Phosphorescent Modulation of Metallophilic Clusters and Recognition of Solvents through a Flexible Host-Guest Assembly: A Theoretical Investigation

Abstract
1. Introduction
2. Computational Methodology
3. Results and Discussion
3.1. Ground States Properties
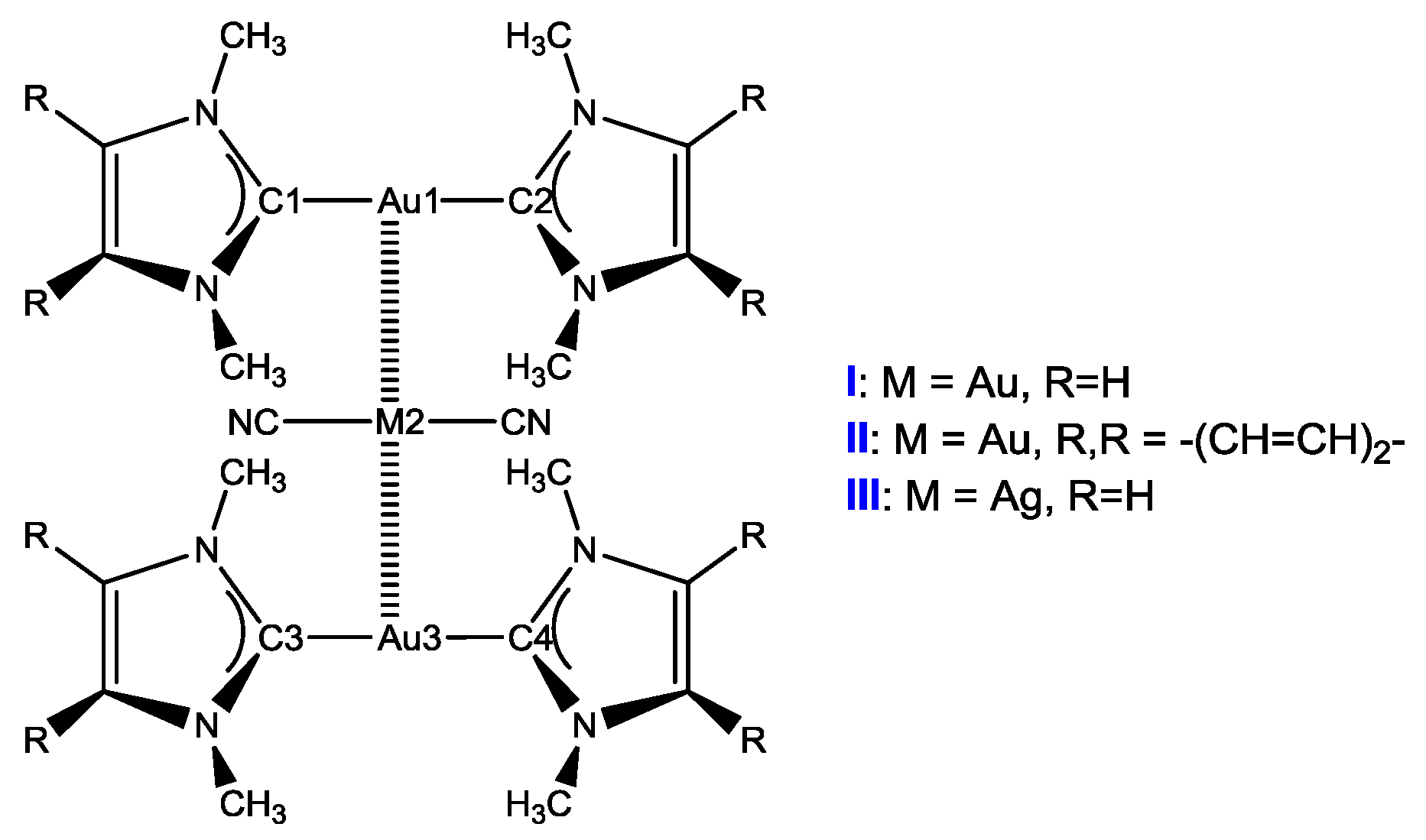
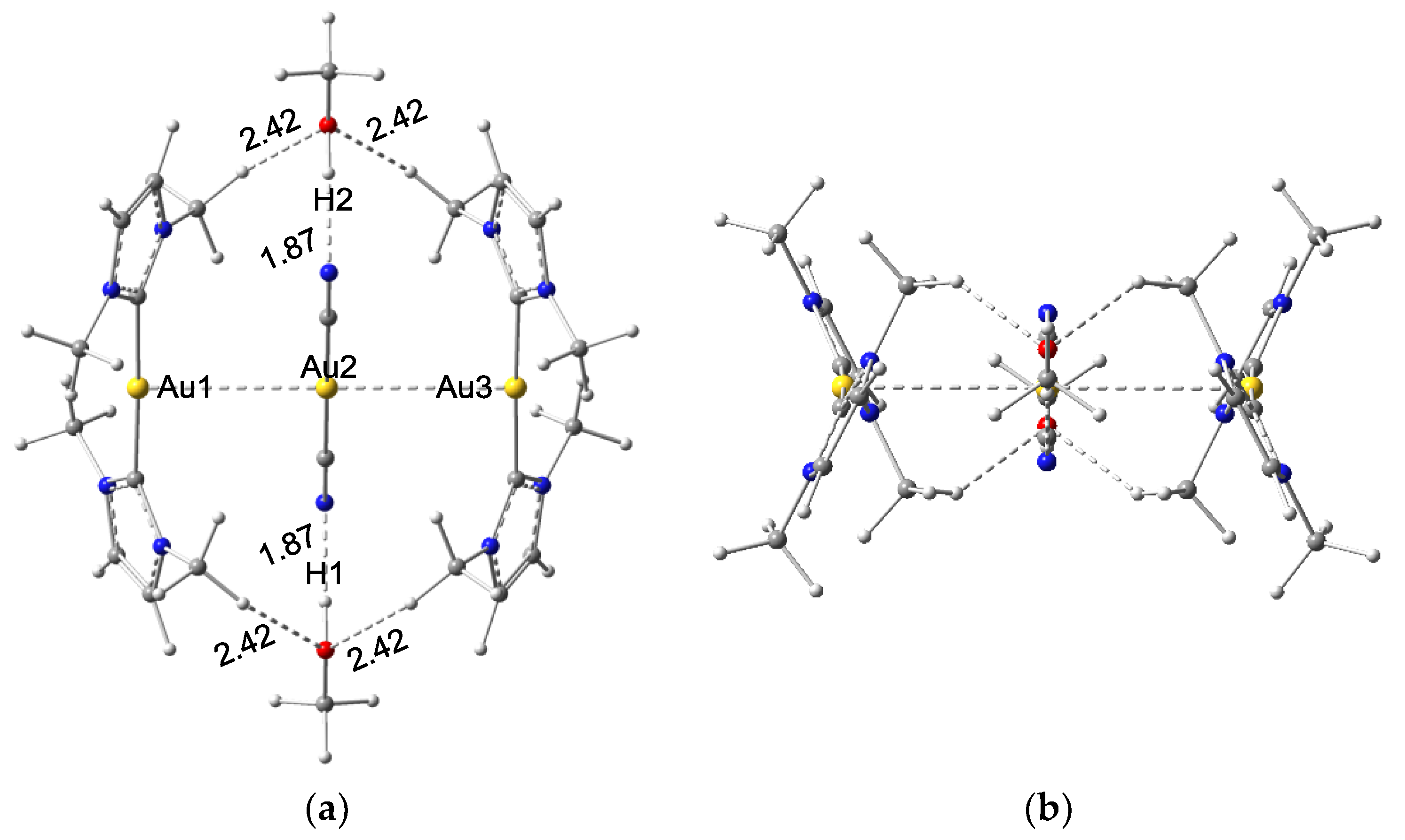
3.1.1. Structures of Clusters X
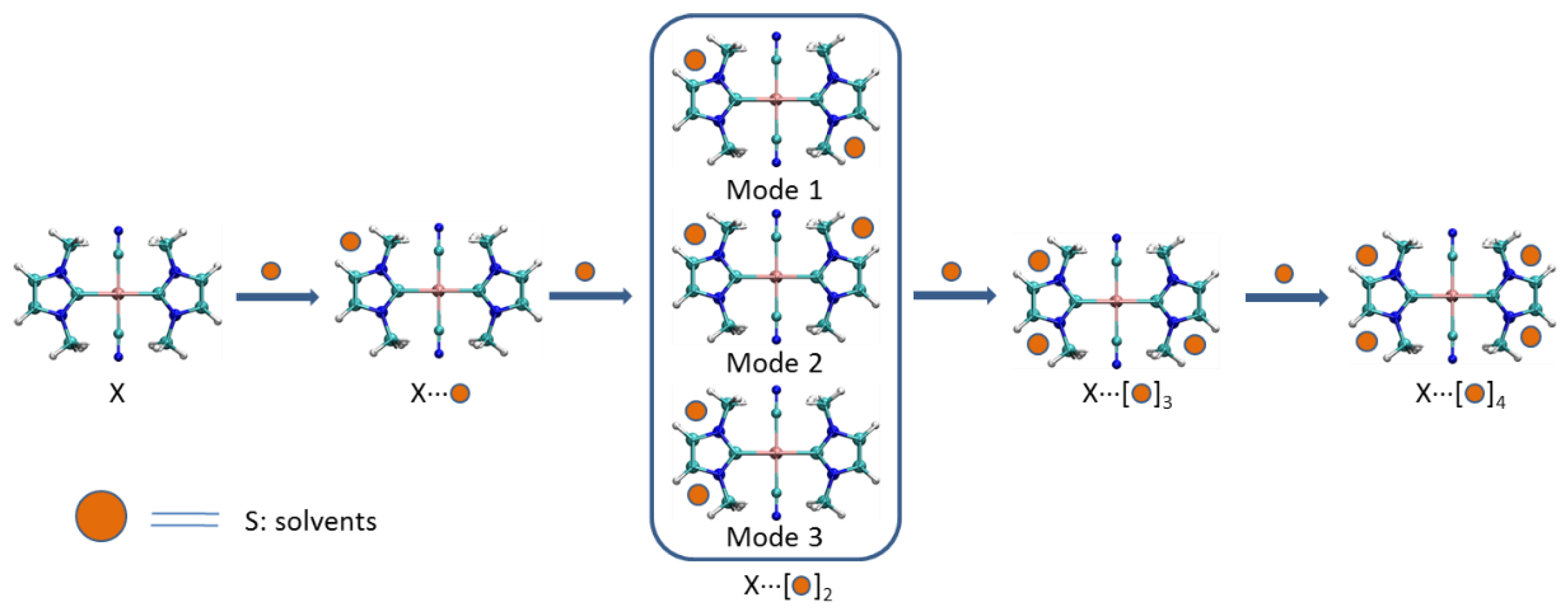
3.1.2. Structures of X⋅⋅⋅(S)x Complexes
3.1.3. Interaction Energies
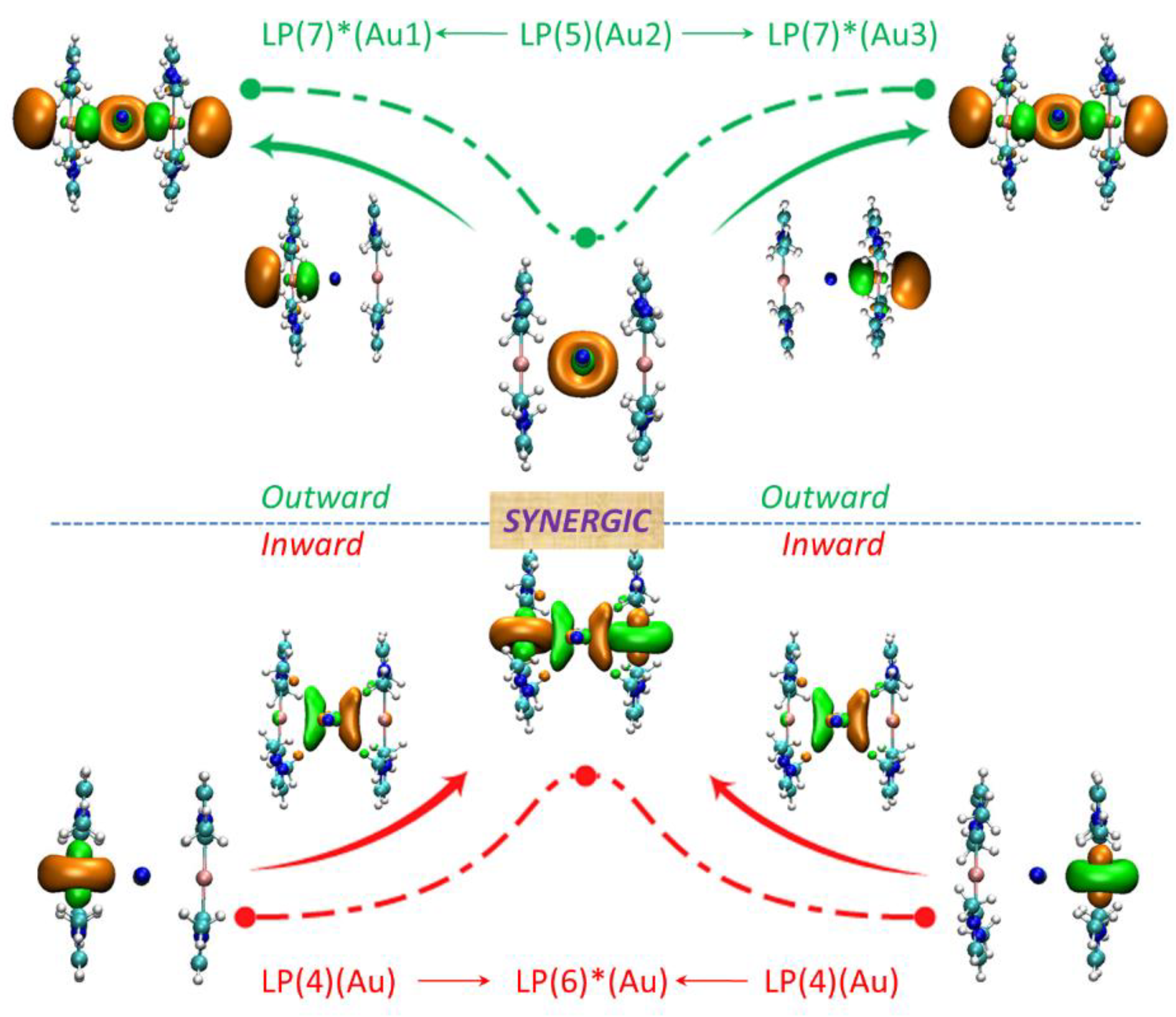
Interaction energies of Au⋅⋅⋅M in clusters X
Interaction energies of complexes X⋅⋅⋅(S)x
3.2. Excited State’s Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Pan, Q.-J.; Zhang, H.-X. An ab initio study on luminescent properties and aurophilic attraction of binuclear gold(I) complexes with phosphinothioether ligands. Inorg. Chem. 2004, 43, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cheng, G.; Li, K.; Shelar, D.P.; Lu, W.; Che, C.-M. Phosphorescent polymeric nanomaterials with metallophilic d10···d10 interactions self-assembled from [Au(NHC)2]+ and [M(CN)2]. Chem. Sci. 2014, 5, 1348–1353. [Google Scholar] [CrossRef]
- López-de-Luzuriaga, J.M.; Monge, M.; Olmos, M.E.; Pascual, D.; Rodrı́guez-Castillo, M.A. Very short metallophilic interactions induced by three-center–two-electron perhalophenyl ligands in phosphorescent Au–Cu complexes. Organometallics 2012, 31, 3720–3729. [Google Scholar] [CrossRef]
- Chen, K.; Strasser, C.E.; Schmitt, J.C.; Shearer, J.; Catalano, V.J. Modulation of luminescence by subtle anion–cation and anion−π interactions in a trigonal AuI···CuI complex. Inorg. Chem. 2012, 51, 1207–1209. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-F.; Yang, X.-P.; Hui-Xue, L.; Guo, Z. Electronic structure of gold carbonyl compounds RAu-L (R = CF3, BO, Br, Cl, CH3, HCC, Mes3P, SIDipp; L = CO, N2, BO) and origins of aurophilic interactions in the clusters [RAuL]n (n = 2–4): A theoretical study. Organometallics 2014, 33, 5101–5110. [Google Scholar] [CrossRef]
- Tang, S.S.; Chang, C.-P.; Lin, I.J.B.; Liou, L.-S.; Wang, J.-C. Molecular aggregation of annular dinuclear gold(I) compounds containing bridging diphosphine and dithiolate ligands. Inorg. Chem. 1997, 36, 2294–2300. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.H.; Phillips, D.L.; Tse, M.-C.; Che, C.-M.; Miskowski, V.M. Resonance raman investigation of the Au(I)−Au(I) interaction of the 1[dσ*pσ] excited state of Au2(dcpm)2(ClO4)2 (dcpm = bis(dicyclohexylphosphine)methane). J. Am. Chem. Soc. 1999, 121, 4799–4803. [Google Scholar] [CrossRef]
- Fernández, E.J.; Gimeno, M.C.; Laguna, A.; López-de-Luzuriaga, J.M.; Monge, M.; Pyykkö, P.; Sundholm, D. Luminescent characterization of solution oligomerization process mediated gold−gold interactions. Dft calculations on [Au2Ag2R4L2]n moieties. J. Am. Chem. Soc. 2000, 122, 7287–7293. [Google Scholar] [CrossRef]
- Rawashdeh-Omary, M.A.; Omary, M.A.; Patterson, H.H.; Fackler, J.P. Excited-state interactions for [Au(CN)2−]n and [Ag(CN)2−]n oligomers in solution. Formation of luminescent gold−gold bonded excimers and exciplexes. J. Am. Chem. Soc. 2001, 123, 11237–11247. [Google Scholar] [CrossRef] [PubMed]
- Adachi, C.; Baldo, M.A.; Thompson, M.E.; Forrest, S.R. Nearly 100% internal phosphorescence efficiency in an organic light-emitting device. J. Appl. Phys. 2001, 90, 5048–5051. [Google Scholar] [CrossRef]
- Baldo, M.A.; Lamansky, S.; Burrows, P.E.; Thompson, M.E.; Forrest, S.R. Very high-efficiency green organic light-emitting devices based on electrophosphorescence. Appl. Phys. Lett. 1999, 75, 4–6. [Google Scholar] [CrossRef]
- Tsuboyama, A.; Iwawaki, H.; Furugori, M.; Mukaide, T.; Kamatani, J.; Igawa, S.; Moriyama, T.; Miura, S.; Takiguchi, T.; Okada, S.; et al. Homoleptic cyclometalated iridium complexes with highly efficient red phosphorescence and application to organic light-emitting diode. J. Am. Chem. Soc. 2003, 125, 12971–12979. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.Y.; Lee, C.Y.; Huang, T.-H.; Lin, J.T.; Tao, Y.-T.; Chien, C.-H.; Tsai, C. High Tg blue emitting materials for electroluminescent devices. J. Mater. Chem. 2005, 15, 2455–2463. [Google Scholar] [CrossRef]
- McDowell, J.J.; Gao, D.; Seferos, D.S.; Ozin, G. Synthesis of poly(spirosilabifluorene) copolymers and their improved stability in blue emitting polymer leds over non-spiro analogs. Polym. Chem. 2015, 6, 3781–3789. [Google Scholar] [CrossRef]
- Chung, Y.-H.; Sheng, L.; Xing, X.; Zheng, L.; Bian, M.; Chen, Z.; Xiao, L.; Gong, Q. A pure blue emitter (CIEy [approximate] 0.08) of chrysene derivative with high thermal stability for OLED. J. Mater. Chem. C 2015, 3, 1794–1798. [Google Scholar] [CrossRef]
- Kim, S.O.; Lee, K.H.; Kim, G.Y.; Seo, J.H.; Kim, Y.K.; Yoon, S.S. A highly efficient deep blue fluorescent OLED based on diphenylaminofluorenylstyrene-containing emitting materials. Synth. Met. 2010, 160, 1259–1265. [Google Scholar] [CrossRef]
- Yam, V.W.-W.; Au, V.K.-M.; Leung, S.Y.-L. Light-emitting self-assembled materials based on d8 and d10 transition metal complexes. Chem. Rev. 2015, 115, 7589–7728. [Google Scholar] [CrossRef] [PubMed]
- Sluch, I.M.; Miranda, A.J.; Elbjeirami, O.; Omary, M.A.; Slaughter, L.M. Interplay of metallophilic interactions, π–π stacking, and ligand substituent effects in the structures and luminescence properties of neutral PtII and PdII aryl isocyanide complexes. Inorg. Chem. 2012, 51, 10728–10746. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Sanders, J.K.M. The nature of π-π Interactions. J. Am. Chem. Soc. 1990, 112, 5525–5534. [Google Scholar] [CrossRef]
- Lindeman, S.V.; Rathore, R.; Kochi, J.K. Silver(I) complexation of (poly)aromatic ligands. Structural criteria for depth penetration into cis-stilbenoid cavities. Inorg. Chem. 2000, 39, 5707–5716. [Google Scholar] [CrossRef] [PubMed]
- Aguilo, E.; Gavara, R.; Baucells, C.; Guitart, M.; Lima, J.C.; Llorca, J.; Rodriguez, L. Tuning supramolecular aurophilic structures: The effect of counterion, positive charge and solvent. Dalton T. 2016, 45, 7328–7339. [Google Scholar] [CrossRef] [PubMed]
- Penney, A.A.; Sizov, V.V.; Grachova, E.V.; Krupenya, D.V.; Gurzhiy, V.V.; Starova, G.L.; Tunik, S.P. Aurophilicity in action: Fine-tuning the gold(I)–gold(I) distance in the excited state to modulate the emission in a series of dinuclear homoleptic gold(I)–NHC complexes. Inorg. Chem. 2016, 55, 4720–4732. [Google Scholar] [CrossRef] [PubMed]
- Deák, A.; Jobbágy, C.; Marsi, G.; Molnár, M.; Szakács, Z.; Baranyai, P. Anion-, solvent-, temperature-, and mechano-responsive photoluminescence in gold(I) diphosphine-based dimers. Chem.-Eur. J. 2015, 21, 11495–11508. [Google Scholar] [CrossRef] [PubMed]
- Berenguer, J.R.; Lalinde, E.; Martín, A.; Moreno, M.T.; Ruiz, S.; Sánchez, S.; Shahsavari, H.R. Photophysical responses in Pt2Pb clusters driven by solvent interactions and structural changes in the PbII environment. Inorg. Chem. 2014, 53, 8770–8785. [Google Scholar] [CrossRef] [PubMed]
- Romanova, J.; Ranga Prabhath, M.R.; Sadik, Y.; Jarowski, P.D. Molecular design of organometallic materials: Effect of the metallophilic interactions, ligand, metal, and oxidation state. In Quantum Systems in Physics, Chemistry, and Biology: Advances in Concepts and Applications; Tadjer, A., Pavlov, R., Maruani, J., Brändas, E.J., Delgado-Barrio, G., Eds.; Springer International Publishing: Cham, Zuerich, Switzerland, 2017; pp. 139–158. [Google Scholar]
- Kruppa, S.V.; Bappler, F.; Klopper, W.; Walg, S.P.; Thiel, W.R.; Diller, R.; Riehn, C. Ultrafast excited-state relaxation of a binuclear Ag(I) phosphine complex in gas phase and solution. Phys. Chem. Chem. Phys. 2017, 19, 22785–22800. [Google Scholar] [CrossRef] [PubMed]
- Imoto, H.; Nishiyama, S.; Yumura, T.; Watase, S.; Matsukawa, K.; Naka, K. Control of aurophilic interaction: Conformations and electronic structures of one-dimensional supramolecular architectures. Dalton T. 2017, 46, 8077–8082. [Google Scholar] [CrossRef] [PubMed]
- Koshevoy, I.O.; Chang, Y.-C.; Karttunen, A.J.; Haukka, M.; Pakkanen, T.; Chou, P.-T. Modulation of metallophilic bonds: Solvent-induced isomerization and luminescence vapochromism of a polymorphic Au–Cu cluster. J. Am. Chem. Soc. 2012, 134, 6564–6567. [Google Scholar] [CrossRef] [PubMed]
- Mirzadeh, N.; Drumm, D.W.; Wagler, J.; Russo, S.P.; Bhargava, S. Different solvates of the dinuclear cyclometallated gold(i) complex [Au2(μ2-C6H4AsMe2)2]: A computational study insight into solvent-effected optical properties. Dalton T. 2013, 42, 12883–12890. [Google Scholar] [CrossRef] [PubMed]
- Laguna, A.; Lasanta, T.; López-de-Luzuriaga, J.M.; Monge, M.; Naumov, P.; Olmos, M.E. Combining aurophilic interactions and halogen bonding to control the luminescence from bimetallic gold−silver clusters. J. Am. Chem. Soc. 2010, 132, 456–457. [Google Scholar] [CrossRef] [PubMed]
- Donamaría, R.; Gimeno, M.C.; Lippolis, V.; López-de-Luzuriaga, J.M.; Monge, M.; Olmos, M.E. Tuning the luminescent properties of a Ag/Au tetranuclear complex featuring metallophilic interactions via solvent-dependent structural isomerization. Inorg. Chem. 2016, 55, 11299–11310. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, R.; Maeba, J.; Nozaki, K.; Iwamura, M. Considerable enhancement of emission yields of [Au(CN)2−] oligomers in aqueous solutions by coexisting cations. Inorg. Chem. 2016, 55, 7739–7746. [Google Scholar] [CrossRef] [PubMed]
- Pyykkö, P. Strong closed-shell interactions in inorganic chemistry. Chem. Rev. 1997, 97, 597–636. [Google Scholar] [CrossRef] [PubMed]
- Runeberg, N.; Schütz, M.; Werner, H.-J. The aurophilic attraction as interpreted by local correlation methods. J. Chem. Phys. 1999, 110, 7210–7215. [Google Scholar] [CrossRef]
- O’Grady, E.; Kaltsoyannis, N. Does metallophilicity increase or decrease down group 11? Computational investigations of [Cl-M-PH3]2 (M = Cu, Ag, Au, [111]). Phys. Chem. Chem. Phys. 2004, 6, 680–687. [Google Scholar] [CrossRef]
- Fernández, E.J.; Laguna, A.; López-de-Luzuriaga, J.M.; Monge, M.; Olmos, M.E.; Puelles, R.C. Au(I)···Ag(I) metallophilic interactions between anionic units: Theoretical studies on a AuAg4 square pyramidal arrangement. J. Phys. Chem. B 2005, 109, 20652–20656. [Google Scholar] [CrossRef] [PubMed]
- Otero-de-la-Roza, A.; Mallory, J.D.; Johnson, E.R. Metallophilic interactions from dispersion-corrected density-functional theory. J. Chem. Phys. 2014, 140, 18A504. [Google Scholar] [CrossRef] [PubMed]
- Latouche, C.; Skouteris, D.; Palazzetti, F.; Barone, V. TD-DFT benchmark on inorganic Pt(II) and Ir(III) complexes. J. Chem. Theory Comput. 2015, 11, 3281–3289. [Google Scholar] [CrossRef] [PubMed]
- Tsipis, A.C. Dft challenge of intermetallic interactions: From metallophilicity and metallaromaticity to sextuple bonding. Coordin. Chem. Rev. 2017, 345, 229–262. [Google Scholar] [CrossRef]
- Paenurk, E.; Gershoni-Poranne, R.; Chen, P. Trends in metallophilic bonding in Pd–Zn and Pd–Cu complexes. Organometallics 2017, 36, 4854–4863. [Google Scholar] [CrossRef]
- Kelly, J.T.; McClellan, A.K.; Joe, L.V.; Wright, A.M.; Lloyd, L.T.; Tschumper, G.S.; Hammer, N.I. Competition between hydrophilic and argyrophilic interactions in surface enhanced raman spectroscopy. ChemPhysChem 2016, 17, 2782–2786. [Google Scholar] [CrossRef] [PubMed]
- Amar, A.; Meghezzi, H.; Boixel, J.; Le Bozec, H.; Guerchais, V.; Jacquemin, D.; Boucekkine, A. Aggregation effect on the luminescence properties of phenylbipyridine Pt(II) acetylide complexes. A theoretical prediction with experimental evidence. J. Phys. Chem. A 2014, 118, 6278–6286. [Google Scholar] [CrossRef] [PubMed]
- Prabhath, M.R.R.; Romanova, J.; Curry, R.J.; Silva, S.R.P.; Jarowski, P.D. The role of substituent effects in tuning metallophilic interactions and emission energy of bis-4-(2-pyridyl)-1,2,3-triazolatoplatinum(ii) complexes. Angew. Chem. Int. Ed. 2015, 54, 7949–7953. [Google Scholar] [CrossRef] [PubMed]
- Sivchik, V.V.; Grachova, E.V.; Melnikov, A.S.; Smirnov, S.N.; Ivanov, A.Y.; Hirva, P.; Tunik, S.P.; Koshevoy, I.O. Solid-state and solution metallophilic aggregation of a cationic [Pt(NCN)L]+ cyclometalated complex. Inorg. Chem. 2016, 55, 3351–3363. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Pople, J.A.; Frisch, M.J. MP2 energy evaluation by direct methods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Pyykkö, P.; Runeberg, N.; Mendizabal, F. Theory of the d10–d10 closed-shell attraction: 1. Dimers near equilibrium. Chem.-Eur. J. 1997, 3, 1451–1457. [Google Scholar] [CrossRef]
- Li, Z.F.; Fan, Y.Z.; DeYonker, N.J.; Zhang, X.T.; Su, C.Y.; Xu, H.Y.; Xu, X.Y.; Zhao, C.Y. Platinum(II)-catalyzed cyclization sequence of aryl alkynes via C(sp3)-H activation: A DFT study. J. Org. Chem. 2012, 77, 6076–6086. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.-F.; Franzese, C.A.; Malek, R.; Żuchowski, P.S.; Ángyán, J.G.; Szczȩśniak, M.M.; Chałasiński, G. Aurophilic interactions from wave function, symmetry-adapted perturbation theory, and rangehybrid approaches. J. Chem. Theory Comput. 2011, 7, 2399–2407. [Google Scholar] [CrossRef] [PubMed]
- Granatier, J.; Lazar, P.; Otyepka, M.; Hobza, P. The nature of the binding of au, ag, and pd to benzene, coronene, and graphene: From benchmark CCSD(T) calculations to plane-wave DFT calculations. J. Chem. Theory Comput. 2011, 7, 3743–3755. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.E.; Pitoňák, M.; Jurečka, P.; Hobza, P. Stabilization and structure calculations for noncovalent interactions in extended molecular systems based on wave function and density functional theories. Chem. Rev. 2010, 110, 5023–5063. [Google Scholar] [CrossRef] [PubMed]
- Vener, M.V.; Egorova, A.N.; Churakov, A.V.; Tsirelson, V.G. Intermolecular hydrogen bond energies in crystals evaluated using electron density properties: DFT computations with periodic boundary conditions. J. Comput. Chem. 2012, 33, 2303–2309. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Pan, X.; Yan, H.; Tan, N. Multiple hydrogen-bonding interactions between macrocyclic triurea and F−, Cl−, Br−, I− and NO3−: A theoretical investigation. Phys. Chem. Chem. Phys. 2011, 13, 7384–7395. [Google Scholar] [CrossRef] [PubMed]
- Peterson, K.A.; Puzzarini, C. Systematically convergent basis sets for transition metals. II. Pseudopotential-based correlation consistent basis sets for the group 11 (Cu, Ag, Au) and 12 (Zn, Cd, Hg) elements. Theor. Chem. Acc. 2005, 114, 283–296. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted gaussian basis sets for molecular calculations. I. Second row atoms, Z=11-18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Su, P.; Jiang, Z.; Chen, Z.; Wu, W. Energy decomposition scheme based on the generalized kohn-sham scheme. J. Phys. Chem. A 2014, 118, 2531–2542. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Miyoshi, E.; Mori, H.; Hirayama, R.; Osanai, Y.; Noro, T.; Honda, H.; Klobukowski, M. Compact and efficient basis sets of s- and p-block elements for model core potential method. J. Chem. Phys. 2005, 122, 074104–074108. [Google Scholar] [CrossRef] [PubMed]
- Sakai, Y.; Miyoshi, E.; Tatewaki, H. Model core potentials for the lanthanides. J. Mol. Struc.-Theochem. 1998, 451, 143–150. [Google Scholar] [CrossRef]
- Miyoshi, E.; Sakai, Y.; Tanaka, K.; Masamura, M. Relativistic dsp-model core potentials for main group elements in the fourth, fifth and sixth row and their applications. J. Mol. Struc.-Theochem. 1998, 451, 73–79. [Google Scholar] [CrossRef]
- Sakai, Y.; Miyoshi, E.; Klobukowski, M.; Huzinaga, S. Model potentials for molecular calculations. I. The sd-MP set for transition metal atoms Sc through Hg. J. Comput. Chem. 1987, 8, 226–255. [Google Scholar] [CrossRef]
- Nakashima, H.; Mori, H.; Mon, M.S.; Miyoshi, E. Theoretical study on structures of gold, silver and copper clusters using relativistic model core potentials. In Proceedings of the 1st WSEAS International Conference on Computational Chemistry, Cairo, Egypt, 29 –31 December 2007; World Scientific and Engineering Academy and Society (WSEAS): Cairo, Egypt, 2007; pp. 11–13. [Google Scholar]
- Sakai, Y.; Miyoshi, E.; Klobukowski, M.; Huzinaga, S. Model potentials for main group elements Li through Rn. J. Chem. Phys. 1997, 106, 8084–8092. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, G.W.; Scuseria, G.W. Gaussian 09, Revision d.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Weinhold, F. NBO 5.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2001. [Google Scholar]
- Pérez Paz, A.; Espinosa Leal, L.A.; Azani, M.-R.; Guijarro, A.; Sanz Miguel, P.J.; Givaja, G.; Castillo, O.; Mas-Ballesté, R.; Zamora, F.; Rubio, A. Supramolecular assembly of diplatinum species through weak PtII⋅⋅⋅PtII intermolecular interactions: A combined experimental and computational study. Chem.-Eur. J. 2012, 18, 13787–13799. [Google Scholar] [CrossRef] [PubMed]
- Romanova, J.; Ranga Prabhath, M.R.; Jarowski, P.D. Relationship between metallophilic interactions and luminescent properties in Pt(II) complexes: TD-DFT guide for the molecular design of light-responsive materials. J. Phys. Chem. C 2016, 120, 2002–2012. [Google Scholar] [CrossRef]
- Martínez-Salvador, S.; Forniés, J.; Martín, A.; Menjón, B. [Au(CF3)(CO)]: A gold carbonyl compound stabilized by a trifluoromethyl group. Angew. Chem. Int. Ed. 2011, 50, 6571–6574. [Google Scholar] [CrossRef] [PubMed]
- Frontera, A.; Quinonero, D.; Costa, A.; Ballester, P.; Deya, P.M. MP2 study of cooperative effects between cation-π, anion-π and π-π interactions. New J. Chem. 2007, 31, 556–560. [Google Scholar] [CrossRef]
- Vijay, D.; Sastry, G.N. The cooperativity of cation–π and π–π interactions. Chem. Phys. Lett. 2010, 485, 235–242. [Google Scholar] [CrossRef]
- Li, Z.-F.; Li, H.-X.; Yang, X.-P. The mutual interactions based on amphipathic tetraoxacalix[2]arene[2]triazine: Recognition cases of anion and cation investigated by computational study. Phys. Chem. Chem. Phys. 2014, 16, 25876–25882. [Google Scholar] [CrossRef] [PubMed]
- Because whatever S1, S2 or S3, the corresponding energy components of Ecpn is comparable and therefore, the GKS-EDA analysis is selected and the results listed.
- Godefroid, G.; Yuqi, L.; Yanling, S.; Juanjuan, S.; Xiaochun, Q.; Xiaohong, S.; Zhijian, W. Enhancing the blue phosphorescence of iridium complexes with a dicyclometalated phosphite ligand via aza-substitution: A density functional theory investigation. J. Mater. Chem. C 2014, 2, 8364–8372. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| I | II | III | |||||
|---|---|---|---|---|---|---|---|
| ECPadd | ECPtot | ECPadd | ECPtot | ECPadd | ECPtot | ||
| −29.0 | −102.5 | −29.7 | −101.2 | −29.7 | −103.6 | ||
| Mode | Ees | Eex | Erep | Epol | Edisp | Ecorr | Etot | |
|---|---|---|---|---|---|---|---|---|
| I | Eadd | −23.3 | −46.0 | 74.8 | −15.6 | −14.0 | −5.6 | −29.6 |
| Etot | −95.8 | −99.5 | 159.8 | −29.9 | −26.2 | −12.5 | −104.0 | |
| II | Eadd | −22.5 | −43.0 | 69.7 | −15.1 | −14.8 | −5.1 | −30.7 |
| Etot | −92.2 | −94.0 | 150.7 | −29.4 | −27.0 | −11.2 | −103.0 | |
| III * | Eadd | −23.7 | −36.7 | 60.3 | −11.7 | −16.9 | −28.7 | |
| Etot | −96.1 | −78.9 | 127.8 | −22.0 | −31.6 | −100.9 |
| (S)x | I | II | III | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ECP1 | ECP2 | ECP3 | ECP4 | ECP | ECP1 | ECP2 | ECP3 | ECP4 | ECP | ECP1 | ECP2 | ECP3 | ECP4 | ECP | |||
| S1 | −16.6 | −111.6 | −15.8 | −109.8 | −17.1 | −111.5 | |||||||||||
| (S1)2 | −15.3 | −30.7 | −124.8 | −15.4 | −30.9 | −123.9 | −15.8 | −31.5 | −124.7 | ||||||||
| (S1)3 | −12.5 | −25.7 | −42.0 | −136.2 | −13.7 | −30.0 | −44.0 | −137.7 | −12.9 | −26.4 | −43.1 | −136.6 | |||||
| (S1)4 | −11.8 | −23.6 | −37.1 | −50.7 | −146.3 | −13.0 | −26.0 | −40.6 | −55.2 | −150.5 | −12.2 | −24.3 | −38.2 | −52.0 | −147.5 | ||
| S2 | −16.7 | −112.0 | −16.4 | −110.2 | −17.3 | −111.8 | |||||||||||
| (S2)2 | −15.6 | −31.2 | −125.4 | −16.0 | −32.1 | −125.0 | −16.0 | −32.0 | −125.2 | ||||||||
| (S2)3 | −12.8 | −26.2 | −42.8 | −179.8 | −14.1 | −28.3 | −45.2 | −139.1 | −13.3 | −30.2 | −43.8 | −137.3 | |||||
| (S2)4 | −12.2 | −25.4 | −38.0 | −51.6 | −147.5 | −13.4 | −26.7 | −41.6 | −56.5 | −152.0 | −12.5 | −25.0 | −39.0 | −53.0 | −148.4 | ||
| S3 | −15.2 | −110.5 | −14.5 | −108.4 | −15.8 | −110.4 | |||||||||||
| (S3)2 | −14.1 | −28.3 | −122.5 | −14.0 | −28.0 | −121.4 | −14.6 | −29.2 | −122.3 | ||||||||
| (S3)3 | −10.6 | −23.5 | −38.8 | −132.6 | −10.9 | −27.7 | −41.4 | −133.4 | −10.9 | −24.1 | −39.8 | −133.0 | |||||
| (S3)4 | −10.5 | −20.9 | −33.3 | −45.7 | −141.8 | −11.9 | −23.9 | −37.7 | −51.5 | −147.5 | −10.8 | −21.5 | −34.3 | −47.0 | −142.3 | ||
| State | Cplx b | Au-C1 | Au-C2 | M-C3 | M-C4 | Au-C5 | Au-C6 | Au1-M2 | M2-Au3 | Au1M2Au3 |
|---|---|---|---|---|---|---|---|---|---|---|
| I | 2.04 | 2.04 | 2.00 | 2.00 | 2.04 | 2.04 | 3.12 | 3.11 | 179.98 | |
| S0 | II | 2.04 | 2.04 | 2.00 | 2.00 | 2.04 | 2.04 | 3.15 | 3.15 | 180.00 |
| III | 2.04 | 2.04 | 2.05 | 2.05 | 2.04 | 2.04 | 3.06 | 3.06 | 180.00 | |
| I | 2.03 | 2.03 | 1.99 | 1.99 | 2.03 | 2.03 | 2.77 | 2.77 | 179.99 | |
| T1 | II | 2.03 | 2.03 | 1.99 | 1.99 | 2.03 | 2.03 | 2.76 | 2.75 | 179.98 |
| III | 2.03 | 2.03 | 2.02 | 2.02 | 2.03 | 2.03 | 2.76 | 2.76 | 179.99 | |
| I | −0.01 | −0.01 | −0.01 | −0.01 | −0.01 | −0.01 | −0.35 | −0.34 | 0.01 | |
| Δ | II | −0.01 | −0.01 | −0.01 | −0.01 | −0.01 | −0.01 | −0.39 | −0.40 | −0.02 |
| III | −0.01 | −0.01 | −0.03 | −0.03 | −0.01 | −0.01 | −0.30 | −0.01 | −0.30 |
| Complex | λ/E(eV) | Configuration | Character | Exp a | δ |
|---|---|---|---|---|---|
| I | 457/2.72 | HOMO-LUMO (98%) | LMCT | 448 | 9 |
| II | 483/2.57 | HOMO-LUMO (97%) | LMCT | 465 | 18 |
| III | 417/2.98 | HOMO-LUMO (97%) | LMCT | 446 | -29 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.-F.; Yang, X.-P.; Li, H.-X.; Zuo, G.-F. Phosphorescent Modulation of Metallophilic Clusters and Recognition of Solvents through a Flexible Host-Guest Assembly: A Theoretical Investigation. Nanomaterials 2018, 8, 685. https://doi.org/10.3390/nano8090685
Li Z-F, Yang X-P, Li H-X, Zuo G-F. Phosphorescent Modulation of Metallophilic Clusters and Recognition of Solvents through a Flexible Host-Guest Assembly: A Theoretical Investigation. Nanomaterials. 2018; 8(9):685. https://doi.org/10.3390/nano8090685
Chicago/Turabian StyleLi, Zhi-Feng, Xiao-Ping Yang, Hui-Xue Li, and Guo-Fang Zuo. 2018. "Phosphorescent Modulation of Metallophilic Clusters and Recognition of Solvents through a Flexible Host-Guest Assembly: A Theoretical Investigation" Nanomaterials 8, no. 9: 685. https://doi.org/10.3390/nano8090685
APA StyleLi, Z.-F., Yang, X.-P., Li, H.-X., & Zuo, G.-F. (2018). Phosphorescent Modulation of Metallophilic Clusters and Recognition of Solvents through a Flexible Host-Guest Assembly: A Theoretical Investigation. Nanomaterials, 8(9), 685. https://doi.org/10.3390/nano8090685