Idebenone: Novel Strategies to Improve Its Systemic and Local Efficacy



Abstract
:1. Introduction
2. Drug Delivery Systems
2.1. Liposomes
2.2. Cyclodextrins
2.3. Microemulsions and Self-Microemulsifying Drug Delivery System SMEDDS
2.4. Polymeric Nanoparticles
2.5. Solid Lipid Nanoparticles and Nanostructured Lipid Carriers
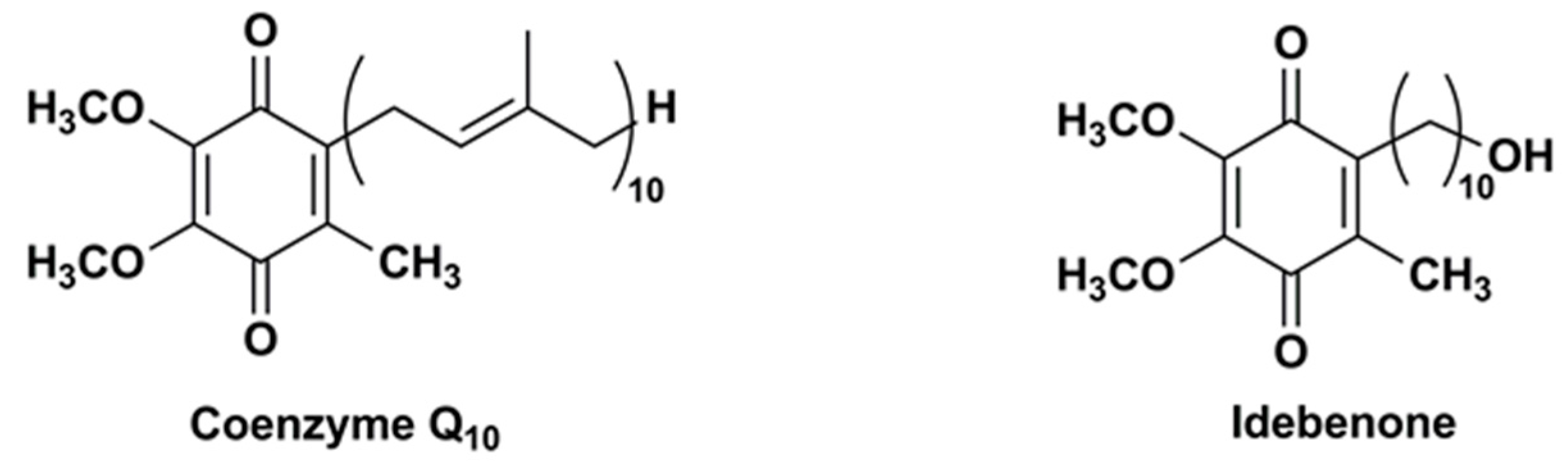
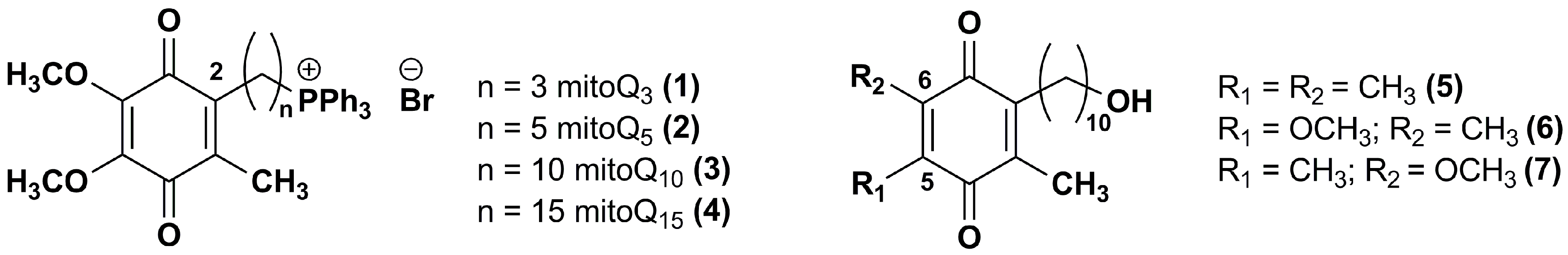
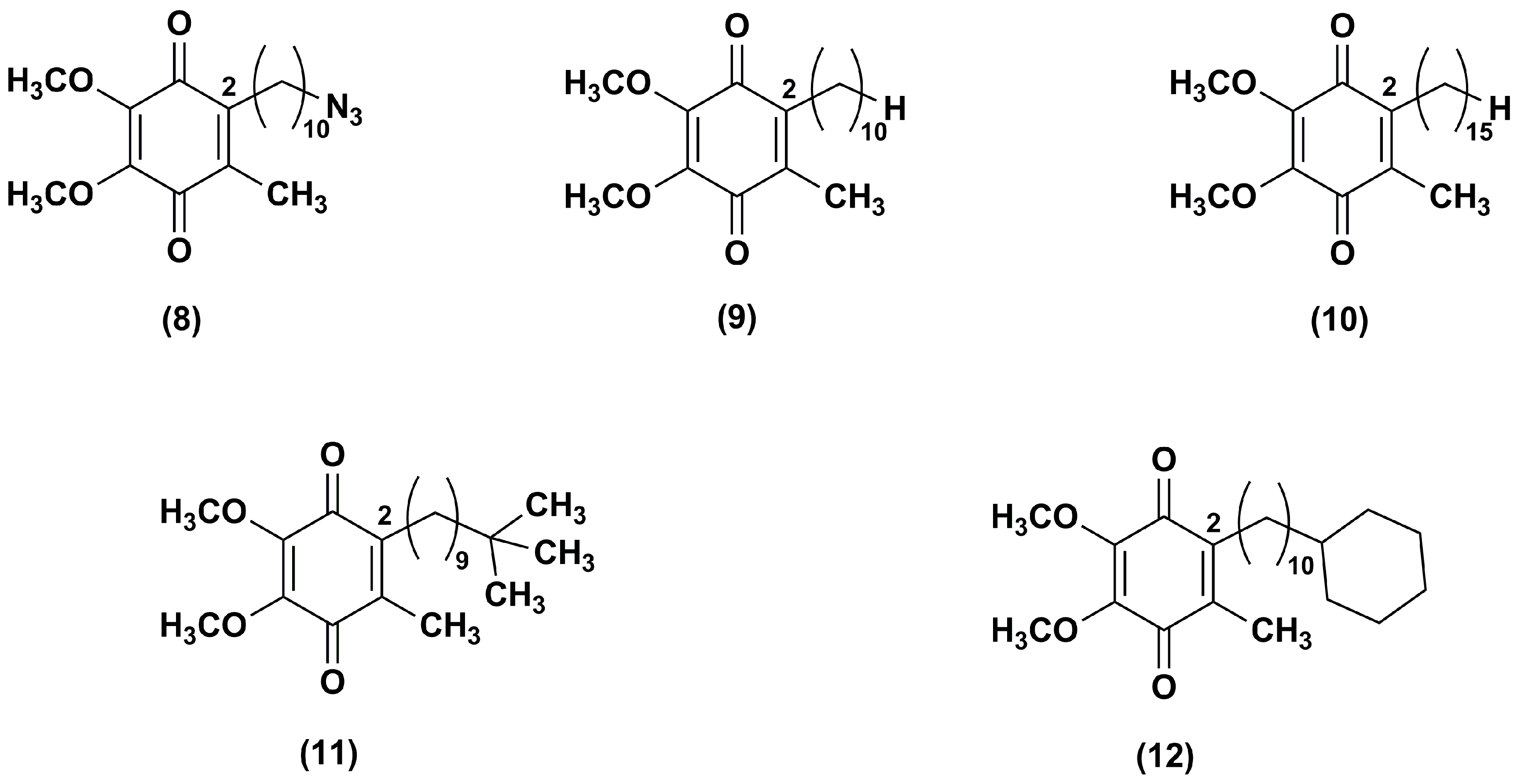
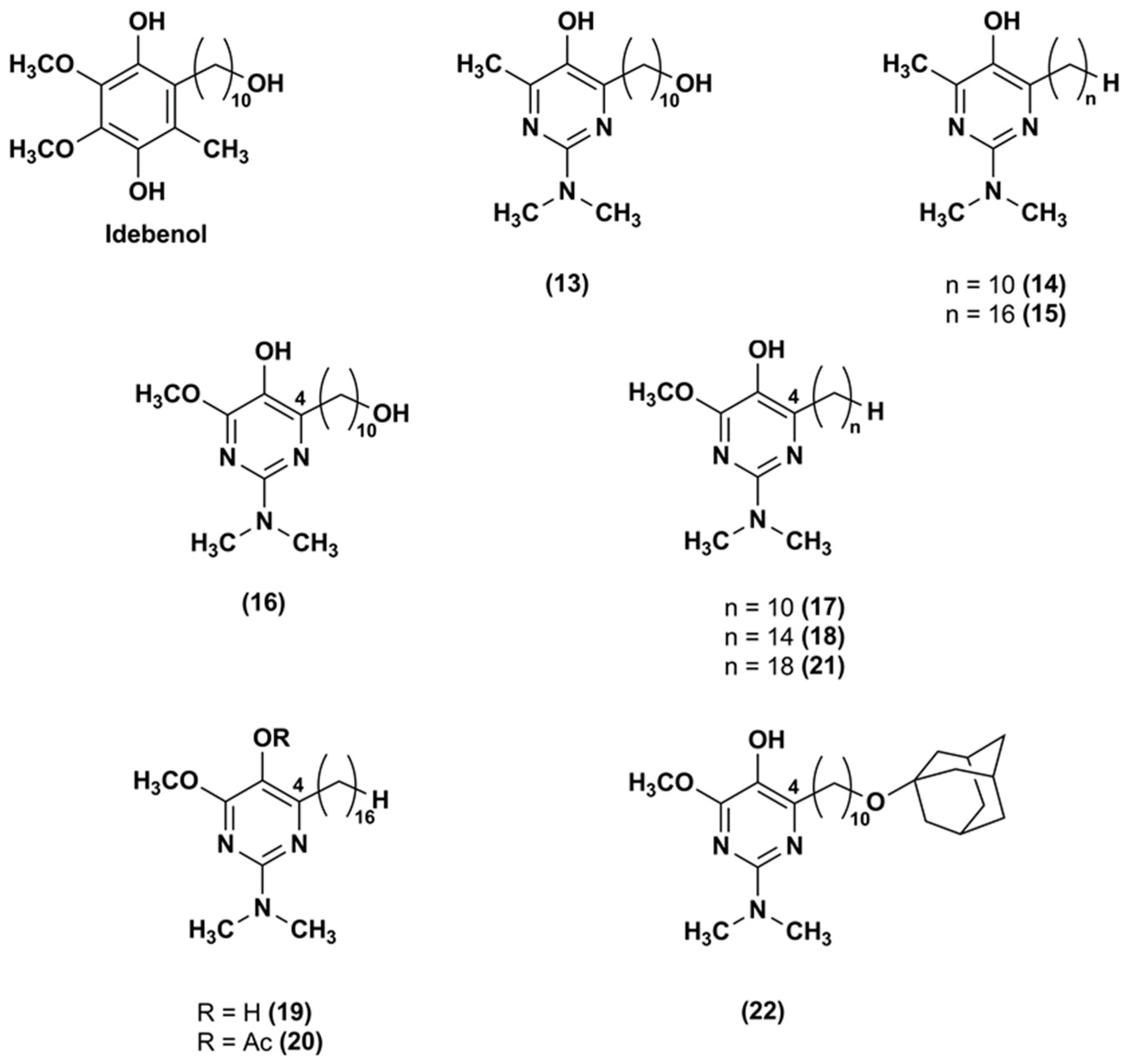
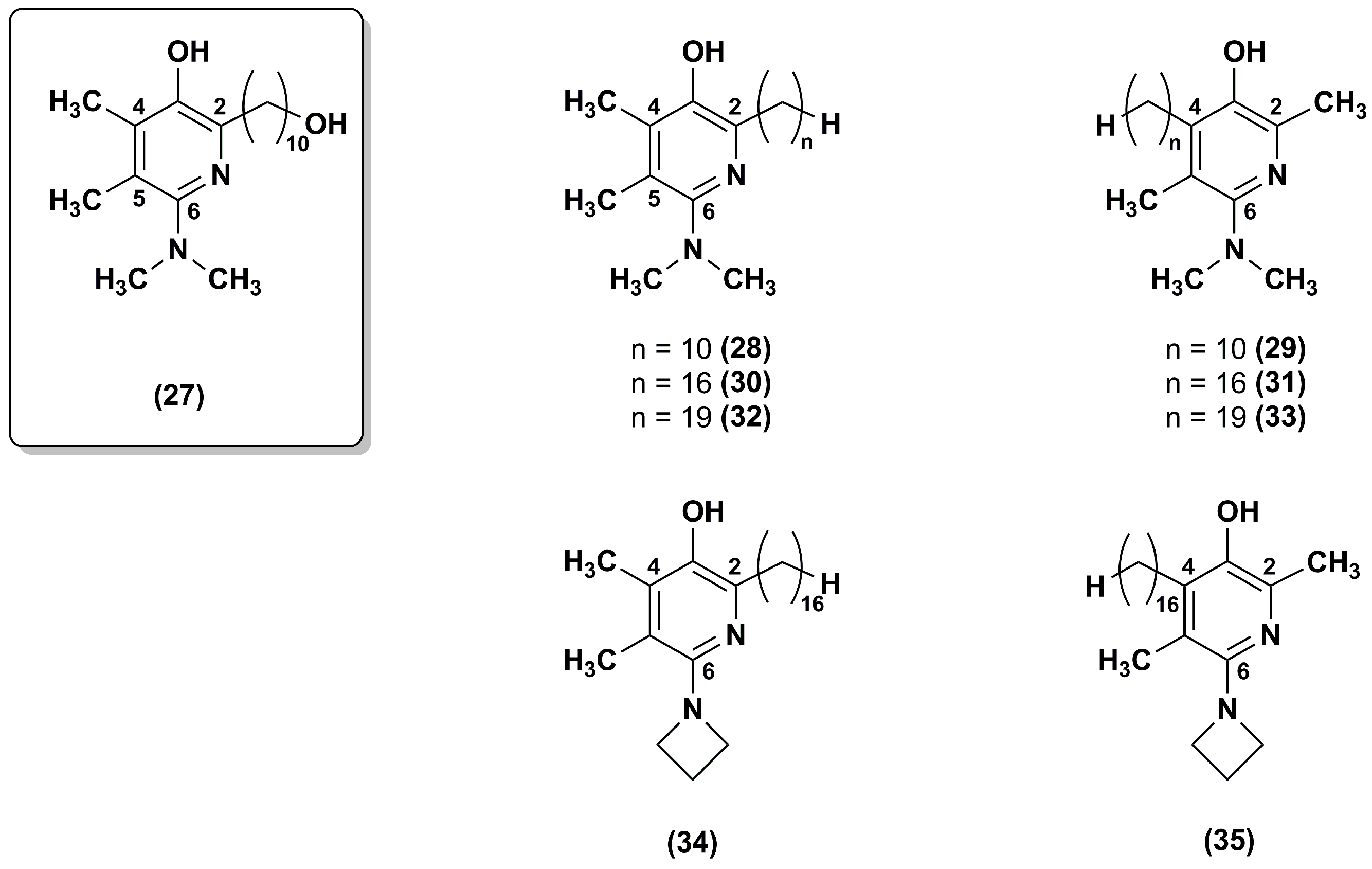
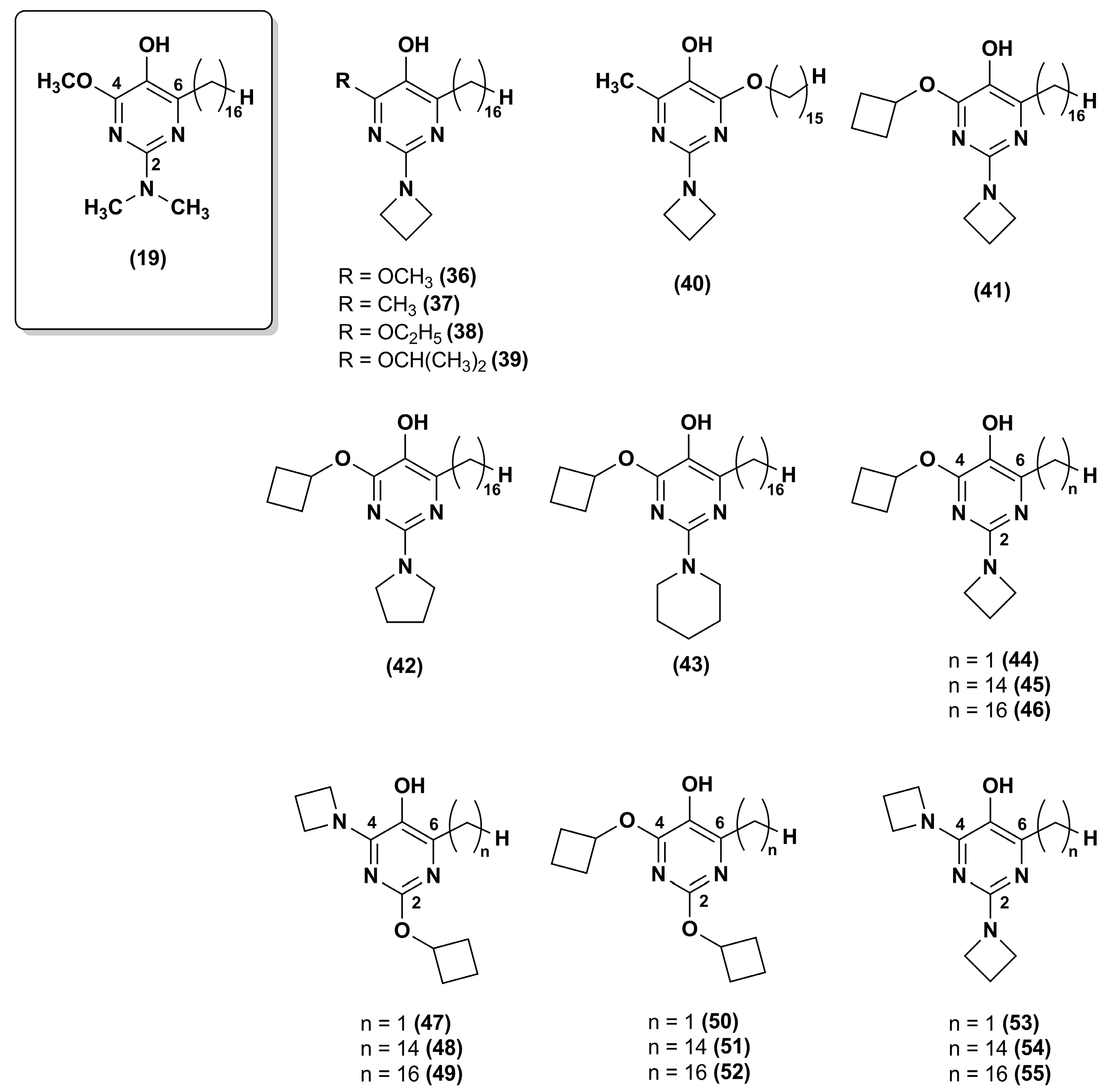
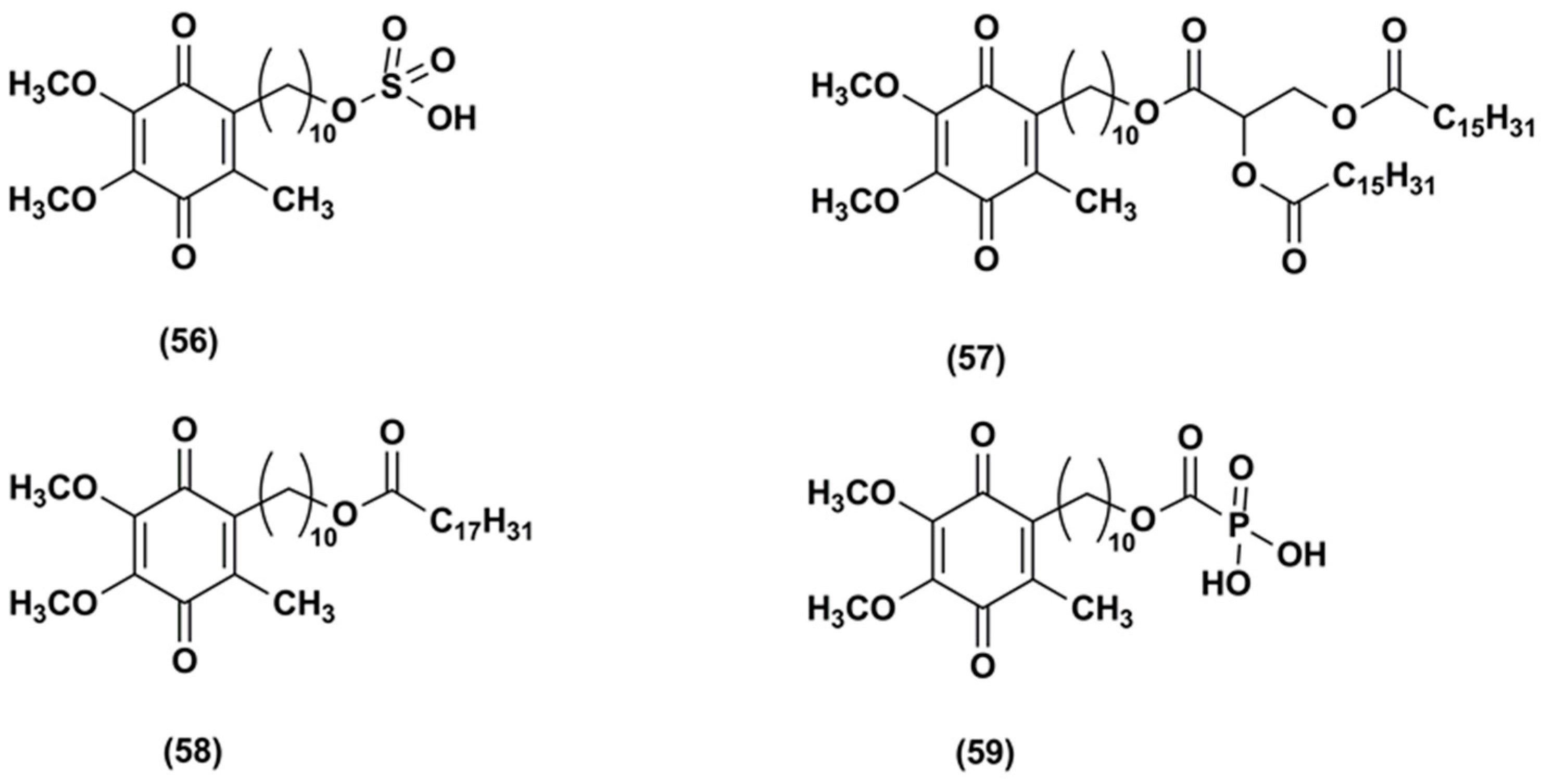
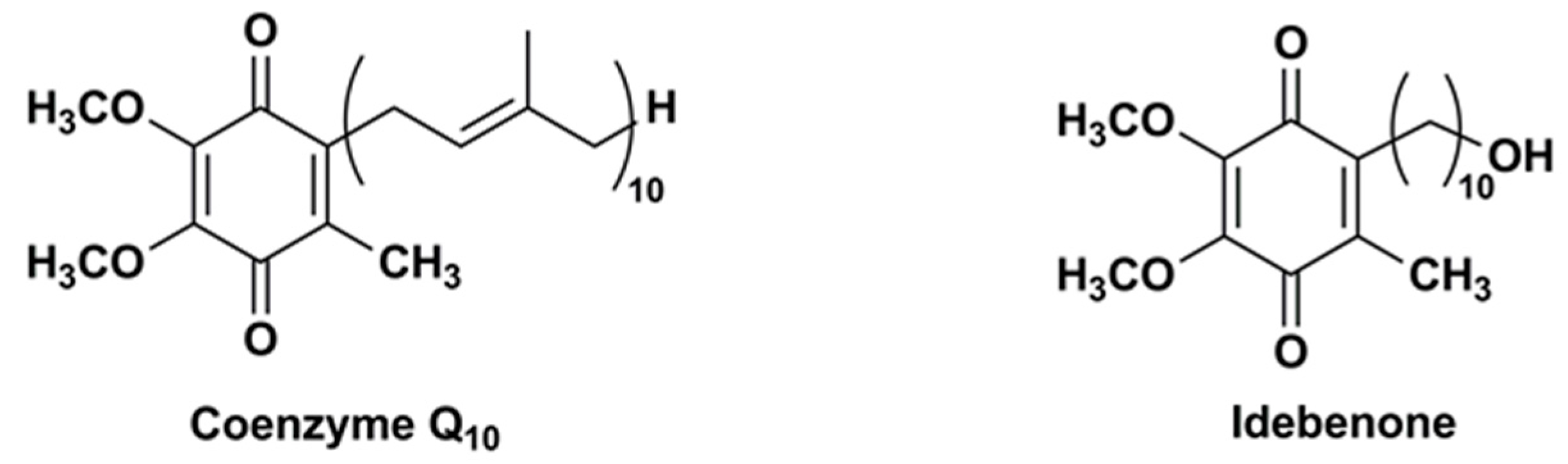
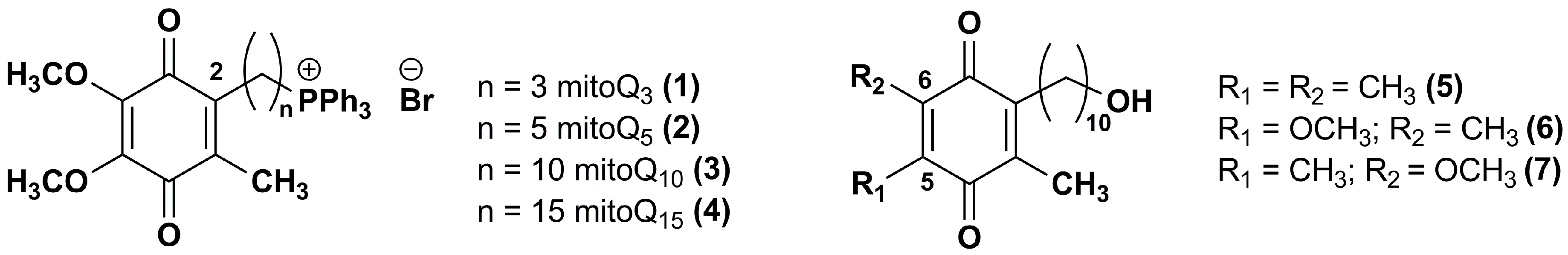
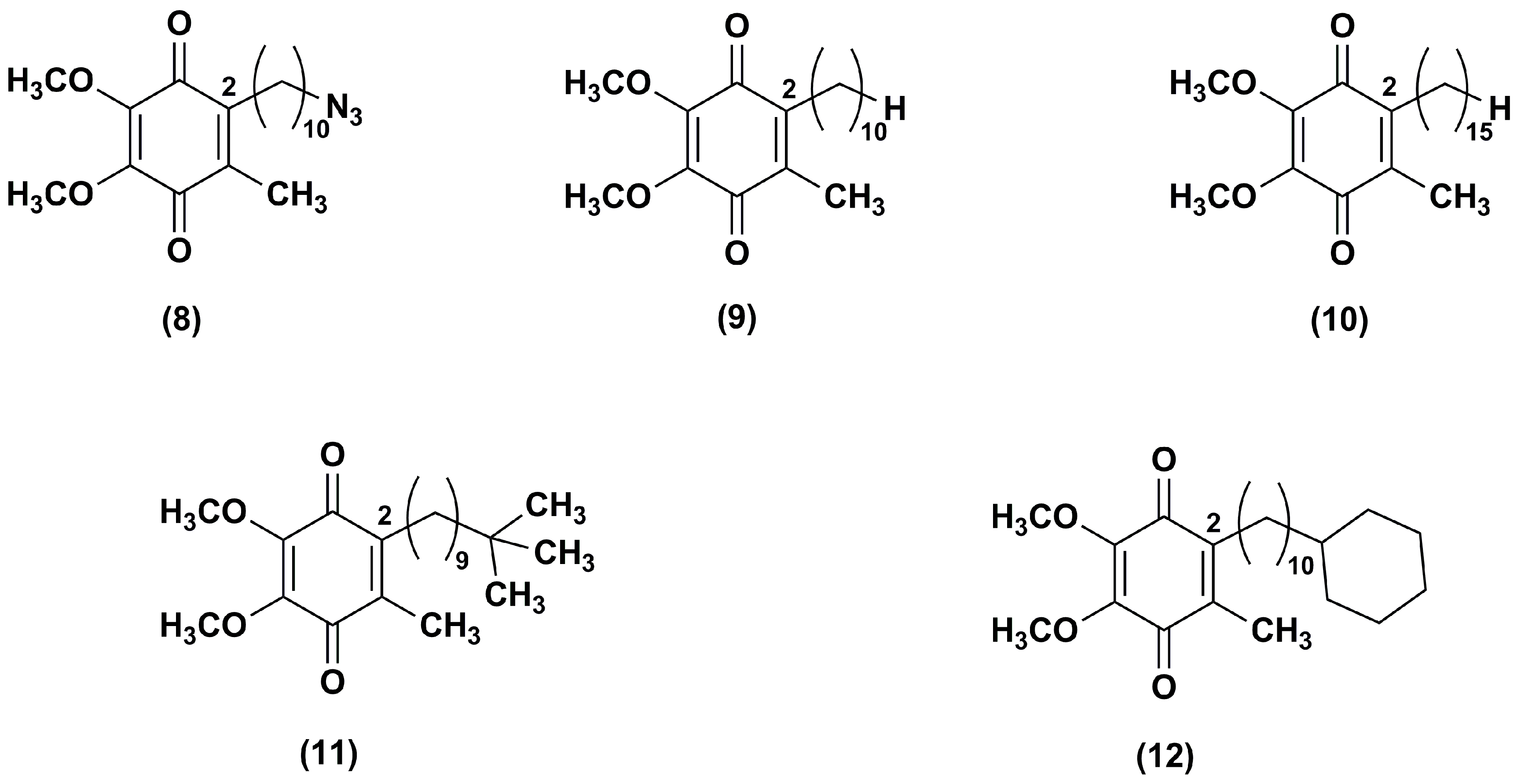
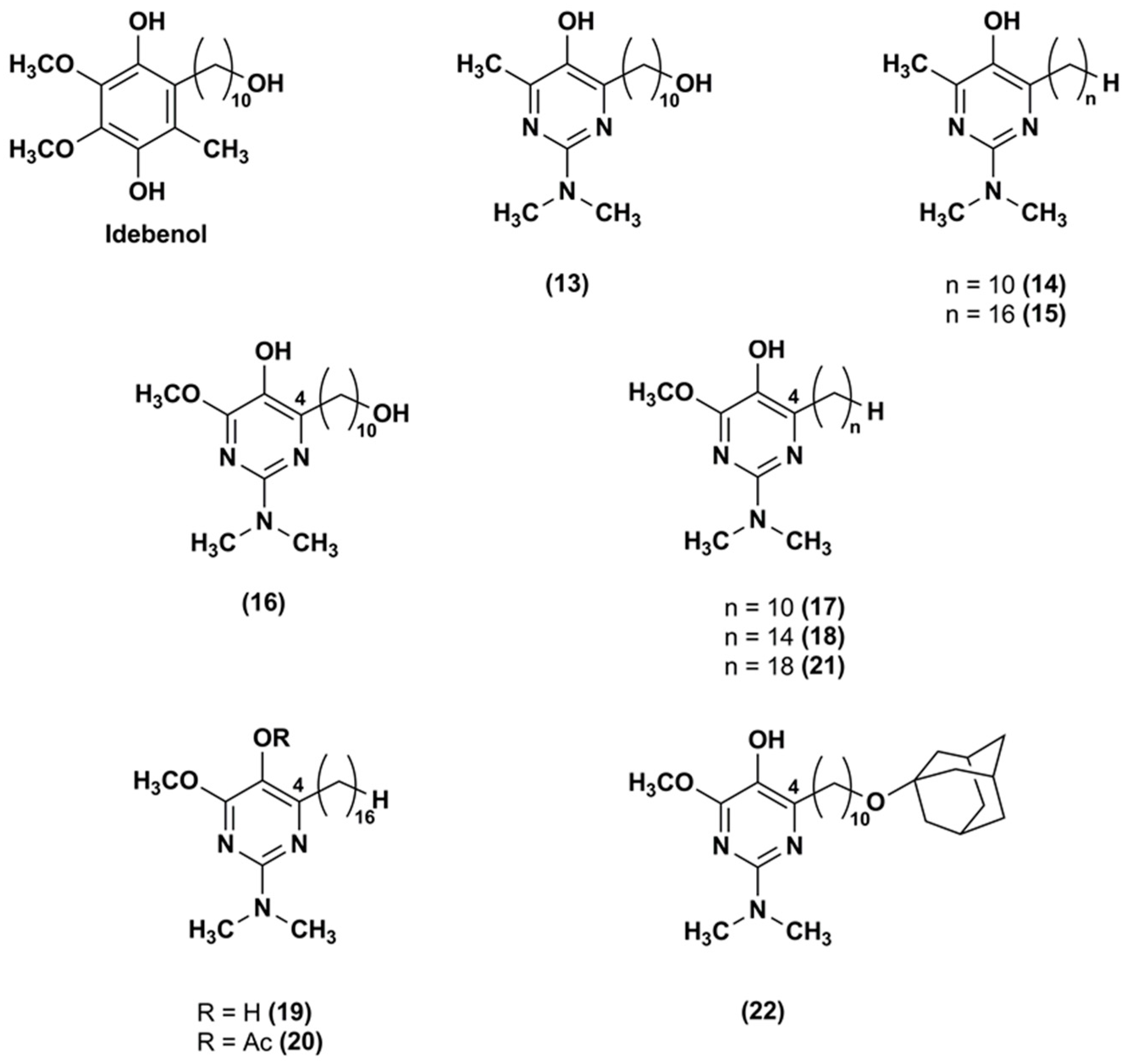
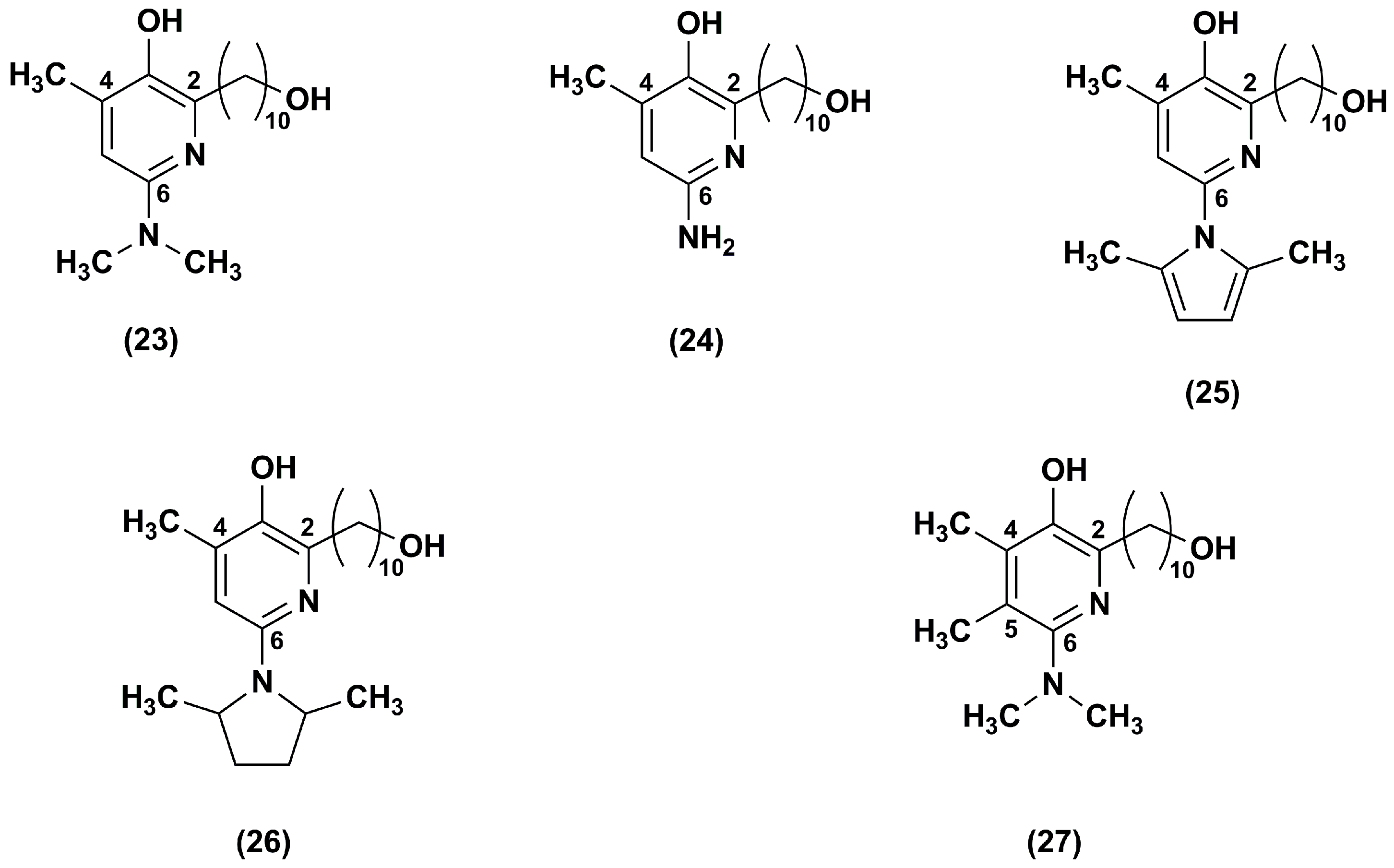
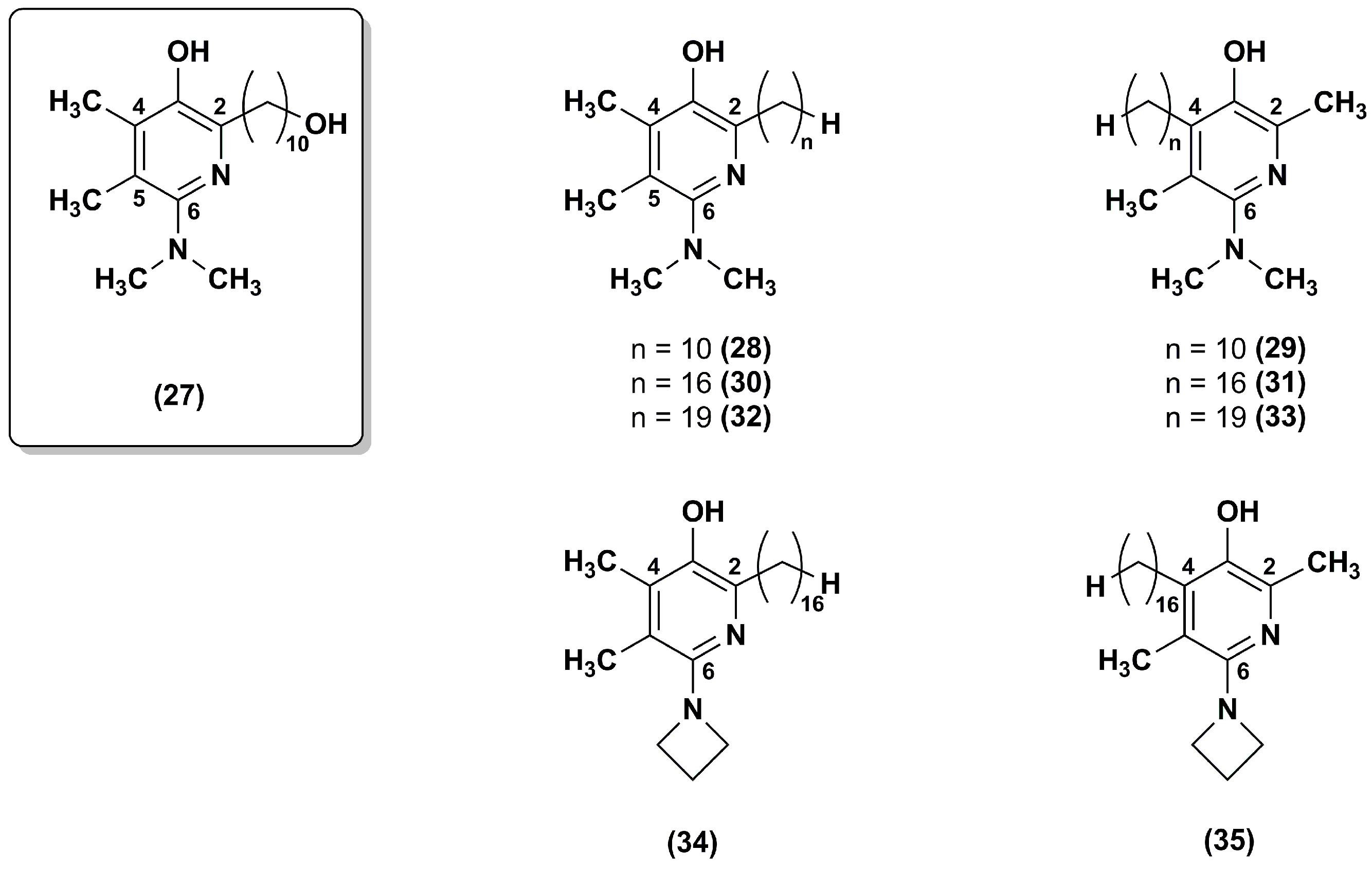
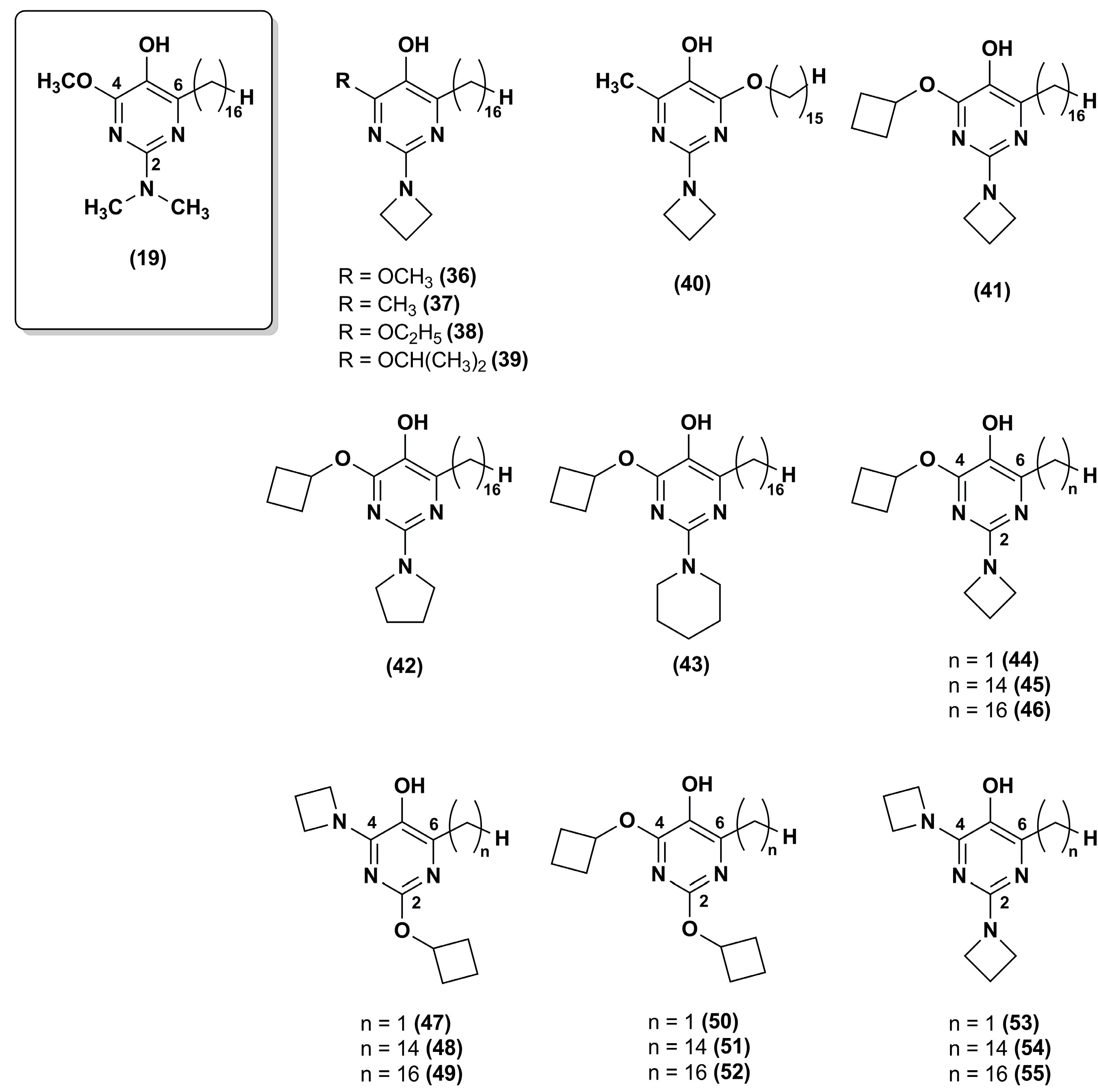
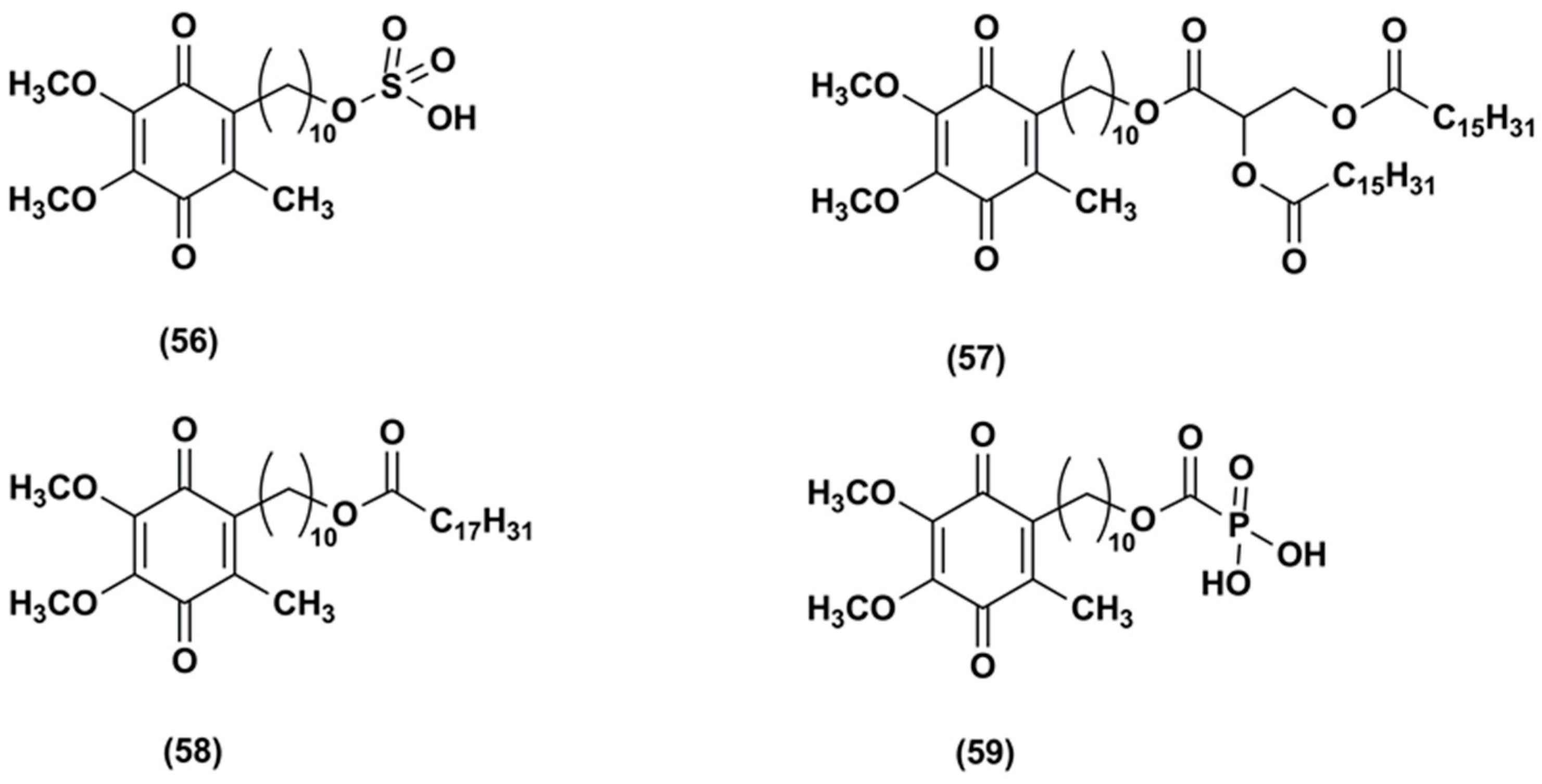
3. Idebenone Analogues
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Meier, T.; Buyse, G. Idebenone: An emerging therapy for Friedreich ataxia. J. Neurol. 2009, 256, 25–30. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Zarante, A.M.; Almannai, M.; Scaglia, F. Therapies for mitochondrial diseases and current clinical trials. Mol. Genet. Metab. 2017, 122, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Erb, M.; Hoffmann-Enger, B.; Deppe, H.; Soeberdt, M.; Haefeli, R.H.; Rummey, C.; Feurer, A.; Gueven, N. Features of idebenone and related short-chain quinones that rescue ATP levels under conditions of impaired mitochondrial complex I. PLoS ONE 2012, 7, e36153. [Google Scholar] [CrossRef] [PubMed]
- Jaber, S.; Polster, B.M. Idebenone and Neuroprotection: Antioxidant, Pro-oxidant, or Electron Carrier? J. Bioenerg. Biomembr. 2015, 47, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.W.; Xu, X.C.; Liu, T.; Yuan, S. Mitochondrion-permeable antioxidants to treat ROS-burst-mediated acute diseases. Oxidative Med. Cell. Longev. 2016, 2016, 6859523. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, M.H.; Schulz, J.B.; Giunti, P. Co-enzyme Q10 and idebenone use in Friedreich’s ataxia. J. Neurochem. 2013, 1, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Strawser, C.; Schadt, K.; Hauser, L.; McCormick, A.; Wells, M.; Larkindale, J.; Lin, H.; Lynch, D.R. Pharmacological therapeutics in Friedreich ataxia: The present state. Expert Rev. Neurother. 2017, 17, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Strawser, C.J.; Schadt, K.A.; Lynch, D.R. Therapeutic approaches for the treatment of Friedreich’s ataxia. Expert Rev. Neurother. 2014, 14, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Kearney, M.; Orrell, R.W.; Fahey, M.; Brassington, R.; Pandolfo, M. Pharmacological treatments for Friedreich ataxia. Cochrane Database Syst. Rev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.P.; Folker, J.; Poole, M.L. Treatment for speech disorder in Friedreich ataxia and other hereditary ataxia syndromes. Cochrane Database Syst. Rev. 2014. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A. A review on mitochondrial restorative mechanism of antioxidants in Alzheimer’s disease and other neurological conditions. Front. Pharmacol. 2015, 6, 206. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Tanaka, T.; Han, D.; Senzaki, K.; Kameyama, T.; Nabeshima, T. Protective effects of idebenone and α-tocopherol on β-amyloid-(1–42)-induced learning and memory deficits in rats: Implication of oxidative stress in β-amyloid-induced neurotoxicity in vivo. Eur. J. Neurosci. 1999, 11, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Gutzmann, H.; Hadler, D. Sustained efficacy and safety of idebenone in the treatment of Alzheimer’s disease: Update on a 2-year double-blind multicentre study. In Alzheimer’s Disease—From Basic Research to Clinical Applications; Springer: Vienna, Austria, 1998; Volume 54, pp. 301–310. [Google Scholar]
- Thal, L.J.; Grundman, M.; Berg, J.; Ernstrom, K.; Margolin, R.; Pfeiffer, E.; Weiner, M.F.; Zamrini, E.; Thomas, R.G. Idebenone treatment fails to slow cognitive decline in Alzheimer’s disease. Neurology 2003, 61, 1498–1502. [Google Scholar] [CrossRef] [PubMed]
- Fiebiger, S.M.; Bros, H.; Grobosch, T.; Janssen, A.; Chanvillard, C.; Paul, F.; Dörr, J.; Millward, J.; Infante-Duarte, C. The antioxidant idebenone fails to prevent or attenuate chronic experimental auto-immune encephalomyelitis in the mouse. J. Neuroimmunol. 2013, 262, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Shirani, A.; Okuda, D.T.; Stüve, O. Therapeutic advances and Future Prospects in Progressive Forms of Multiple Sclerosis. Neurotherapeutics 2016, 13, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Carelli, V.; Carbonelli, M.; de Coo, I.F.; Kawasaki, A.; Klopstock, T.; Lagrèze, W.A.; La Morgia, C.; Newman, N.J.; Orssaud, C.; Pott, J.W.R.; et al. International Consensus Statement on the Clinical and Therapeutic Management of Leber Hereditary Optic Neuropathy. J. Neuroophthalmol. 2017, 37, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Lyseng-Williamson, K.A. Idebenone: A Review in Leber’s Hereditary Optic Neuropathy. Drugs 2016, 76, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Soiferman, D.; Moore, D.G.; Burté, F.; Saada, A. Evaluating the therapeutic potential of idebenone and related quinone analogues in Leber hereditary optic neuropathy. Mitochondrion 2017, 36, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Lekoubou, A.; Kouamé-Assouan, A.E.; Cho, T.H.; Luauté, J.; Nighoghossian, N.; Derex, L. Effect of long-term oral treatment with L-arginine and idebenone on the prevention of stroke-like episodes in an adult MELAS patient. Rev. Neurol. 2011, 167, 852–855. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M.; Meier, T.; Voit, T.; Schara, U.; Straathof, C.S.; D’Angelo, M.G.; Bernert, G.; Cuisset, J.M.; Finkel, R.S.; Goemans, N.; et al. Idebenone reduces respiratory complications in patients with Duchenne muscular dystrophy. Neuromuscul. Disord. 2016, 26, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Liu, J.; Ren, M.; Ji, K.; Li, L.; Zhang, B.; Gong, Y.; Yan, C. Idebenone protects against oxidized low density lipoprotein induced mitochondrial dysfunction in vascular endothelial cells via GSK3β/β-catenin signalling pathways. Biochem. Biophys. Res. Commun. 2015, 465, 548–555. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, D.H.; Neudecker, B.A.; DiNardo, J.C.; Lewis, J.A., II; Maibach, H.I. Clinical efficacy assessment in photodamaged skin of 0.5% and 1.0% idebenone. J. Cosmet. Dermatol. 2005, 4, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Bray-French, K.; Drewe, J. Pharmacokinetic evaluation of idebenone. Expert Opin. Drug Metab. Toxicol. 2010, 6, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Helfand, W.H.; Cowen, D.L. Evolution of pharmaceutical oral dosage forms. Pharm. Hist. 1983, 25, 3–18. [Google Scholar] [PubMed]
- Park, K. Controlled drug delivery systems: Past forward and future back. J. Control. Release 2014, 190, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Patel, B.B.; Tiwari, S. Colloidal nanocarriers: A review on formulation technology, types and applications toward targeted drug delivery. Nanomed. Nanotechnol. Biol. Med. 2010, 6, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Schuber, F.; Kichler, A.; Boeckler, C.; Frisch, B. Liposomes: From membrane models to gene therapy. Pure Appl. Chem. 1998, 70, 89–96. [Google Scholar] [CrossRef]
- Bangham, A.D.; Standish, M.M.; Watkins, J.C. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 1965, 13, 238–252. [Google Scholar] [CrossRef]
- Zylberberg, C.; Matosevic, S. Pharmaceutical liposomal drug delivery: A review of new delivery systems and a look at the regulatory landscape. Drug Deliv. 2016, 23, 3319–3329. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.I.; Yeh, M.K. Clinical development of liposome-based drugs: Formulation, characterization, and therapeutic efficacy. Int. J. Nanomed. 2012, 7, 49–60. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal formulations in clinical use: An updated review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Muscoli, C.; Fresta, M.; Cardile, V.; Palumbo, M.; Renis, M.; Puglisi, G.; Paolino, D.; Nisticò, S.; Rotiroti, D.; Mollace, V. Ethanol-induced injury in rat primary cortical astrocytes involves oxidative stress: Effect of idebenone. Neurosci. Lett. 2002, 329, 21–24. [Google Scholar] [CrossRef]
- Paolino, D.; Iannone, M.; Cardile, V.; Renis, M.; Puglisi, G.; Rotiroti, D.; Fresta, M. Tolerability and improved protective action of idebenone-loaded pegylated liposomes on ethanol-induced injury in primary cortical astrocytes. J. Pharm. Sci. 2004, 93, 1815–1827. [Google Scholar] [CrossRef] [PubMed]
- Pignatello, R.; Acquaviva, R.; Campisi, A.; Raciti, G.; Musumeci, T.; Puglisi, G. Effects of liposomal encapsulation on the antioxidant activity of lipophilic prodrugs of idebenone. J. Liposome Res. 2011, 21, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, L. Nanocarriers for skin delivery of cosmetic antioxidants. J. Pharm. Pharmacogn. Res. 2014, 2, 73–92. [Google Scholar]
- Shah, S.M.; Ashtikar, M.; Jain, A.S.; Makhija, D.T.; Nikam, Y.; Gude, R.P.; Steiniger, F.; Jagtap, A.A.; Nagarsenker, M.S.; Fahr, A. LeciPlex, invasomes, and liposomes: A skin penetration study. Int. J. Pharm. 2015, 490, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Cagno, M.; Stein, P.C.; Skalko-Basnet, N.; Brandl, M.; Bauer-Brandl, A. Solubilization of ibuprofen with β-cyclodextrin derivatives: Energetic and structural studies. J. Pharm. Biomed. Anal. 2011, 55, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Del Valle, E.M.M. Cyclodextrins and their uses: A review. Process Biochem. 2004, 39, 1033–1046. [Google Scholar] [CrossRef]
- Szejtli, J. Medicinal Applications of Cyclodextrins. Med. Res. Rev. 1994, 14, 353–386. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins. 1. Drug solubilization and stabilization. J. Pharm. Sci. 1996, 85, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Challa, R.; Ahuja, A.; Ali, J.; Khar, R.K. Cyclodextrins in drug delivery: An updated review. AAPS PharmSciTech 2005, 6, E329–E357. [Google Scholar] [CrossRef] [PubMed]
- Carrier, R.L.; Miller, L.A.; Ahmed, I. The utility of cyclodextrins for enhancing oral bioavailability. J. Control. Release 2007, 123, 78–99. [Google Scholar] [CrossRef] [PubMed]
- Menezes, P.P.; Serafini, M.R.; Santana, B.V.; Nunes, R.S.; Quintans, L.J.; Silva, G.F.; Medeiros, I.A.; Marchioro, M.; Fraga, B.P.; Santos, M.R.V.; et al. Solid-state β-cyclodextrin complexes containing geraniol. Thermochim. Acta 2012, 548, 45–50. [Google Scholar] [CrossRef]
- Sobrinho, J.L.S.; Soares, M.F.; de La Roca, M.F.; Labandeira, J.J.T.; Alves, L.D.S.; Rolim Neto, P.J. Improving the solubility of the antichagasic drug benznidazole through formation of inclusion complexes with cyclodextrins. Quim. Nova 2011, 34, 1534–1538. [Google Scholar] [CrossRef]
- Ventura, C.A.; Fresta, M.; Giovinazzo, C.; Puglisi, G. Solid state characterization and in solution studies of idebenone-β-cyclodextrin inclusion complex. Acta Technol. Legis Medicam. 1995, VI, 55–66. [Google Scholar]
- Puglisi, G.; Ventura, C.A.; Fresta, M.; Vandelli, M.A.; Cavallaro, G.; Zappalà, M. Preparation and physico-chemical study of inclusion complexes between idebenone and modified β-cyclodextrins. J. Incl. Phenom. Mol. Recognit. Chem. 1996, 24, 193–210. [Google Scholar] [CrossRef]
- Rathi, A.A.; Dhamecha, D.L.; Patel, K.A.; Saifee, M.; Dehghan, M.H.G. Effect of permeation enhancers on permeation kinetics of idebenone through the bovine buccal mucosa. Indian J. Pharm. Educ. Res. 2011, 45, 370–374. [Google Scholar]
- Li, S.C.; Han, M. Idebenone Injection Containing 2-Hydroxylpropyl-beta-cyclodextrin and Preparation Method Thereof. Patent CN 101,926,759, 29 December 2010. [Google Scholar]
- Lauro, F.; Ilari, S.; Giancotti, L.A.; Ventura, C.A.; Morabito, C.; Gliozzi, M.; Malafoglia, V.; Palma, E.; Paolino, D.; Mollace, V.; et al. Pharmacological effect of a new idebenone formulation in a model of carrageenan-induced inflammatory pain. Pharmacol. Res. 2016, 111, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Cannavà, C.; Crupi, V.; Guardo, M.; Majolino, D.; Stancanelli, R.; Tommasini, S.; Ventura, C.A.; Venuti, V. Phase solubility and FTIR-ART studies of idebenone/sulfobutylether-β-cyclodextrin inclusion complex. J. Incl. Phenom. Mol. Recognit. Chem. 2013, 75, 255–262. [Google Scholar] [CrossRef]
- Lauro, M.R.; Carbone, C.; Sansone, F.; Ruozi, B.; Chillemi, R.; Sciuto, S.; Aquino, R.P.; Puglisi, G. Innovative oral spray-dried Idebenone systems to improve patient compliance. Drug Dev. Ind. Pharm. 2015, 42, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Danielsson, I.; Lindman, B. The definition of microemulsion. Colloid Surf. 1981, 3, 391–392. [Google Scholar] [CrossRef]
- Gasco, M.R. Microemulsions in the pharmaceutical field: Perspective and application. In Industrial Applications of Microemulsions; Solans, C., Kunieda, H., Eds.; Marcel Dekker: New York, NY, USA, 1997; pp. 97–122. ISBN 0-8247-9795-7. [Google Scholar]
- Santos, P.; Watkinson, A.C.; Hadgraft, J.; Lane, M.E. Application of microemulsions in dermal and transdermal drug delivery. Skin Pharmacol. Physiol. 2008, 21, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Schmalfub, U.; Neubert, R.; Wohlrab, W. Modification of drug penetration into human skin using microemulsions. J. Control. Release 1997, 46, 279–285. [Google Scholar] [CrossRef]
- Montenegro, L.; Lai, F.; Offerta, A.; Sarpietro, M.G.; Micicchè, L.; Maccioni, A.M.; Valenti, D.; Fadda, A.M. From nanoemulsions to nanostructured lipid carriers: A relevant development in dermal delivery of drugs and cosmetics. J. Drug Deliv. Sci. Technol. 2016, 32, 100–112. [Google Scholar] [CrossRef]
- Nastiti, C.M.R.R.; Ponto, T.; Abd, E.; Grice, J.E.; Benson, H.A.E.; Roberts, M.S. Topical Nano and Microemulsions for Skin Delivery. Pharmaceutics 2017, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, L.; Carbone, C.; Condorelli, G.; Drago, R.; Puglisi, G. Effect of oil phase lipophilicity on in vitro drug release from O/W microemulsions with low surfactant content. Drug Dev. Ind. Pharm. 2006, 32, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Giri, T.K.; Tripathi, D.K.; Ajazuddin, M.; Alexander, A. A review on novel therapeutic strategies for the enhancement of solubility for hydrophobic drugs through lipid and surfactant-based self micro emulsifying drug delivery system: A novel approach. Am. J. Drug Discov. Dev. 2012, 2, 143–183. [Google Scholar] [CrossRef]
- Khedekar, K.; Mittal, S. Self-emulsifying drug delivery system: A review. Int. J. Pharm. Sci. Res. 2013, 4, 4494–4507. [Google Scholar] [CrossRef]
- Kim, H.J.; Yoon, K.A.; Hahn, M.; Park, E.S.; Chi, S.C. Preparation and In Vitro Evaluation of Self-Microemulsifying Drug Delivery Systems Containing Idebenone. Drug Dev. Ind. Pharm. 2000, 26, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Banik, B.L.; Fattahi, P.; Brown, J.L. Polymeric nanoparticles: The future of nanomedicine. WIREs Nanomed. Nanobiotechnol. 2016, 8, 271–299. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Yadav, S.K.; Yadav, S.C. Biodegradable polymeric nanoparticles-based drug delivery systems. Colloids Surf. B Biointerfaces 2010, 75, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Guterres, S.S.; Alves, M.P.; Pohlmann, A.R. Polymeric nanoparticles, nanospheres and nanocapsules, for cutaneous applications. Drug Target Insights 2007, 2, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, M.; Russo, A.; Cardile, V.; Renis, M.; Paolino, D.; Puglisi, G.; Fresta, M. Improved antioxidant effect of idebenone-loaded polyethyl-2-cyanoacrylate nanocapsules tested on human fibroblasts. Pharm. Res. 2002, 19, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Amorim, C.D.M.; Couto, A.G.; Netz, D.J.; de Freitas, R.A.; Bresolin, T.M. Antioxidant idebenone-loaded nanoparticles based on chitosan and N-carboxymethylchitosan. Nanomedicine 2010, 6, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, L.; Trapani, A.; Fini, P.; Mandracchia, D.; Latrofa, A.; Cioffi, N.; Chiarantini, L.; Picceri, G.G.; Brundu, S.; Puglisi, G. Chitosan nanoparticles for topical co-administration of the antioxidants glutathione and idebenone: Characterization and in vitro release. Br. J. Pharm. Res. 2014, 4, 2387–2406. [Google Scholar] [CrossRef]
- Müller, R.H.; Mäder, K.; Gohla, S. Solid lipid nanoparticles (SLN) for controlled drug delivery—A review of the state of the art. Eur. J. Pharm. Biopharm. 2000, 50, 161–177. [Google Scholar] [CrossRef]
- Nair, R.; Arun Kumar, K.S.; Vishnu Priya, K.; Sevukarajan, M. Recent advances in solid lipid nanoparticle-based drug delivery systems. J. Biomed. Sci. Res. 2011, 3, 368–384. [Google Scholar]
- Radtke, M.; Müller, R.H. Nanostructured lipid carriers: The new generation of lipid drug carriers. New Drugs 2001, 2, 48–52. [Google Scholar]
- Singhal, G.B.; Patel, R.P.; Prajapati, B.G.; Patel, N.A. Solid lipid nanoparticles and nano lipid carriers: As novel solid lipid-based drug carrier. Int. Res. J. Pharm. 2011, 2, 40–52. [Google Scholar]
- Pardeike, J.; Hommoss, A.; Müller, R.H. Lipid nanoparticles (SLN, NLC) in cosmetic and pharmaceutical dermal products. Int. J. Pharm. 2009, 366, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, L. Lipid-based nanoparticles as carriers for dermal delivery of antioxidants. Curr. Drug Metab. 2017, 18, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, L.; Parenti, C.; Turnaturi, R.; Pasquinucci, L. Resveratrol-loaded lipid nanocarriers: Correlation between in vitro occlusion factor and in vivo skin hydrating effect. Pharmaceutics 2017, 9, 58. [Google Scholar] [CrossRef] [PubMed]
- Blasi, P.; Giovagnoli, S.; Schoubben, A.; Ricci, M.; Rossi, C. Solid lipid nanoparticles for targeted brain drug delivery. Adv. Drug Deliv. Rev. 2007, 59, 454–477. [Google Scholar] [CrossRef] [PubMed]
- Stancampiano, A.H.; Acquaviva, R.; Campisi, A.; Vanella, L.; Ventura, C.A.; Puglisi, G.; Pignatello, R. Technological and biological characterization of idebenone-loaded solid lipid nanoparticles prepared by a modified solvent injection technique. J. Biomed. Nanotechnol. 2006, 2, 253–260. [Google Scholar] [CrossRef]
- Montenegro, L.; Campisi, A.; Sarpietro, M.G.; Carbone, C.; Acquaviva, R.; Raciti, G.; Puglisi, G. In vitro evaluation of idebenone-loaded solid lipid nanoparticles for drug delivery to the brain. Drug Dev. Ind. Pharm. 2011, 37, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, L.; Trapani, A.; Latrofa, A.; Puglisi, G. In vitro evaluation on a model of blood brain barrier of idebenone-loaded solid lipid nanoparticles. J. Nanosci. Nanotechnol. 2012, 12, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, L.; Sinico, C.; Castangia, I.; Carbone, C.; Puglisi, G. Idebenone-loaded solid lipid nanoparticles for drug delivery to the skin: In vitro evaluation. Int. J. Pharm. 2012, 434, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ge, Z.Q. Nanostructured lipid carriers improve skin permeation and chemical stability of idebenone. AAPS PharmSciTech 2012, 13, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Kyadarkunte, A.Y.; Patole, M.S.; Pokharkar, V.B. Cellular interactions and photoprotective effects of idebenone-loaded nanostructured lipid carriers stabilized using PEG-free surfactant. Int. J. Pharm. 2015, 479, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Fresta, M.; Ventura, C.A.; Mezzasalma, E.; Puglisi, G. A calorimetric study on the idebenone-phospholipid membrane interaction. Int. J. Pharm. 1998, 163, 133–143. [Google Scholar] [CrossRef]
- Montenegro, L.; Ottimo, S.; Puglisi, G.; Castelli, F.; Sarpietro, M.G. Idebenone loaded solid lipid nanoparticles interact with biomembrane models: Calorimetric evidence. Mol. Pharm. 2012, 9, 2534–2541. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, L.; Sarpietro, M.G.; Ottimo, S.; Puglisi, G.; Castelli, F. Differential scanning calorimetry studies on sunscreen loaded solid lipid nanoparticles prepared by the phase inversion temperature method. Int. J. Pharm. 2011, 415, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Sarpietro, M.G.; Accolla, M.L.; Puglisi, G.; Castelli, F.; Montenegro, L. Idebenone loaded solid lipid nanoparticles: Calorimetric studies on surfactant and drug loading effects. Int. J. Pharm. 2014, 471, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, A.; Crasci’, L.; Panico, A.; Pignatello, R. Antioxidant activity of idebenone-loaded neutral and cationic solid-lipid nanoparticles. Pharm. Dev. Technol. 2015, 20, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, L.; Modica, M.N.; Salerno, L.; Panico, A.M.; Crascì, L.; Puglisi, G.; Romeo, G. In Vitro Antioxidant Activity of Idebenone Derivative-Loaded Solid Lipid Nanoparticles. Molecules 2017, 22, 887. [Google Scholar] [CrossRef] [PubMed]
- Duveau, D.Y.; Arce, P.M.; Schoenfeld, R.A.; Raghav, N.; Cortopassi, G.A.; Hecht, S.M. Synthesis and characterization of mitoQ and idebenone analogues as mediators of oxygen consumption in mitochondria. Bioorg. Med. Chem. 2010, 18, 6429–6441. [Google Scholar] [CrossRef] [PubMed]
- Fash, D.M.; Khdour, O.M.; Sahdeo, S.J.; Goldschmidt, R.; Jaruvangsanti, J.; Dey, S.; Arce, P.M.; Collin, V.C.; Cortopassi, G.A.; Hecht, S.M. Effects of alkyl side chain modification of coenzyme Q10 on mitochondrial respiratory chain function and cytoprotection. Bioorg. Med. Chem. 2013, 21, 2346–2354. [Google Scholar] [CrossRef] [PubMed]
- Arce, P.M.; Khdour, O.M.; Goldschmidt, R.; Armstrong, J.S.; Hecht, S.M. A strategy for suppressing redox stress within mitochondria. ACS Med. Chem. Lett. 2011, 2, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Pasquinucci, L.; Turnaturi, R.; Prezzavento, O.; Arena, E.; Aricò, G.; Georgoussi, Z.; Parenti, R.; Cantarella, G.; Parenti, C. Development of novel LP1-based analogues with enhanced delta opioid receptor profile. Bioorg. Med. Chem. 2017, 25, 4745–4752. [Google Scholar] [CrossRef] [PubMed]
- Turnaturi, R.; Oliveri, V.; Viale, M.; Monticone, M.; Vecchio, G. Antiproliferative and antioxidant activity of glycoconjugates of dithiocarbamates and their copper(II) and zinc(II) complexes. ChemPlusChem 2015, 80, 1786–1792. [Google Scholar] [CrossRef]
- Goldschmidt, R.; Arce, P.M.; Khdour, O.M.; Collin, V.C.; Dey, S.; Jaruvangsanti, J.; Fash, D.M.; Hecht, S.M. Effects of cytoprotective antioxidants olymphocytes from representative mitochondrial neurodegenerative diseases. Bioorg. Med. Chem. 2013, 21, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Khdour, O.M.; Arce, P.M.; Roy, B.; Hecht, S.M. An optimized pyrimidinol multifunctional radical quencher. ACS Med. Chem. Lett. 2013, 4, 724–729. [Google Scholar] [CrossRef] [PubMed]
- Arce, P.M.; Goldschmidt, R.; Khdour, O.M.; Madathil, M.M.; Jaruvangsanti, J.; Dey, S.; Fash, D.M.; Armstrong, J.S.; Hecht, S.M. Analysis of the structural and mechanistic factors in antioxidants that preserve mitochondrial function and confer cytoprotection. Bioorg. Med. Chem. 2012, 20, 5188–5201. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.P.; Khdour, O.M.; Arce, P.M.; Chen, Y.; Roy, B.; Johnson, W.G.; Dey, S.; Hecht, S.M. Cytoprotective pyridinol antioxidants as potential therapeutic agents for neurodegenerative and mitochondrial diseases. Bioorg. Med. Chem. 2014, 22, 4935–4947. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, A.; Alam, M.P.; Khdour, O.M.; Schmierer, M.; Arce, P.M.; Cripe, C.D.; Hecht, S.M. Optimization of pyrimidinol antioxidants as mitochondrial protective agents: ATP production and metabolic stability. Bioorg. Med. Chem. 2016, 24, 5206–5220. [Google Scholar] [CrossRef] [PubMed]
- Prezzavento, O.; Arena, E.; Parenti, C.; Pasquinucci, L.; Aricò, G.; Scoto, G.M.; Grancara, S.; Toninello, A.; Ronsisvalle, S. Design and synthesis of new bifunctional sigma-1 selective ligands with antioxidant activity. J. Med. Chem. 2013, 56, 2447–2455. [Google Scholar] [CrossRef] [PubMed]
- Turnaturi, R.; Oliveri, V.; Vecchio, G. Biotin-8-hydroxyquinoline conjugates and their metal complexes: Exploring the chemical properties and the antioxidant activity. Polyhedron 2016, 110, 254–260. [Google Scholar] [CrossRef]
- Chevalier, A.; Khdour, O.M.; Schmierer, M.; Bandyopadhyay, I.; Hecht, S.M. Influence of substituent heteroatoms on the cytoprotective properties of pyrimidinol antioxidants. Bioorg. Med. Chem. 2017, 25, 1703–1716. [Google Scholar] [CrossRef] [PubMed]
- Neudecker, B.; Wieland, E.; Diedrich, F. Topically Applied Idebenone Containing Agent with Protective and Regenerative Effect. U.S. Patent 6,756,045 B1, 29 June 2004. [Google Scholar]
- Diedrich, F.; Neudecker, B.; Wieland, E.; Joseph, A.L.; DiNardo, J.C.; Thompson, A.S.; Kerschen, J.A.; Wade, P.C. Skin Treatments with Carboxylic Acid-Substituted Idebenone Derivatives. U.S. Patent 8,173,703 B2, 8 May 2012. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Delivery System | Target | Investigated Properties and Results | Ref. |
|---|---|---|---|
| Liposomes | Brain delivery | IDE loaded liposomes reduced ethanol-induced injury on rat astroglial cell cultures and improved cell viability compared to free IDE. These liposomes were not suitable for in vivo systemic administration due to their uptake by the reticulo-endothelial system. | [34] |
| Liposomes (PEGylated large unilamellar vesicles) | Brain delivery Systemic administration | IDE loaded liposomes were more effective than the free drug in reducing ethanol-induced injury in rat primary cortical astrocyte cultures. A concentration-dependent toxic effect on cortical astrocytes was observed. These liposomes were supposed to be suitable for in in vivo systemic administration as they could escape the uptake by the reticulo-endothelial system. | [35] |
| Liposomes (neutral or negatively charged small unilamellar vesicles) loaded with IDE or IDE prodrugs | Brain delivery | These liposomes showed poor release of the encapsulated IDE prodrugs. Negative liposomes loading IDE and IDE prodrugs were less effective at reducing lactic dehydrogenase production and at protecting against oxidative damages in rat astrocyte cultures than the corresponding neutral liposomes. | [36] |
| Liposomes (conventional, cationic, invasome) | Skin delivery Topical administration | Cationic liposomes provided the highest IDE skin delivery in ex vivo human skin penetration studies and the highest in vitro cytotoxicity on B16F10 melanoma. | [38] |
| β-cyclodextrins | Not specified | IDE inclusion in β-cyclodextrins showed a linear increase in drug solubility and an enhancement of dissolution rate in comparison with free IDE. | [47] |
| Modified-β-cyclodextrin Dimethyl-β-cyclodextrin Hydroxypropyl-β-cyclodextrins | Brain delivery Oral administration | Dimethyl-β-cyclodextrins and hydroxypropyl-β-cyclodextrins showed the best ability to increase IDE water solubility and to enhance IDE dissolution rate. | [48] |
| β-cyclodextrins hydroxypropyl-β-cyclodextrins | Brain delivery Buccal administration | Cyclodextrins enhanced IDE water solubility, dissolution rate and permeability through the buccal mucosa. IDE complex with hydroxypropyl-β-cyclodextrins acted as a penetration enhancer for IDE buccal delivery, providing the best enhancement ratio (45.93) in comparison with all other penetration enhancers tested. | [49] |
| Hydroxypropyl-β-cyclodextrins | Systemic administration | Enhanced IDE solubility in aqueous vehicles | [50] |
| Hydroxypropyl-β-cyclodextrins | Systemic administration | Intraperitoneal pretreatment with hydroxypropyl-β-cyclodextrins complexed IDE inhibited hyperalgesia and edema in an animal model (rat) of carrageenan induced thermal hyperalgesia. This complex highlighted the analgesic and anti-inflammatory activity of IDE. | [51] |
| Sulfobutyl ether-β-cyclodextrins | Brain delivery Oral administration | IDE complexation with sulfobutyl ether-β-cyclodextrins increased its water solubility and dissolution rate. | [52] |
| β-cyclodextrins polymer | Brain delivery Oral administration | Loading IDE into microparticles containing a β-cyclodextrins polymer and an enhancer of dissolution rate increased its water solubility, wettability and dissolution rate. | [53] |
| Microemulsions | Skin delivery Topical administration | IDE release depended on the type of surfactant and on the lipophilicity of the oils used to prepare the microemulsion. | [60] |
| Self-microemulsifying drug delivery systems | Brain delivery Oral administration | IDE release rate from optimized SMEDDS was two-fold higher than that obtained from conventional tablets. | [63] |
| Polymeric nanoparticles (polyethyl-2-cyanoacrylates) | Brain delivery Oral and systemic administration | IDE loaded nanoparticles showed higher in vitro antioxidant effects in human fibroblasts than free IDE. | [68] |
| Polymeric nanoparticles (chitosan and N-carboxymethylchitosan) | Skin or nasal delivery Topical administration | These nanoparticles increased IDE stability while preserving its in vitro antioxidant activity and reducing mucous membrane irritation in comparison with the free drug. | [69] |
| Polymeric nanoparticles (Glutathionylchitosan) | Skin delivery Topical administration | IDE loaded nanoparticles showed a strong in vitro antioxidant activity while IDE in aqueous vehicle showed no activity. These nanoparticles were not cytotoxic in human keratinocytes (HaCaT) cell lines. | [70] |
| Solid lipid nanoparticles | Brain delivery Systemic administration | These solid lipid nanoparticles provided a slow and prolonged IDE in vitro release and maintained or increased IDE protective effect against free radical-induced oxidative stress in astrocyte cell cultures. | [79] |
| Solid lipid nanoparticles | Brain delivery Systemic administration | IDE in vitro release from these carriers depended on the type of surfactant used and the amount of loaded drug. IDE loaded solid lipid nanoparticles were more effective than free IDE at inhibiting free radical-induced oxidative stress in primary cultures of astrocytes obtained from rat cerebral cortex. | [80] |
| Solid lipid nanoparticles | Brain delivery Systemic administration | IDE permeability across a model of blood brain barrier (MDCKII-MDR1 cell monolayers) from these solid lipid nanoparticles was slightly lower than free IDE but IDE could be administered in aqueous media. | [81] |
| Solid lipid nanoparticles | Skin delivery Topical administration | These solid lipid nanoparticles provided an accumulation of IDE into the upper skin layers without any significant permeation through pig skin, depending on their composition and IDE loading. | [82] |
| Solid lipid nanoparticles (cationic) | Ocular delivery Topical administration | Cationic solid lipid nanoparticles provided an increase of IDE stability in comparison with the free drug while preserving its in vitro antioxidant activity. | [89] |
| Solid lipid nanoparticles IDE and IDE derivatives | Brain delivery Systemic administration | IDE and IDE derivatives loaded solid lipid nanoparticles showed prolonged in vitro antioxidant activity and increased water solubility. | [90] |
| Nanostructured lipid carriers | Skin delivery Topical administration | IDE loaded nanostructured lipid carriers increased IDE in vitro permeation through guinea pig skin and improved IDE chemical stability. | [83] |
| Nanostructured lipid carriers | Skin delivery Topical administration | IDE loaded nanostructured lipid carriers increased in vitro IDE skin deposition and cellular uptake (HaCaT cells), showing photo-protective effects against UVB-mediated oxidative stress in HaCaT cells. | [84] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montenegro, L.; Turnaturi, R.; Parenti, C.; Pasquinucci, L. Idebenone: Novel Strategies to Improve Its Systemic and Local Efficacy. Nanomaterials 2018, 8, 87. https://doi.org/10.3390/nano8020087
Montenegro L, Turnaturi R, Parenti C, Pasquinucci L. Idebenone: Novel Strategies to Improve Its Systemic and Local Efficacy. Nanomaterials. 2018; 8(2):87. https://doi.org/10.3390/nano8020087
Chicago/Turabian StyleMontenegro, Lucia, Rita Turnaturi, Carmela Parenti, and Lorella Pasquinucci. 2018. "Idebenone: Novel Strategies to Improve Its Systemic and Local Efficacy" Nanomaterials 8, no. 2: 87. https://doi.org/10.3390/nano8020087
APA StyleMontenegro, L., Turnaturi, R., Parenti, C., & Pasquinucci, L. (2018). Idebenone: Novel Strategies to Improve Its Systemic and Local Efficacy. Nanomaterials, 8(2), 87. https://doi.org/10.3390/nano8020087