Characterization and Applications of Nanoparticles Modified in-Flight with Silica or Silica-Organic Coatings

Abstract

1. Introduction
2. Materials and Methods
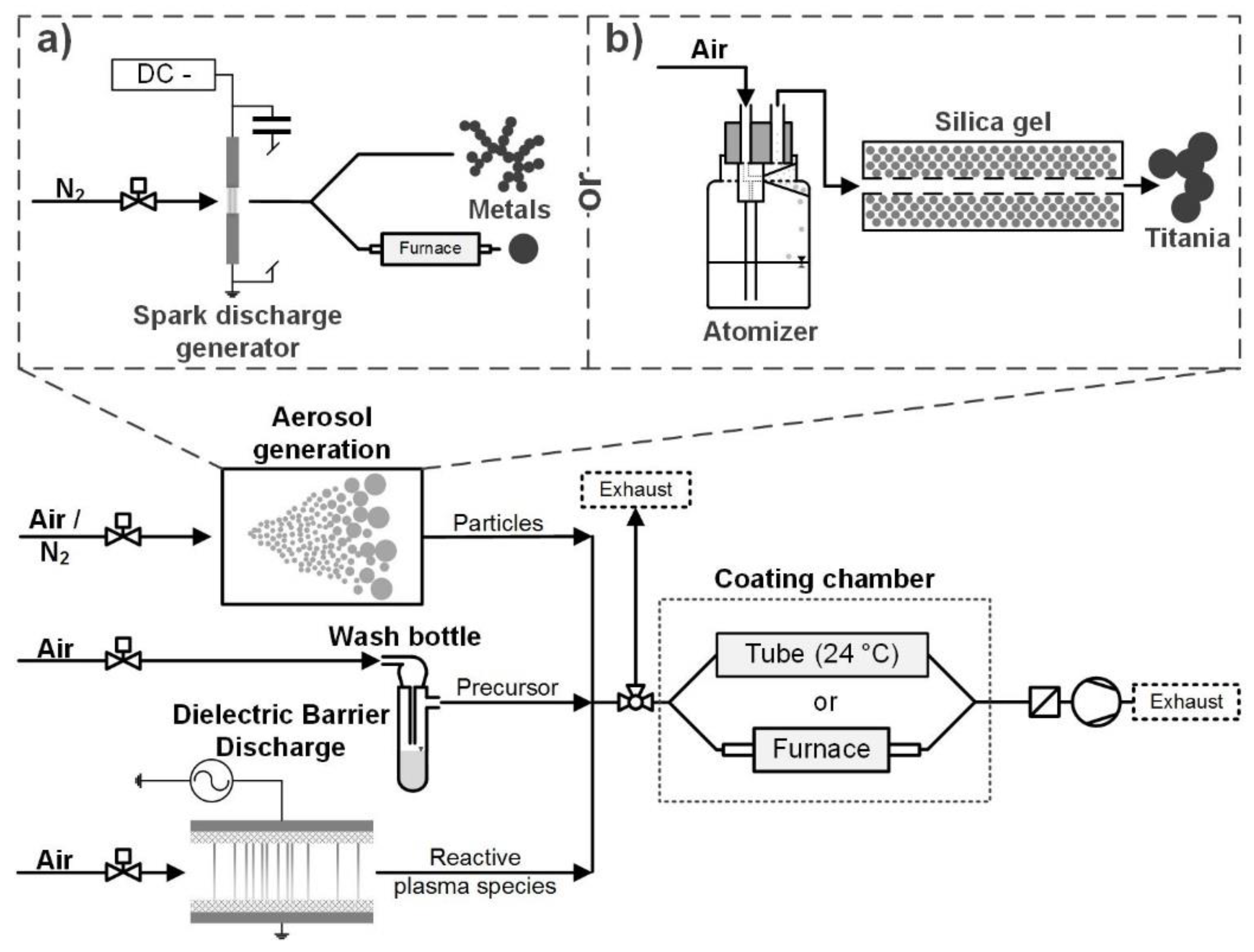
2.1. Aerosol Generation
2.2. Particle Coating
2.3. Coating Analysis
2.4. Photocatalytic Characterization
3. Results and Discussion
3.1. Coating Morphology
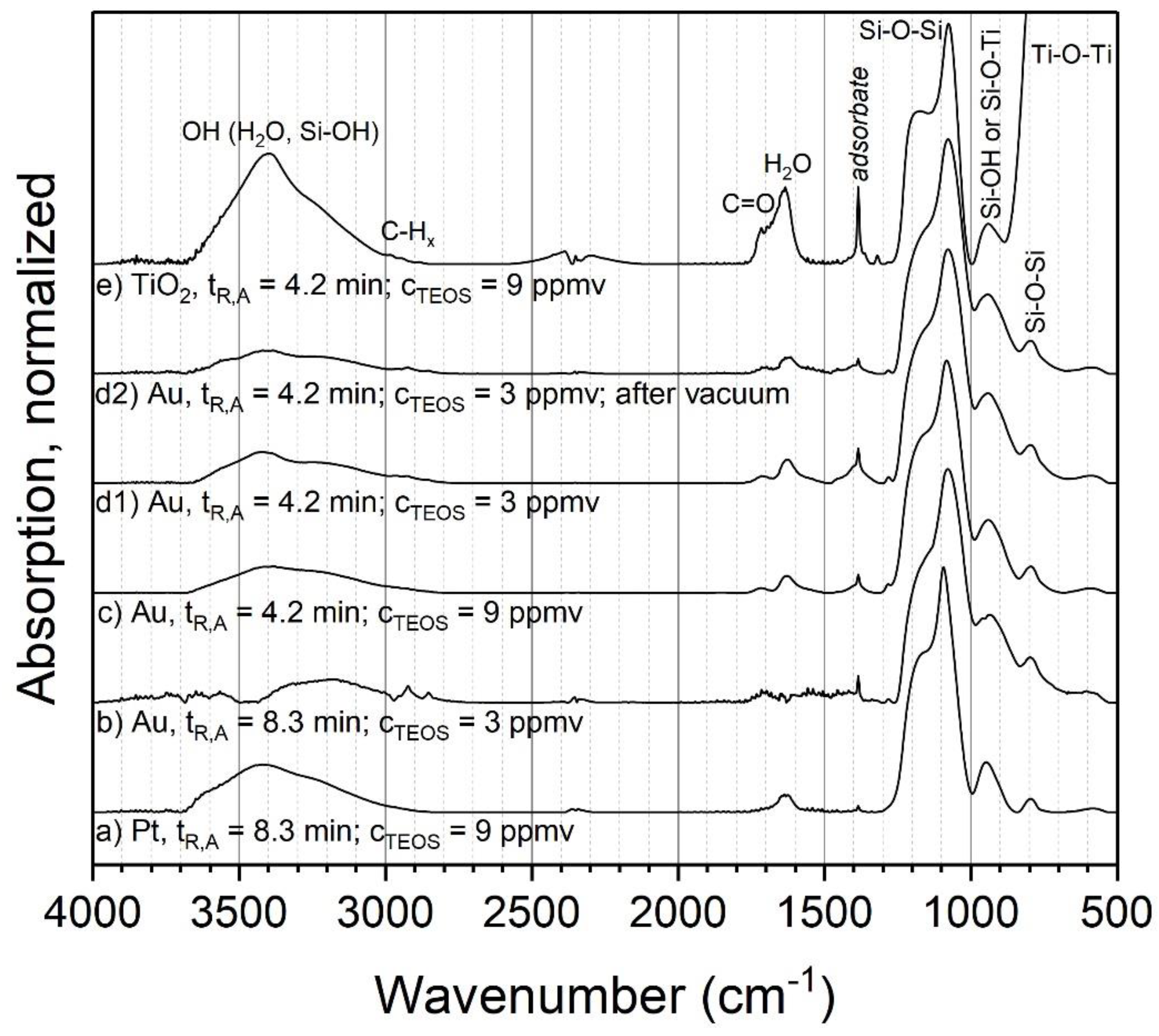
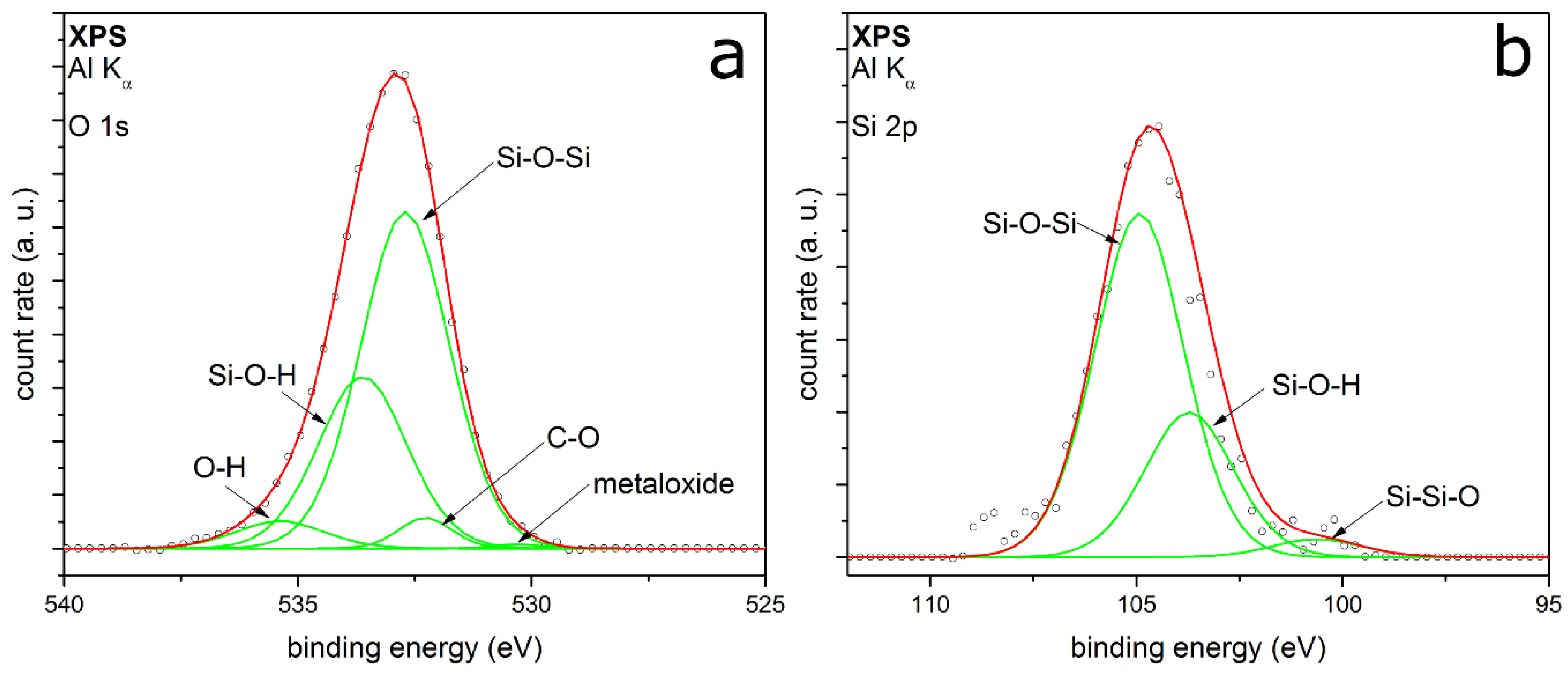
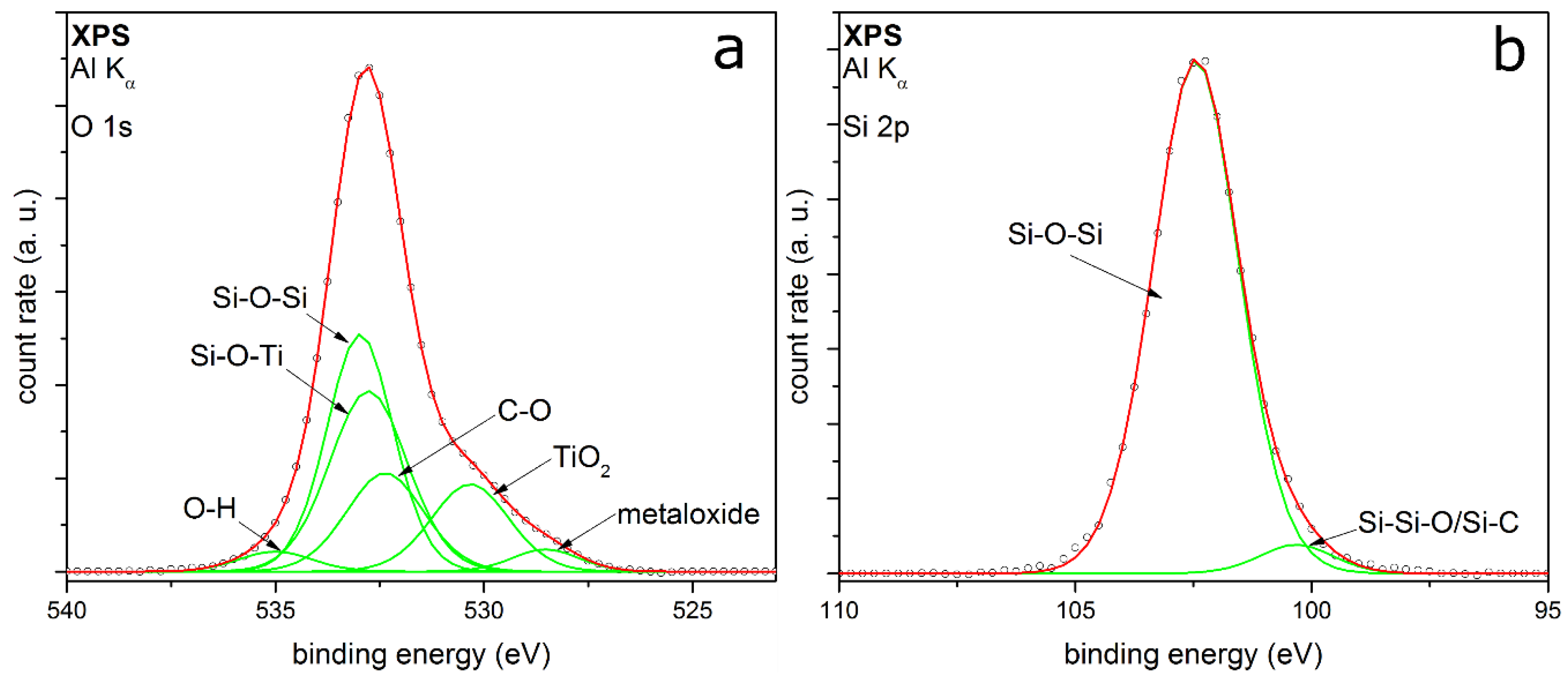
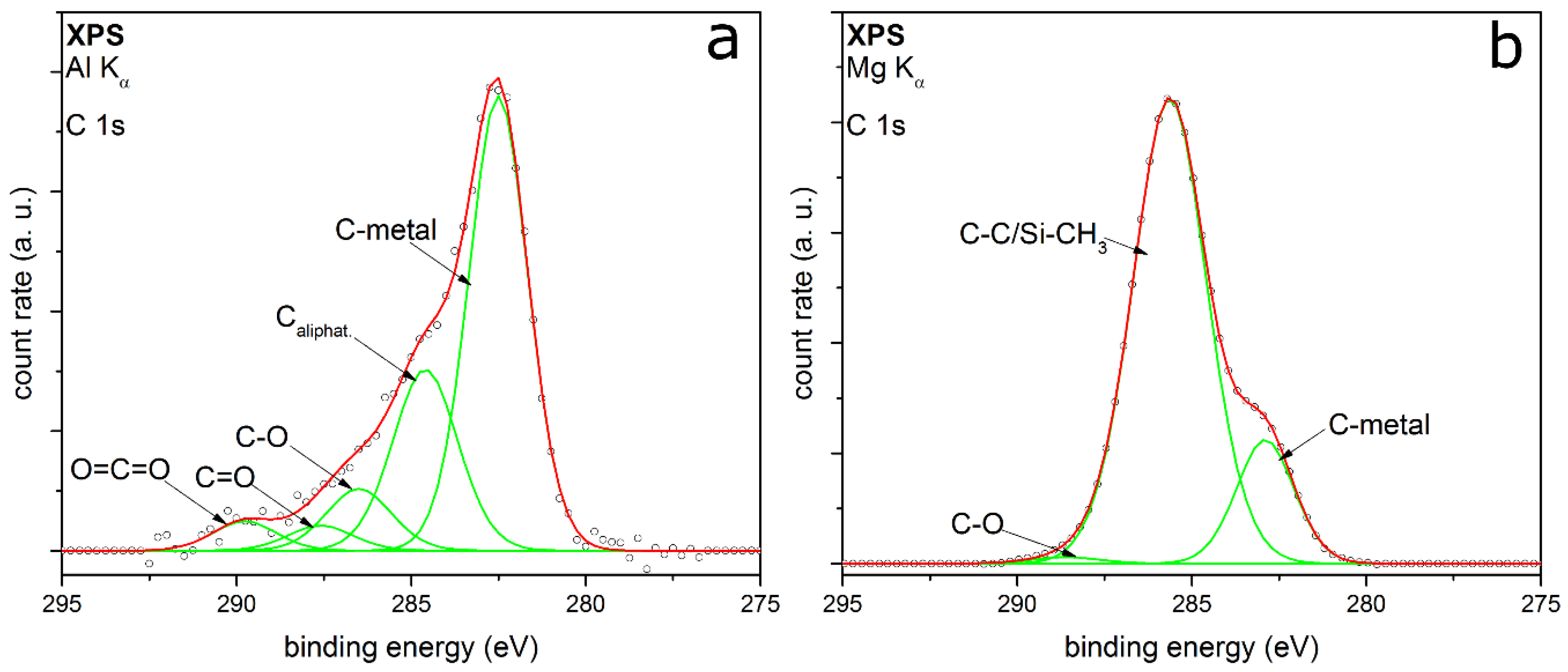
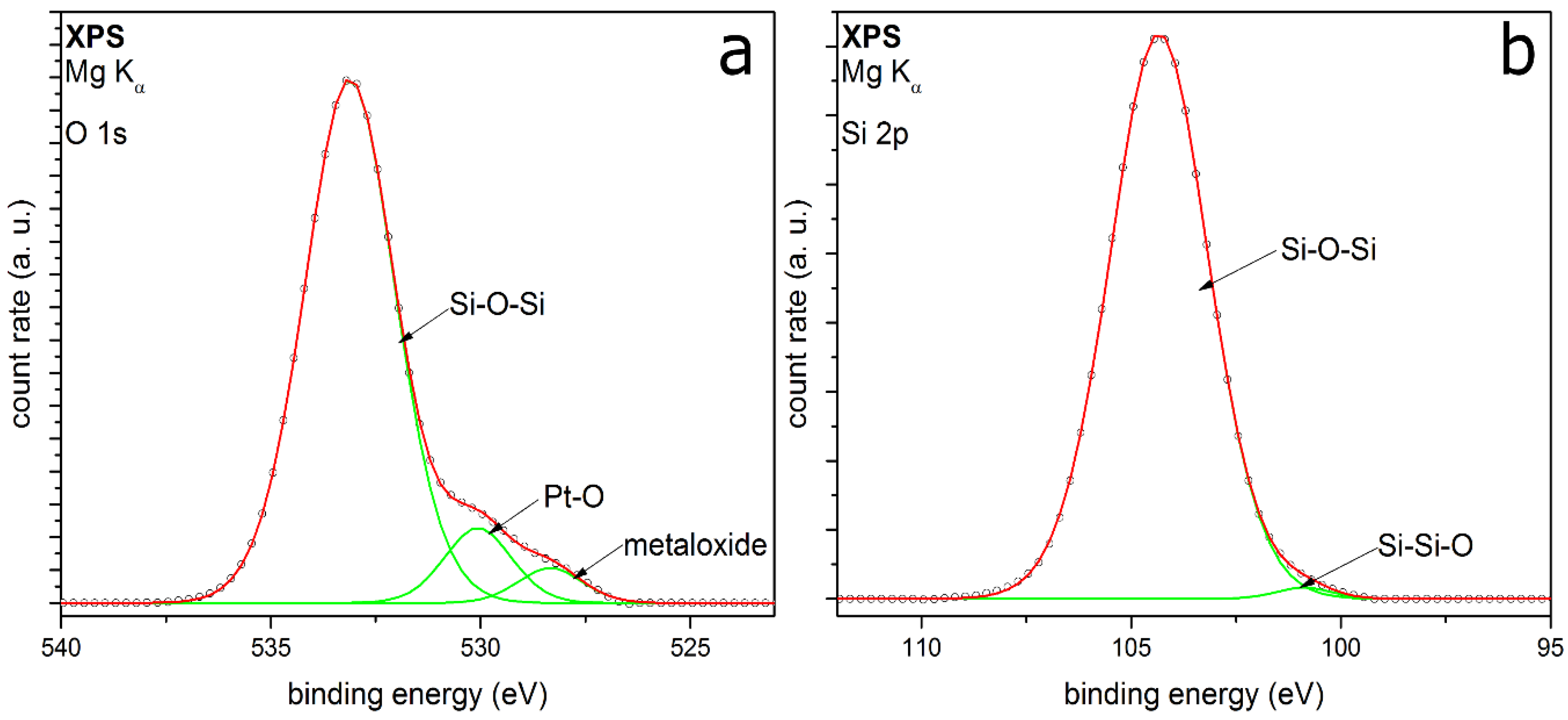
3.2. Coating Composition with Different Precursors and Process Conditions
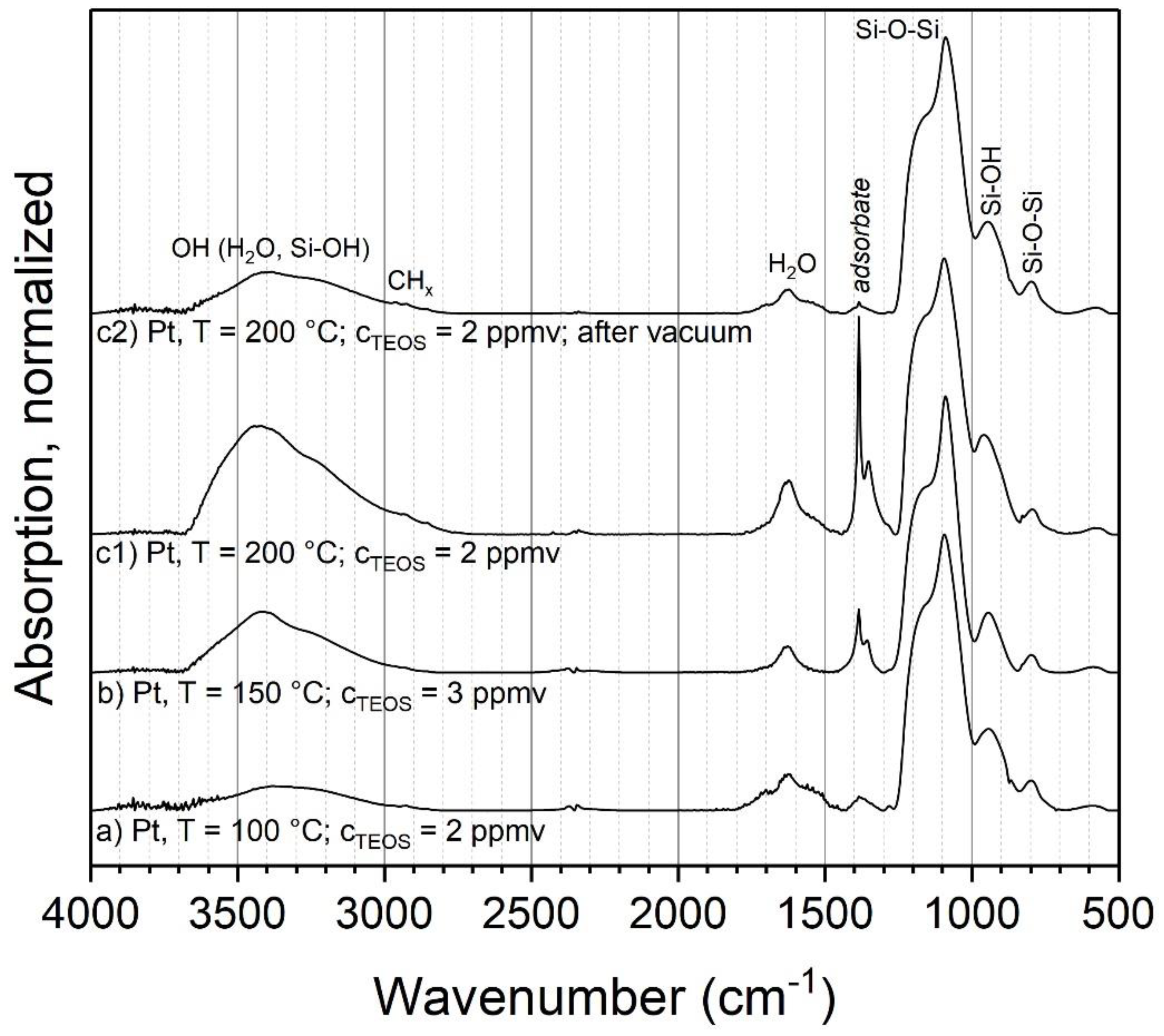
3.2.1. TEOS at Ambient Temperature
3.2.2. TEOS at Elevated Temperatures
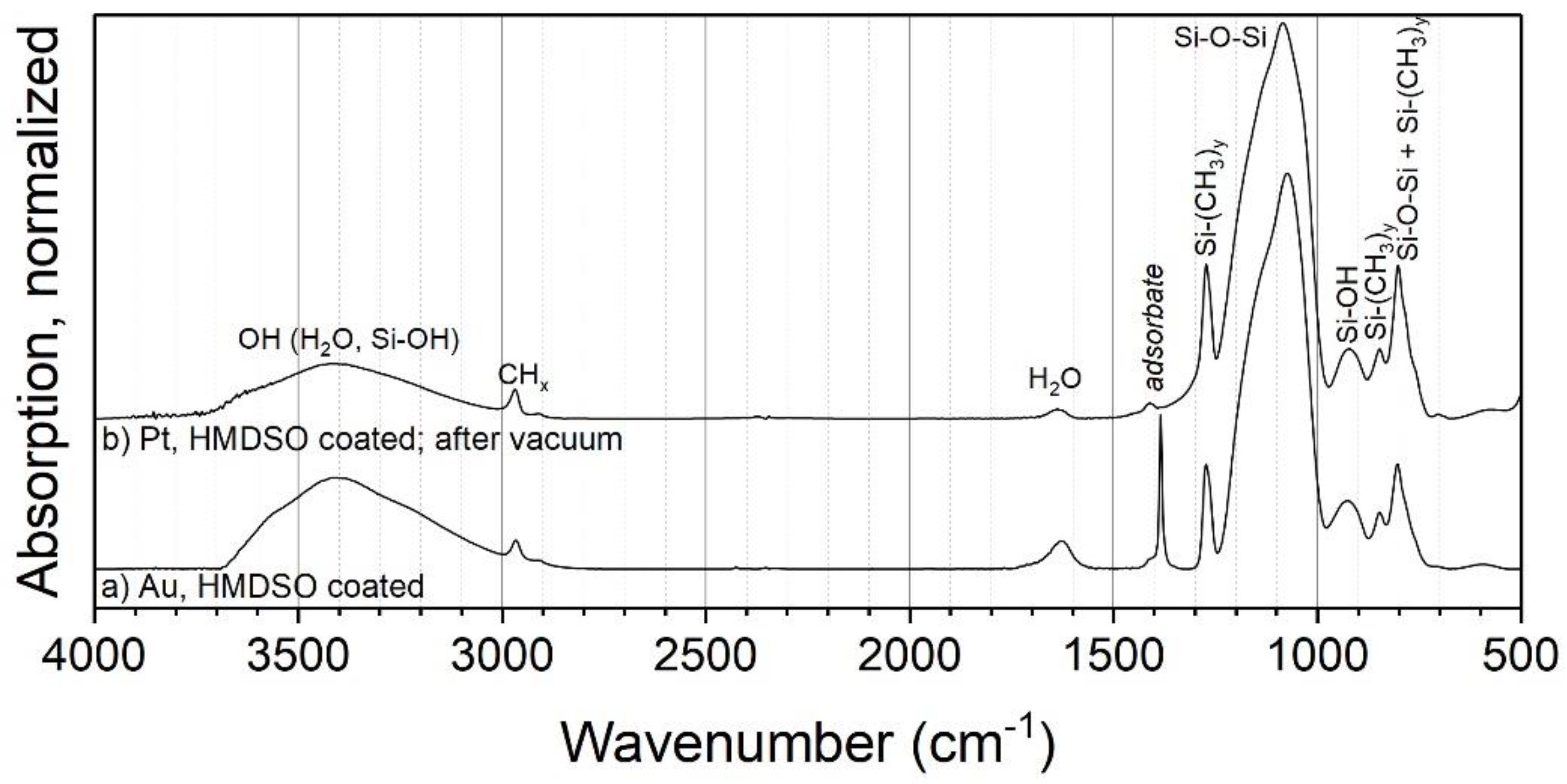
3.2.3. HMDSO
3.3. Application Examples for the Coatings
3.3.1. Modification of Powder Wettability
3.3.2. Photocatalysis Effects of TEOS-Coated P25 Titania
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ghosh Chaudhuri, R.; Paria, S. Core/Shell Nanoparticles: Classes, Properties, Synthesis Mechanisms, Characterization, and Applications. Chem. Rev. 2012, 112, 2373–2433. [Google Scholar] [CrossRef] [PubMed]
- Egerton, T. The Modification of Fine Powders by Inorganic Coatings. KONA Powder Part. J. 1998, 16, 46–59. [Google Scholar] [CrossRef]
- King, D.M.; Liang, X.; Burton, B.B.; Kamal Akhtar, M.; Weimer, A.W. Passivation of pigment-grade TiO2 particles by nanothick atomic layer deposited SiO2 films. Nanotechnology 2008, 19, 25. [Google Scholar] [CrossRef] [PubMed]
- Croissant, J.G.; Fatieiev, Y.; Almalik, A.; Khashab, N.M. Mesoporous Silica and Organosilica Nanoparticles: Physical Chemistry, Biosafety, Delivery Strategies, and Biomedical Applications. Adv. Healthc. Mater. 2018, 7, 1700831. [Google Scholar] [CrossRef] [PubMed]
- Kruis, F.E.; Fissan, H.; Peled, A. Synthesis of nanoparticles in the gas phase for electronic, optical and magnetic applications—A review. J. Aerosol Sci. 1998, 29, 511–535. [Google Scholar] [CrossRef]
- Teleki, A.; Heine, M.C.; Krumeich, F.; Akhtar, M.K.; Pratsinis, S.E. In Situ Coating of Flame-Made TiO2 Particles with Nanothin SiO2 Films. Langmuir 2008, 24, 12553–12558. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Moiseev, A.; Deubener, J.; Weber, A. Thermostable photocatalytically active TiO2 anatase nanoparticles. J. Nanoparticle Res. 2011, 13, 1325–1334. [Google Scholar] [CrossRef]
- George, S.M. Atomic Layer Deposition: An Overview. Chem. Rev. 2010, 110, 111–131. [Google Scholar] [CrossRef] [PubMed]
- Powell, Q.H.; Fotou, G.P.; Kodas, T.T.; Anderson, B.M.; Guo, Y. Gas-phase coating of TiO2 with SiO2 in a continuous flow hot-wall aerosol reactor. J. Mater. Res. 1997, 12, 552–559. [Google Scholar] [CrossRef]
- Kogelschatz, U. Dielectric-barrier discharges: Their history, discharge physics, and industrial applications. Plasma Chem. Plasma Process. 2003, 23, 1–46. [Google Scholar] [CrossRef]
- Vons, V.; Creyghton, Y.; Schmidt-Ott, A. Nanoparticle production using atmospheric pressure cold plasma. J. Nanoparticle Res. 2006, 8, 721–728. [Google Scholar] [CrossRef]
- Nessim, C.; Boulos, M.; Kogelschatz, U. In-flight coating of nanoparticles in atmospheric-pressure DBD torch plasmas. Eur. Phys. J. Appl. Phys. 2009, 47, 22819. [Google Scholar] [CrossRef]
- Post, P.; Jidenko, N.; Weber, A.P.; Borra, J.-P. Post-Plasma SiOx Coatings of Metal and Metal Oxide Nanoparticles for Enhanced Thermal Stability and Tunable Photoactivity Applications. Nanomaterials 2016, 6, 91. [Google Scholar] [CrossRef] [PubMed]
- Post, P.; Weber, A.P. Beschichtung von gasgetragenen Nanopartikeln mit SiO2 mithilfe eines plasma-unterstützten CVD-Prozesses bei Umgebungsbedingungen. Chem. Ing. Tech. 2018, 90, 443–450. [Google Scholar] [CrossRef]
- Adachi, M.; Okuyama, K.; Tohge, N.; Shimada, M.; Sato, J.; Muroyama, M. Particle Generation and Film Formation in an Atmospheric-Pressure Chemical Vapor Deposition Reactor Using the Tetraethylorthosilicate (TEOS)/He, TEOS/O2/He, and TEOS/O3/He Systems. Jpn. J. Appl. Phys. 1993, 32, L748–L751. [Google Scholar] [CrossRef]
- Romet, S.; Couturier, M.F.; Whidden, T.K. Modeling of silicon dioxide chemical vapor deposition from tetraethoxysilane and ozone. J. Electrochem. Soc. 2001, 148, G82–G90. [Google Scholar] [CrossRef]
- Tabrizi, N.S.; Ullmann, M.; Vons, V.A.; Lafont, U.; Schmidt-Ott, A. Generation of nanoparticles by spark discharge. J. Nanoparticle Res. 2009, 11, 315–332. [Google Scholar] [CrossRef]
- Evonik Industries AEROXIDE, AERODISP and AEROPERL Titanium Dioxide as Photocatalyst. Technical Information 1243. Available online: http://www.aerosil.com/sites/lists/RE/DocumentsSI/TI-1243-Titanium-Dioxide-as-Photocatalyst-EN.pdf (accessed on 13 July 2018).
- Krischok, S.; Höfft, O.; Günster, J.; Stultz, J.; Goodman, D.; Kempter, V. H2O interaction with bare and Li-precovered TiO2: Studies with electron spectroscopies (MIES and UPS (HeI and II)). Surf. Sci. 2001, 495, 8–18. [Google Scholar] [CrossRef]
- Klarhöfer, L.; Roos, B.; Viöl, W.; Höfft, O.; Dieckhoff, S.; Kempter, V.; Maus-Friedrichs, W. Valence band spectroscopy on lignin. Holzforschung 2008, 62. [Google Scholar] [CrossRef]
- Heinlin, J.; Isbary, G.; Stolz, W.; Morfill, G.; Landthaler, M.; Shimizu, T.; Steffes, B.; Nosenko, T.; Zimmermann, J.; Karrer, S. Plasma applications in medicine with a special focus on dermatology: Plasma medicine. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Shirley, D.A. High-Resolution X-Ray Photoemission Spectrum of the Valence Bands of Gold. Phys. Rev. B 1972, 5, 4709–4714. [Google Scholar] [CrossRef]
- Scofield, J.H. Hartree-Slater subshell photoionization cross-sections at 1254 and 1487 eV. J. Electron Spectrosc. Relat. Phenom. 1976, 8, 129–137. [Google Scholar] [CrossRef]
- Yeh, J.J.; Lindau, I. Atomic subshell photoionization cross sections and asymmetry parameters: 1 ⩽ Z. ⩽ 103. At. Data Nucl. Data Tables 1985, 32, 1–155. [Google Scholar] [CrossRef]
- Bewig, K.W.; Zisman, W.A. The wetting of gold and platinum by water. J. Phys. Chem. 1965, 69, 4238–4242. [Google Scholar] [CrossRef]
- Moravej, M.; Hicks, R.F. Atmospheric Plasma Deposition of Coatings Using a Capacitive Discharge Source. Chem. Vap. Depos. 2005, 11, 469–476. [Google Scholar] [CrossRef]
- Klapiszewski, Ł.; Siwińska-Stefańska, K.; Kołodyńska, D. Preparation and characterization of novel TiO2/lignin and TiO2-SiO2/lignin hybrids and their use as functional biosorbents for Pb (II). Chem. Eng. J. 2017, 314, 169–181. [Google Scholar] [CrossRef]
- Himpsel, F.J.; McFeely, F.R.; Taleb-Ibrahimi, A.; Yarmoff, J.A.; Hollinger, G. Microscopic structure of the SiO2/Si interface. Phys. Rev. B 1988, 38, 6084–6096. [Google Scholar] [CrossRef]
- Hollinger, G.; Himpsel, F.J. Probing the transition layer at the SiO2-Si interface using core level photoemission. Appl. Phys. Lett. 1984, 44, 93–95. [Google Scholar] [CrossRef]
- Cerofolini, G.F.; Galati, C.; Renna, L. Accounting for anomalous oxidation states of silicon at the Si/SiO2 interface. Surf. Interface Anal. 2002, 33, 583–590. [Google Scholar] [CrossRef]
- McCafferty, E.; Wightman, J.P. Determination of the concentration of surface hydroxyl groups on metal oxide films by a quantitative XPS method. Surf. Interface Anal. 1998, 26, 549–564. [Google Scholar] [CrossRef]
- D’Souza, A.S.; Pantano, C.G. Mechanisms for Silanol Formation on Amorphous Silica Fracture Surfaces. J. Am. Ceram. Soc. 2004, 82, 1289–1293. [Google Scholar] [CrossRef]
- Gustus, R.; Gruber, W.; Wegewitz, L.; Geckle, U.; Prang, R.; Kübel, C.; Schmidt, H.; Maus-Friedrichs, W. Decomposition of amorphous Si2C by thermal annealing. Thin Solid Films 2014, 552, 232–240. [Google Scholar] [CrossRef]
- Bebensee, F.; Voigts, F.; Maus-Friedrichs, W. The adsorption of oxygen and water on Ca and CaO films studied with MIES, UPS and XPS. Surf. Sci. 2008, 602, 1622–1630. [Google Scholar] [CrossRef]
- Barr, T.L. An XPS study of Si as it occurs in adsorbents, catalysts, and thin films. Appl. Surf. Sci. 1983, 15, 1–35. [Google Scholar] [CrossRef]
- Anwar, M.; Hogarth, C.A.; Bulpett, R. An XPS study of amorphous MoO3/SiO films deposited by co-evaporation. J. Mater. Sci. 1990, 25, 1784–1788. [Google Scholar] [CrossRef]
- Dahle, S.; Wegewitz, L.; Qi, F.; Weber, A.P.; Maus-Friedrichs, W. Silicon Dioxide Coating of Titanium Dioxide Nanoparticles from Dielectric Barrier Discharge in a Gaseous Mixture of Silane and Nitrogen. Plasma Chem. Plasma Process. 2013, 33, 839–853. [Google Scholar] [CrossRef]
- Cheung, S.H.; Nachimuthu, P.; Joly, A.G.; Engelhard, M.H.; Bowman, M.K.; Chambers, S.A. N incorporation and electronic structure in N-doped TiO2(110) rutile. Surf. Sci. 2007, 601, 1754–1762. [Google Scholar] [CrossRef]
- Kaufmann, E.N. Common Concepts in Materials Characterization, Introduction. In Characterization of Materials; Kaufmann, E.N., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 747–847. ISBN 978-0-471-26696-9. [Google Scholar]
- Chinh, V.D.; Broggi, A.; Di Palma, L.; Scarsella, M.; Speranza, G.; Vilardi, G.; Thang, P.N. XPS Spectra Analysis of Ti2+, Ti3+ Ions and Dye Photodegradation Evaluation of Titania-Silica Mixed Oxide Nanoparticles. J. Electron. Mater. 2018, 47, 2215–2224. [Google Scholar] [CrossRef]
- Fanelli, F.; Lovascio, S.; d’Agostino, R.; Arefi-Khonsari, F.; Fracassi, F. Ar/HMDSO/O2 Fed Atmospheric Pressure DBDs: Thin Film Deposition and GC-MS Investigation of By-Products. Plasma Process. Polym. 2010, 7, 535–543. [Google Scholar] [CrossRef]
- Ramqvist, L. Charge transfer in transition metal carbides and related compounds studied by ESCA. J. Phys. Chem. Solids 1969, 30, 1835–1847. [Google Scholar] [CrossRef]
- Ichihara, T.; Aizawa, K. 1/f noise in a-Si1–xCx: H thin films as novel thermistor materials for micro-machined IR sensors. J. Non. Cryst. Solids 1998, 227–230, 1345–1348. [Google Scholar]
- Alexander, M.R.; Jones, F.R.; Short, R.D. Mass spectral investigation of the radio-frequency plasma deposition of Hexamethyldisiloxane. J. Phys. Chem. B 1997, 101, 3614–3619. [Google Scholar] [CrossRef]
- Rügner, K.; Reuter, R.; Ellerweg, D.; de los Arcos, T.; von Keudell, A.; Benedikt, J. Insight into the Reaction Scheme of SiO2 Film Deposition at Atmospheric Pressure: Insight into the Reaction Scheme of SiO2 Film Deposition. Plasma Process. Polym. 2013, 10, 1061–1073. [Google Scholar] [CrossRef]
- Alexander, M.R.; Short, R.D.; Jones, F.R.; Stollenwerk, M.; Zabold, J.; Michaeli, W. An X-ray photoelectron spectroscopic investigation into the chemical structure of deposits formed from hexamethyldisiloxane/ oxygen plasmas. J. Mater. Sci. 1996, 31, 1879–1885. [Google Scholar] [CrossRef]
- Shukla, A.; Neergat, M.; Bera, P.; Jayaram, V.; Hegde, M. An XPS study on binary and ternary alloys of transition metals with platinized carbon and its bearing upon oxygen electroreduction in direct methanol fuel cells. J. Electroanal. Chem. 2001, 504, 111–119. [Google Scholar] [CrossRef]
- Kashiwagi, K.; Yoshida, Y.; Murayama, Y. Hybrid Films Formed from Hexamethyldisiloxane and SiO by Plasma Process. Jpn. J. Appl. Phys. 1991, 30, 1803–1807. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Park, S.-J. TiO2 photocatalyst for water treatment applications. J. Ind. Eng. Chem. 2013, 19, 1761–1769. [Google Scholar] [CrossRef]
- Nakata, K.; Fujishima, A. TiO2 photocatalysis: Design and applications. J. Photochem. Photobiol. C Photochem. Rev. 2012, 13, 169–189. [Google Scholar] [CrossRef]
- Hu, S.; Li, F.; Fan, Z. Preparation of SiO2-Coated TiO2 Composite Materials with Enhanced Photocatalytic Activity Under UV Light. Bull. Korean Chem. Soc. 2012, 33, 1895–1899. [Google Scholar] [CrossRef]
- Gholami, T.; Bazarganipour, M.; Salavati-Niasari, M.; Bagheri, S. Photocatalytic degradation of methylene blue on TiO2@SiO2 core/shell nanoparticles: Synthesis and characterization. J. Mater. Sci. Mater. Electron. 2015, 26, 6170–6177. [Google Scholar] [CrossRef]
- Minero, C.; Catozzo, F.; Pelizzetti, E. Role of adsorption in photocatalyzed reactions of organic molecules in aqueous titania suspensions. Langmuir 1992, 8, 481–486. [Google Scholar] [CrossRef]
- Nussbaum, M.; Paz, Y. Ultra-thin SiO2 layers on TiO2: Improved photocatalysis by enhancing products’ desorption. Phys. Chem. Chem. Phys. 2012, 14, 3392–3399. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Ehrman, S.H. Photocatalytic activity of a surface-modified anatase and rutile titania nanoparticle mixture. J. Colloid Interface Sci. 2009, 338, 304–307. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wachs, I.E. Titania–silica as catalysts: Molecular structural characteristics and physico-chemical properties. Catal. Today 1999, 51, 233–254. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Detail Peak | Binding Energy (eV) | FWHM (eV) | Relative Intensity |
|---|---|---|---|---|
| TEOS/Au | O 1s | 530.3 | 1.1 | 0.01 |
| 532.3 | 1.3 | 0.03 | ||
| 532.7 | 2.2 | 0.61 | ||
| 533.6 | 2.2 | 0.30 | ||
| 535.4 | 2.2 | 0.05 | ||
| Si 2p | 100.6 | 2.5 | 0.04 | |
| 103.7 | 2.5 | 0.29 | ||
| 104.9 | 2.5 | 0.68 | ||
| TEOS/TiO2 | O 1s | 528.5 | 1.8 | 0.03 |
| 530.3 | 2.2 | 0.14 | ||
| 532.4 | 2.2 | 0.16 | ||
| 532.8 | 2.2 | 0.30 | ||
| 533.0 | 1.8 | 0.33 | ||
| 535.0 | 2.0 | 0.03 | ||
| Si 2p | 100.7 | 2.1 | 0.06 | |
| 102.5 | 2.1 | 0.93 |
| System | Detail Peak | Binding Energy (eV) | FWHM (eV) | Relative Intensity |
|---|---|---|---|---|
| HMDSO/Metal | O 1s | 528.3 | 1.8 | 0.04 |
| 530.0 | 1.8 | 0.09 | ||
| 533.1 | 2.5 | 0.87 | ||
| Si 2p | 100.8 | 1.5 | 0.01 | |
| 104.3 | 2.7 | 0.99 | ||
| C 1s | 282.9 | 1.9 | 0.17 | |
| 285.6 | 2.5 | 0.81 | ||
| 288.6 | 2.3 | 0.01 | ||
| TEOS/TiO2 | C 1s | 282.5 | 2.0 | 0.58 |
| 284.6 | 2.2 | 0.25 | ||
| 286.5 | 2.2 | 0.09 | ||
| 287.6 | 2.2 | 0.04 | ||
| 289.7 | 2.1 | 0.04 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Post, P.; Wurlitzer, L.; Maus-Friedrichs, W.; Weber, A.P. Characterization and Applications of Nanoparticles Modified in-Flight with Silica or Silica-Organic Coatings. Nanomaterials 2018, 8, 530. https://doi.org/10.3390/nano8070530
Post P, Wurlitzer L, Maus-Friedrichs W, Weber AP. Characterization and Applications of Nanoparticles Modified in-Flight with Silica or Silica-Organic Coatings. Nanomaterials. 2018; 8(7):530. https://doi.org/10.3390/nano8070530
Chicago/Turabian StylePost, Patrick, Lisa Wurlitzer, Wolfgang Maus-Friedrichs, and Alfred P. Weber. 2018. "Characterization and Applications of Nanoparticles Modified in-Flight with Silica or Silica-Organic Coatings" Nanomaterials 8, no. 7: 530. https://doi.org/10.3390/nano8070530
APA StylePost, P., Wurlitzer, L., Maus-Friedrichs, W., & Weber, A. P. (2018). Characterization and Applications of Nanoparticles Modified in-Flight with Silica or Silica-Organic Coatings. Nanomaterials, 8(7), 530. https://doi.org/10.3390/nano8070530