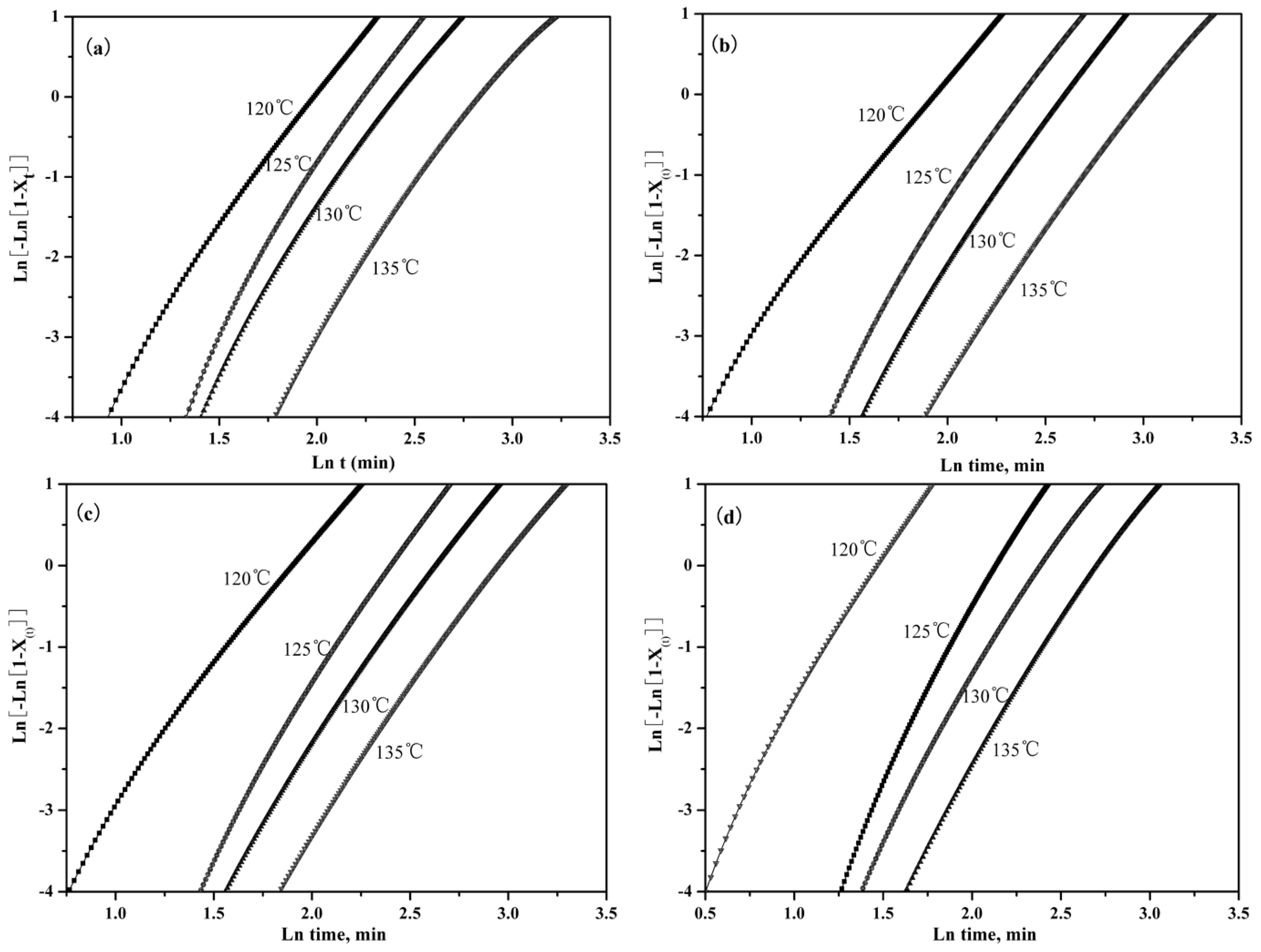
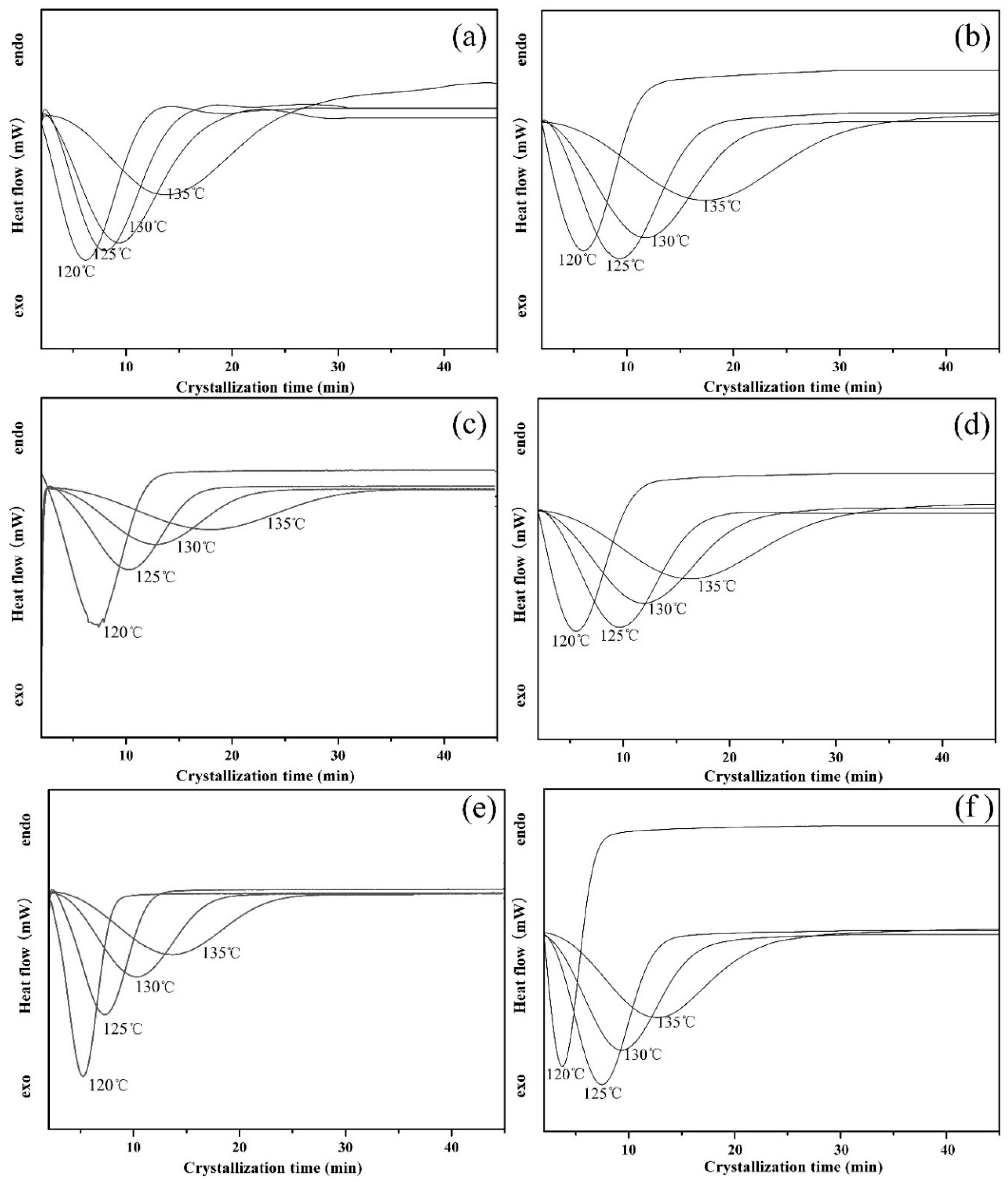
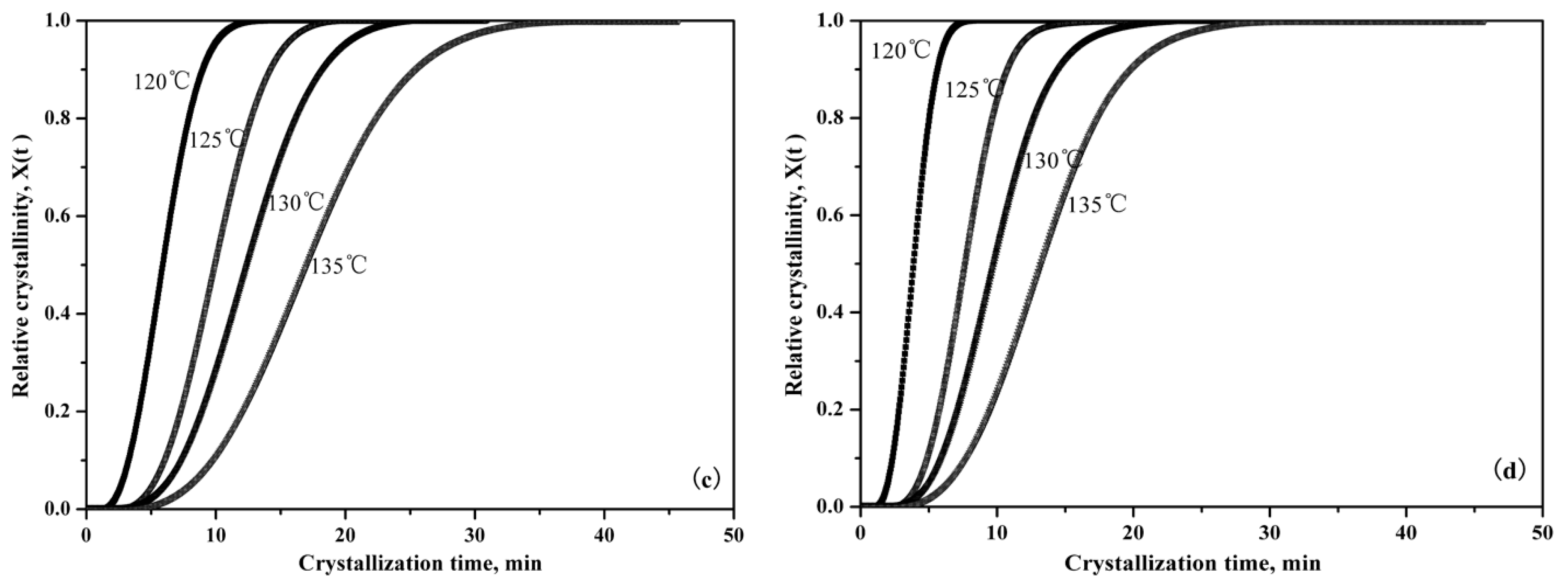
3.2.3. Isothermal Crystallization Kinetics
3.3. Morphological Characterization of Nanocomposites
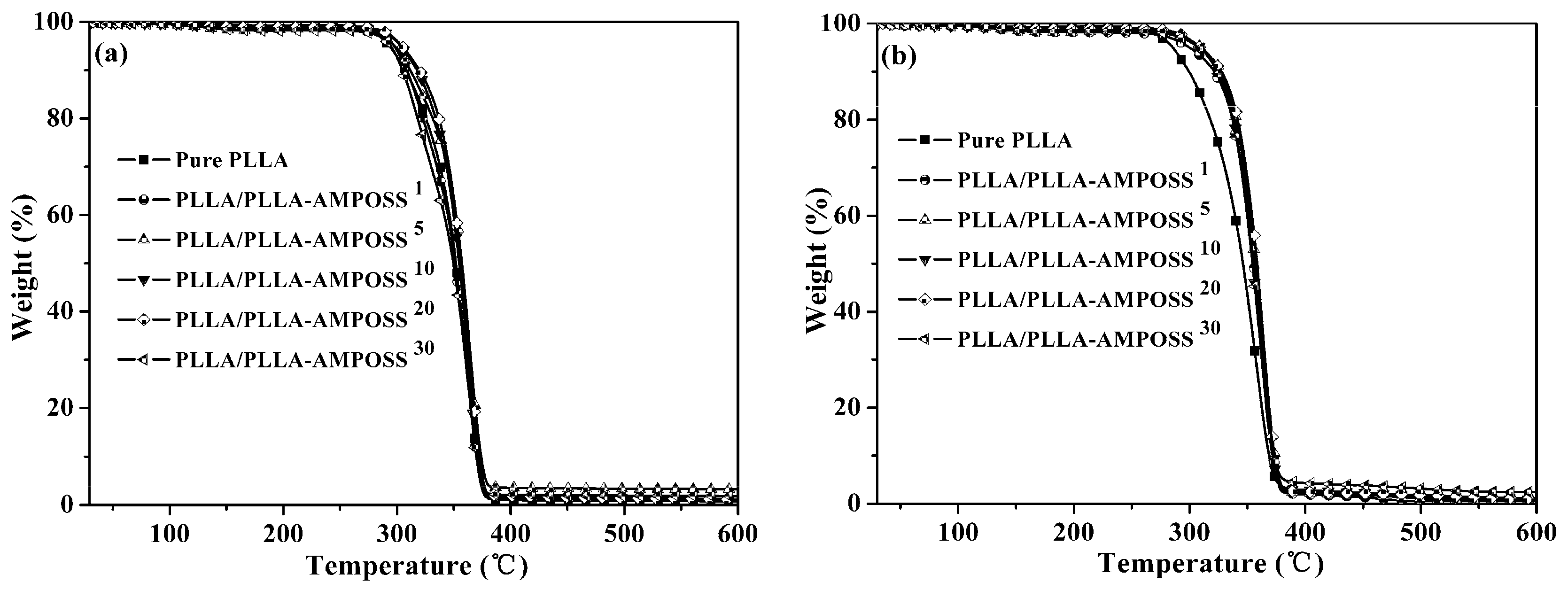
3.4. Thermal Stability of Nanocomposites
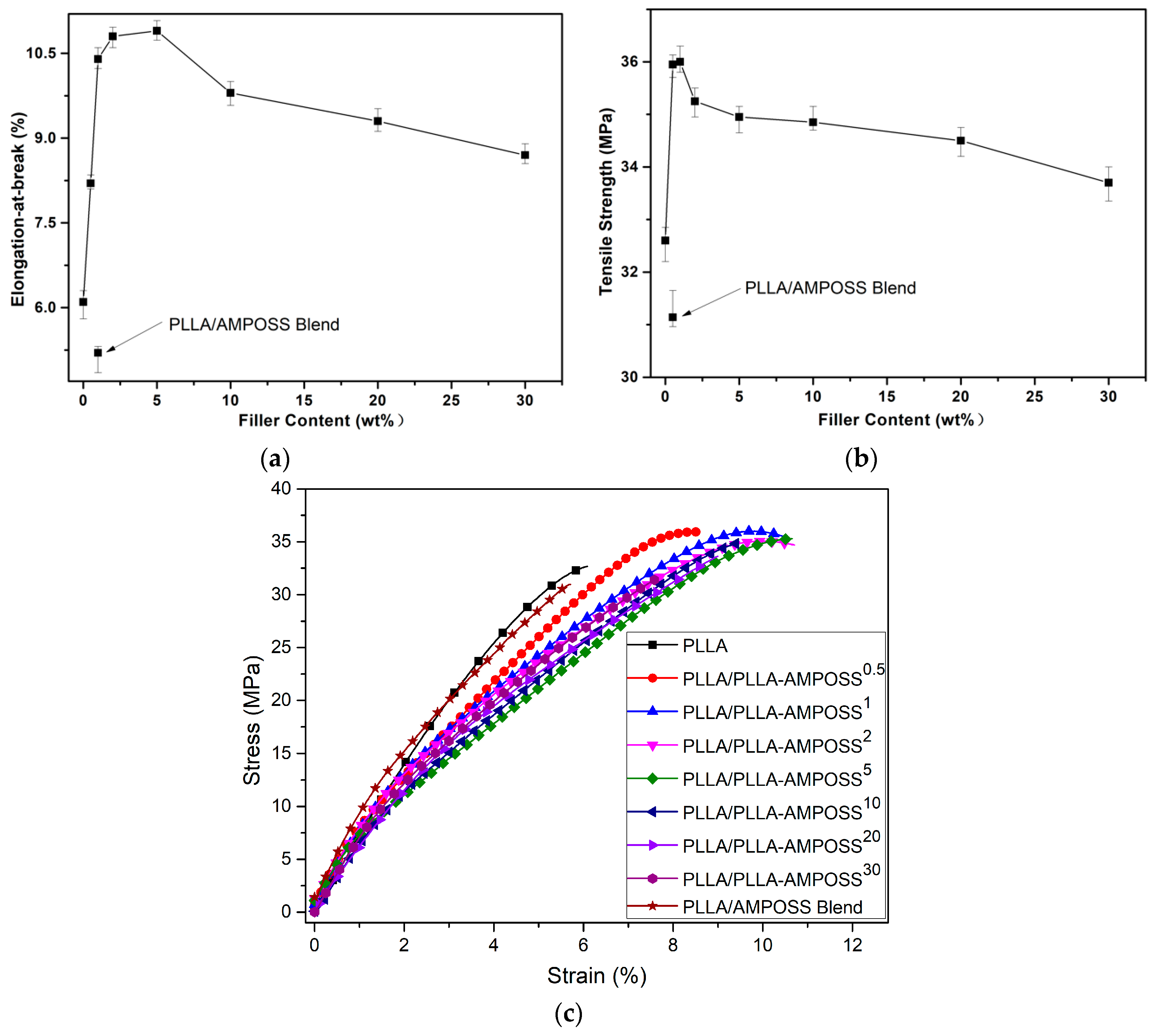
3.5. Mechanical Properties of the Nanocomposites
4. Conclusions
Supplementary Materials
Supplementary File 1Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Puchalski, M.; Siwek, P.; Panayotov, N.; Bereova, M.; Kowalska, S.; Krucinska, I. Influence of Various Climatic Conditions on the Structural Changes of Semicrystalline PLA Spun-Bonded Mulching Nonwovens during Outdoor Composting. Polymers 2019, 11, 559. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Li, Z.; Zhang, F.; Peng, X. A Novel Approach for Fabricating Poly-L-lactide Materials by One-step Bulk Polymerization. Polym. Polym. Compos. 2016, 24, 249–253. [Google Scholar] [CrossRef]
- Sasaki, T. Properties of Porous Poly(L-Lactic Acid) and its Application as a Drug Carrier. J. Soc. Fiber Sci Technol. Jpn. 2016, 72, 215. (In Japanese) [Google Scholar]
- Bian, X.; Zhang, B.; Sun, B.; Sun, Z.; Xiang, S.; Li, G.; Chen, X. Preparation of high toughness and high transparency polylactide blends resin based on multiarmed polycaprolactone-block-poly(l-lactide). Polym. Eng. Sci. 2016, 56, 1125–1137. [Google Scholar] [CrossRef]
- Liu, P.; Wu, J.; Yang, G.; Shao, H. Comparison of static mixing reaction and reactive extrusion technique for ring-opening polymerization of L-lactide. Mater. Lett. 2017, 186, 372–374. [Google Scholar] [CrossRef]
- Liu, P.; Yang, G.; Shao, H. Synthesis of poly(L-lactide) by static mixing reaction technique via ring-opening polymerization of L-lactide. Eur. Polym. J. 2017, 93, 815–821. [Google Scholar] [CrossRef]
- Colomines, G.; Domenek, S.; Ducruet, V.; Guinault, A. Influences of the Crystallization Rate on Thermal and Barrier Properties of Polylactide Acid (PLA) Food Packaging Films. Int. Mater. Form. 2008, 1, 607–610. [Google Scholar] [CrossRef]
- Nilmini, K.A. Amido-Modified Polylactide for Potential Tissue Engineering Applications. J. Biomater. Sci. Polym. Ed. 2004, 15, 595–606. [Google Scholar]
- Zhang, C.; Wang, L.; Zhai, T.; Wang, X.; Dan, Y.; Turng, L.S. The surface grafting of graphene oxide with poly (ethylene glycol) as a reinforcement for poly(lactic acid) nanocomposite scaffolds for potential tissue engineering applications. J. Mech. Behav. Biomed. Mater. 2016, 53, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, H. Physical Properties, Crystallization, and Spherulite Growth of Linear and 3-Arm Poly(L-Lactide)S. Biomacromolecules 2005, 1, 944–947. [Google Scholar] [CrossRef]
- Huang, A.; Kharbas, H.; Ellingham, T.; Mi, H.; Turng, L.S.; Peng, X. Mechanical Properties, Crystallization Characteristics, and Foaming Behavior of Polytetrafluoroethylene-Reinforced Poly(Lactic Acid) Composites. Polym. Eng. Sci. 2017, 57, 570–580. [Google Scholar] [CrossRef]
- Saeidlou, S.; Huneault, M.A.; Li, H.; Park, C.B. Poly(lactic acid) crystallization. Prog. Polym. Sci. 2012, 37, 1657–1677. [Google Scholar] [CrossRef]
- Zhou, Y.G.; Lu, S.; Wang, G.L.; Shi, F.J. Modeling and Numerical Simulation of Injection Molding of Semi-crystalline Polymer Isotactic Polypropylene. J. Comput. Theor. Nanosci. 2012, 9, 1364–1367. [Google Scholar] [CrossRef]
- Zhou, Y.G.; Wu, W.B.; Zou, J.; Turng, L.S. Dual-scale modeling and simulation of film casting of isotactic polypropylene. J. Plast. Film Sheeting 2016, 32, 239–271. [Google Scholar] [CrossRef]
- Su, B.; Zhou, Y.G.; Wu, H.H. Influence of Mechanical Properties of PP/LDPE Nanocomposites: Compatibility and Crystallization. Nanomater. Nanotechnol. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Zhou, Y.G.; Su, B.; Wu, H.H. Effect of Cold-drawn Fibers on the Self-reinforcement of the PP/LDPE Composites. J. Mater. Eng. Perform. 2017, 26, 4072–4082. [Google Scholar] [CrossRef]
- Jenkins, M.J.; Harrison, K.L.; Silva, M.M.C.G.; Whitaker, M.J.; Shakesheff, K.M.; Howdle, S.M. Characterization of Microcellular Foams Produced from Semi-crystalline PCL using Supercritical Carbon Dioxide. Eur. Polym. J. 2006, 42, 3145–3151. [Google Scholar] [CrossRef]
- Behera, K.; Yadav, M.; Chiu, F.-C.; Rhee, K.Y. Graphene Nanoplatelet-Reinforced Poly(vinylidene fluoride)/High Density Polyethylene Blend-Based Nanocomposites with Enhanced Thermal and Electrical Properties. Nanomaterials 2019, 9, 361. [Google Scholar] [CrossRef]
- Su, B.; Zhou, Y.G. Improvement of Transparencies and Mechanical Properties of Poly(cyclohexylene dimethylene cyclohexanedicarboxylate) Parts Using a Compounding Nucleating Agent to Control Crystallization. Materials 2019, 12, 563. [Google Scholar] [CrossRef]
- Zhou, Y.G.; Wu, W.B.; Lu, G.Y.; Zou, J. Isothermal and Non-Isothermal Crystallization Kinetics and Predictive Modeling in the Solidification of Poly(cyclohexylene dimethylene cyclohexanedicarboxylate) Melt. J. Elastom. Plast. 2017, 49, 132–156. [Google Scholar] [CrossRef]
- Gao, X.; Tao, W.; Lu, W.; Zhang, Q.; Zhang, Y.; Jiang, X.; Fu, S. Lectin-Conjugated Peg-Pla Nanoparticles: Preparation and Brain Delivery after Intranasal Administration. Biomaterials 2006, 27, 3482–3490. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chen, H.; Yuan, Y.; Peng, S.; Zhao, X. Synergistic effects of polyethylene glycol and organic montmorillonite on the plasticization and enhancement of poly(lactic acid). J. Appl. Polym. Sci. 2019, 136, 47576. [Google Scholar] [CrossRef]
- Jie, J.; Qiao, Q.A.; Jin, Y.; Ma, C.; Cai, H.; Meng, Y.; Cai, Z.; Feng, D. Molecular and Mesoscopic Dynamics Simulations on the Compatibility of Pla/Plasticizer Blends. Chin. J. Chem. 2012, 30, 133–138. [Google Scholar]
- Zhang, C.; Zhai, T.; Turng, L.S.; Dan, Y. Morphological, Mechanical, and Crystallization Behavior of Polylactide/Polycaprolactone Blends Compatibilized by L-Lactide/Caprolactone Copolymer. Ind. Eng. Chem. Res. 2015, 54, 9505–9511. [Google Scholar] [CrossRef]
- Srisuwan, Y.; Baimark, Y.; Suttiruengwong, S. Toughening of Poly(L-lactide) with Blends of Poly(epsilon-caprolactone-co-L-lactide) in the Presence of Chain Extender. Int. J. Biomater. 2018, 1294397. [Google Scholar] [CrossRef]
- Rahaman, M.H.; Rana, M.M.; Gafur, M.A.; Mohona, A.A. Preparation and analysis of poly(l-lactic acid) composites with oligo(d-lactic acid)-grafted cellulose. J. Appl. Polym. Sci. 2019, 136, 47424. [Google Scholar] [CrossRef]
- Kowalczyk, M.; Pluta, M.; Piorkowska, E.; Krasnikova, N. Plasticization of Polylactide with Block Copolymers of Ethylene Glycol and Propylene Glycol. J. Appl. Polym. Sci. 2012, 125, 4292–4301. [Google Scholar] [CrossRef]
- Nofar, M.; Park, C.B. Poly (lactic acid) foaming. Prog. Polym. Sci. 2014, 39, 1721–1741. [Google Scholar] [CrossRef]
- Volpe, V.; De Filitto, M.; Klofacova, V. Effect of Mold Opening on the Properties of PLA Samples Obtained by Foam Injection Molding. Polym. Eng. Sci. 2018, 58, 475–484. [Google Scholar] [CrossRef]
- Li, H.; Huneault, M.A. Effect of Nucleation and Plasticization on the Crystallization of Poly(Lactic Acid). Polymer 2007, 48, 6855–6866. [Google Scholar] [CrossRef]
- Ye, H.; Hou, K.; Zhou, Q. Improve the thermal and mechanical properties of poly(L-lactide) by forming nanocomposites with pristine vermiculite. Chin. J. Polym. Sci. 2016, 34, 1–12. [Google Scholar] [CrossRef]
- Yu, J.; Qiu, Z. Preparation and Properties of Biodegradable Poly(L-Lactide)/Octamethyl-Polyhedral Oligomeric Silsesquioxanes Nanocomposites with Enhanced Crystallization Rate Via Simple Melt Compounding. ACS Appl. Mater. Interfaces 2011, 3, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Davachi, S.M.; Kaffashi, B. Preparation and Characterization of Poly L-Lactide/Triclosan Nanoparticles for Specific Antibacterial and Medical Applications. Int. J. Polym. Mater. Polym. Biomater. 2015, 64, 497–508. [Google Scholar] [CrossRef]
- Naffakh, M.; Marco, C. Isothermal crystallization kinetics and melting behavior of poly(L-lactic acid)/WS2 inorganic nanotube nanocomposites. J. Mater. Sci. 2015, 50, 6066–6074. [Google Scholar] [CrossRef]
- Yang, J.; Cao, X.; Zhao, Y.; Wang, L.; Liu, B.; Jia, J.; Liang, H.; Chen, M. Enhanced pH stability, cell viability and reduced degradation rate of poly(L-lactide)-based composite in vitro: Effect of modified magnesium oxide nanoparticles. J. Biomater. Sci. Polym. Ed. 2017, 28, 486–503. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.Z.; Yang, X.M.; Li, Q.F. Influences of Terminal Poss on Crystallization and Degradation Behavior of Pcl-Plla Block Copolymer. Polym. Cryst. 2018, 1, 79070–79080. [Google Scholar] [CrossRef]
- Elda, M.; Milena, G.; Stephen, C.; Janis, M.; Manwar, H.; Simon, G.P. Poly(Ethylene Glycol)-Octafunctionalized Polyhedral Oligomeric Silsesquioxane: Synthesis and Thermal Analysis. Macromolecules 2007, 40, 2694–2701. [Google Scholar]
- Pyun, J.; Matyjaszewski, K.; Wu, J.; Kim, G.M.; Chun, S.B.; Mather, P.T. Aba Triblock Copolymers Containing Polyhedral Oligomeric Silsesquioxane Pendant Groups: Synthesis and Unique Properties. Polymer 2003, 44, 2739–2750. [Google Scholar] [CrossRef]
- Li, G.; Wang, L.; Ni, H.; Pittman, C.U. Polyhedral Oligomeric Silsesquioxane (Poss) Polymers and Copolymers: A Review. J. Inorg. Organomet. Polym. 2001, 11, 123–154. [Google Scholar] [CrossRef]
- Sellinger, A.; Laine, R.M.; Chu, V.; Viney, C. Palladium-Catalyzed and Platinum-Catalyzed Coupling Reactions of Allyloxy Aromatics with Hydridosilanes and Hydridosiloxanes-Novel Liquid-Crystalline Organosilane Materials. J. Polym. Sci. Part A Polym. Chem. 1994, 32, 3069–3089. [Google Scholar] [CrossRef]
- Bolln, C.; Tsuchida, A.; Frey, H.; Mülhaupt, R. Thermal Properties of the Homologous Series of 8-Fold Alkyl-Substituted Octasilsesquioxanes. Chem. Mater. 1997, 9, 1475–1479. [Google Scholar] [CrossRef]
- Chen, K.B.; Chen, H.Y.; Yang, S.H.; Hsu, C.S. Synthesis and Opto-Electrical Properties of Stellar Polyfluorene Derivatives Containing Polyhedral Oligomeric Silsesquioxanes as the Center Core. J. Polym. Res. 2006, 13, 237–245. [Google Scholar] [CrossRef]
- Ryo, T.; Yasuyuki, T.; Michael, Z.A.; Jiwon, C.; Richard, M.L. Octa(Aminophenyl)Silsesquioxane as a Nanoconstruction Site. J. Am. Chem. Soc. 2001, 123, 12416–12417. [Google Scholar]
- Kricheldorf, H.R.; Kreiser-Saunders, I.; Boettcher, C. Polylactones: 31. Sn(Ii)Octoate-Initiated Polymerization of L-Lactide: A Mechanistic Study. Polymer 1995, 36, 1253–1259. [Google Scholar] [CrossRef]
- Sun, Y.; Chaobin, H. Biodegradable “Core-Shell” Rubber Nanoparticles and Their Toughening of Poly(Lactides). Macromolecules 2013, 46, 9625–9633. [Google Scholar] [CrossRef]
- Fox, D.M.; Lee, J.; Citro, C.J.; Novy, M. Flame Retarded Poly(Lactic Acid) Using Poss-Modified Cellulose. 1. Thermal and Combustion Properties of Intumescing Composites. Polym. Degrad. Stab. 2013, 98, 590–596. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, X.; Zhang, W.; Zheng, S. Star-Shaped Poly(ε-Caprolactone) with Polyhedral Oligomeric Silsesquioxane Core. Polymer 2006, 47, 6814–6825. [Google Scholar] [CrossRef]
- Liu, Z.; Hu, D.; Lin, H.; Li, W.; Tian, J.; Lu, L.; Zhou, C.J. Simultaneous Improvement in Toughness, Strength and Biocompatibility of Poly(Lactic Acid) with Polyhedral Oligomeric Silsesquioxane. Chem. Eng. J. 2018, 346, 649–661. [Google Scholar] [CrossRef]
- Zhou, Y.G.; Su, B.; Turng, L.S. Depositing—Induced Effects of Isotactic Polypropylene and Polycarbonate Composites During Fused Deposition Modeling. Rapid Prototyp. J. 2017, 23, 869–880. [Google Scholar] [CrossRef]
- Zhou, Y.G.; Su, B.; Turng, L.S. Influence of Processing Conditions on Morphological Structure and Ductility of Water-Foamed Injection Molded PP/LDPE Blended Parts. Cell. Polym. 2017, 36, 51–74. [Google Scholar] [CrossRef]
- Zhou, Y.G.; Su, B.; Turng, L.S. Mechanical Properties, Fiber Orientation, and Length Distribution of Glass Fiber-Reinforced Polypropylene Parts: Influence of Water-Foaming Technology. Polym. Compos. 2018, 39, 4386–4399. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | First Scan | Second Scan | |||||
|---|---|---|---|---|---|---|---|
| Tm (°C) | Tg (°C) | Tcc (°C) | ΔHcc (J/g) | Tm(°C) | ΔHm (J/g) | ||
| Tm1 | Tm2 | ||||||
| Pure PLLA | 164.6 | 59.8 | 110.5 | 38.1 | 160.5 | 167.2 | 42.2 |
| PLLA/PLLA-AMPOSS1 | 167.4 | 60.0 | 112.7 | 41.7 | 160.8 | 168.3 | 42.9 |
| PLLA/PLLA-AMPOSS5 | 169.5 | 61.3 | 113.7 | 41.8 | 163.1 | 169.8 | 46.0 |
| PLLA/PLLA-AMPOSS10 | 170.7 | 60.9 | 115.1 | 44.0 | 163.3 | 169.8 | 47.3 |
| PLLA/PLLA-AMPOSS20 | 173.3 | 60.3 | 114.9 | 44.2 | 163.3 | 170.1 | 48.0 |
| PLLA/PLLA-AMPOSS30 | 156.0 | 59.2 | 111.3 | 40.6 | 160.7 | 170.3 | 48.8 |
| Samples | Tc (°C) | n | k (°C min−1) | t1/2 |
|---|---|---|---|---|
| Pure PLLA | 120 | 3.19 | 1.43 × 10−3 | 6.93 |
| 125 | 3.29 | 2.94 × 10−4 | 10.6 | |
| 130 | 3.15 | 2.08 × 10−4 | 13.2 | |
| 135 | 3.05 | 7.68 × 10−5 | 19.8 | |
| PLLA/PLLA-AMPOSS1 | 120 | 3.01 | 2.95 × 10−3 | 6.15 |
| 125 | 3.38 | 3.18 × 10−4 | 9.74 | |
| 130 | 3.35 | 1.62 × 10−4 | 12.1 | |
| 135 | 3.15 | 7.36 × 10−5 | 18.3 | |
| PLLA/PLLA-AMPOSS5 | 120 | 2.98 | 3.13 × 10−3 | 6.12 |
| 125 | 3.42 | 2.80 × 10−4 | 9.82 | |
| 130 | 3.31 | 1.71 × 10−4 | 12.3 | |
| 135 | 3.15 | 8.27 × 10−5 | 17.6 | |
| PLLA/PLLA-AMPOSS10 | 120 | 2.95 | 3.63 × 10−3 | 5.93 |
| 125 | 3.51 | 2.16 × 10−4 | 9.99 | |
| 130 | 3.28 | 1.78 × 10−4 | 12.5 | |
| 135 | 3.16 | 8.67 × 10−5 | 17.2 | |
| PLLA/PLLA-AMPOSS20 | 120 | 3.17 | 7.02 × 10−3 | 3.82 |
| 125 | 3.68 | 3.75 × 10−4 | 7.58 | |
| 130 | 3.29 | 3.39 × 10−4 | 9.69 | |
| 135 | 3.21 | 2.18 × 10−4 | 13.03 | |
| PLLA/PLLA-AMPOSS30 | 120 | 3.38 | 6.90 × 10−3 | 3.91 |
| 125 | 3.77 | 3.13 × 10−4 | 7.72 | |
| 130 | 3.31 | 3.60 × 10−4 | 9.84 | |
| 135 | 3.19 | 1.75 × 10−4 | 13.4 |
| Sample | Nitrogen Gas Condition | Air Gas Condition | ||||
|---|---|---|---|---|---|---|
| T0.05 (°C) | T0.50 (°C) | Residue (%) | T0.05 (°C) | T0.50 (°C) | Residue (%) | |
| Pure PLLA | 295 | 352 | 1.11 | 286 | 346 | 0.73 |
| PLLA/PLLA-AMPOSS1 | 298 | 352 | 0.59 | 302 | 355 | 1.05 |
| PLLA/PLLA-AMPOSS5 | 299 | 357 | 0.78 | 311 | 357 | 3.23 |
| PLLA/PLLA-AMPOSS10 | 305 | 355 | 0.91 | 311 | 355 | 1.01 |
| PLLA/PLLA-AMPOSS20 | 306 | 357 | 1.08 | 311 | 358 | 1.12 |
| PLLA/PLLA-AMPOSS30 | 296 | 350 | 2.44 | 310 | 354 | 1.84 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, X.-X.; Lu, H.; Lu, L.; Xu, H.-Q.; Zhou, Y.-G.; Zou, J. Improving the Thermal and Mechanical Properties of Poly(l-lactide) by Forming Nanocomposites with an in Situ Ring-Opening Intermediate of Poly(l-lactide) and Polyhedral Oligomeric Silsesquioxane. Nanomaterials 2019, 9, 748. https://doi.org/10.3390/nano9050748
Lei X-X, Lu H, Lu L, Xu H-Q, Zhou Y-G, Zou J. Improving the Thermal and Mechanical Properties of Poly(l-lactide) by Forming Nanocomposites with an in Situ Ring-Opening Intermediate of Poly(l-lactide) and Polyhedral Oligomeric Silsesquioxane. Nanomaterials. 2019; 9(5):748. https://doi.org/10.3390/nano9050748
Chicago/Turabian StyleLei, Xiu-Xiu, Hao Lu, Lei Lu, Hai-Qing Xu, Ying-Guo Zhou, and Jun Zou. 2019. "Improving the Thermal and Mechanical Properties of Poly(l-lactide) by Forming Nanocomposites with an in Situ Ring-Opening Intermediate of Poly(l-lactide) and Polyhedral Oligomeric Silsesquioxane" Nanomaterials 9, no. 5: 748. https://doi.org/10.3390/nano9050748
APA StyleLei, X.-X., Lu, H., Lu, L., Xu, H.-Q., Zhou, Y.-G., & Zou, J. (2019). Improving the Thermal and Mechanical Properties of Poly(l-lactide) by Forming Nanocomposites with an in Situ Ring-Opening Intermediate of Poly(l-lactide) and Polyhedral Oligomeric Silsesquioxane. Nanomaterials, 9(5), 748. https://doi.org/10.3390/nano9050748