2. Surface Ionization: Principle Sensing Mechanism
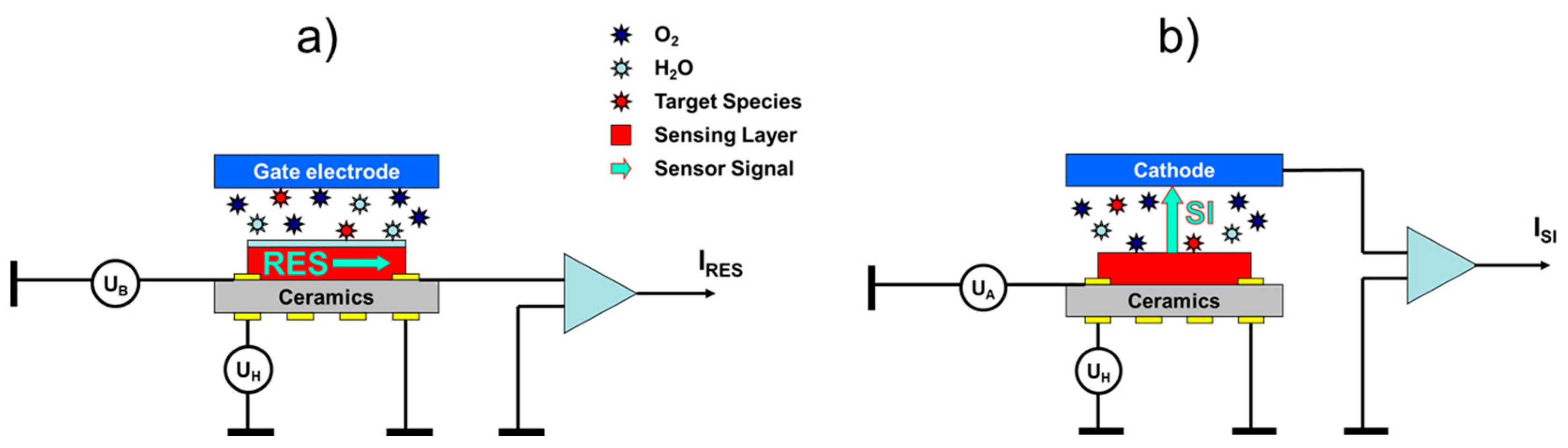
Surface ionization gas detectors show some resemblance to the more conventional metal oxide (MOX) gas sensors. This fact is shown in
Figure 1, where we have schematically drawn the architectures of a conventional MOX gas sensor with a resistive (RES) readout (
Figure 1a) and a surface ionization (SI) gas sensor with a vertical SI current readout (
Figure 1b). Both kinds of sensors rely on functional materials deposited on heatable substrates. In RES gas sensors, the back-surface heater heats the MOX sensing layer on the front side to its optimum working temperature to induce gas surface interactions. While oxidizing gases, such as O
2, NO
2, and O
3, withdraw electrons from n-type MOX materials reducing ones consume negative oxygen ion adsorbates, thereby forming neutral reaction products, such as H
2O and CO
2, and returning electrons to the MOX adsorbent layers [
14,
15]. The resulting resistivity changes are detected by external electronic circuits. In
Figure 1a, we have additionally drawn an optional gate electrode, positioned above an air gap on top of the MOX sensing layer. Bias voltages applied to this electrode can generate electrical fields acting vertically to the electrical current flow inside the MOX layer. Such fields can influence both the charge transport inside the MOX sensing layer itself as well as the rates of charge exchange between the adsorbates and the sub-surface MOX layer. In this way, electro-adsorption and -desorption phenomena can be utilized to influence the sensitivity-selectivity characteristics of MOX gas sensing layers [
16,
17]. Inducing such effects requires surface electrical fields of more than
and small air gaps in the order of
or below, if battery-compatible low-voltage operation is desired. As generating such small air gaps requires methods of multi-wafer micro-assembly, this latter option is not usually followed [
18].
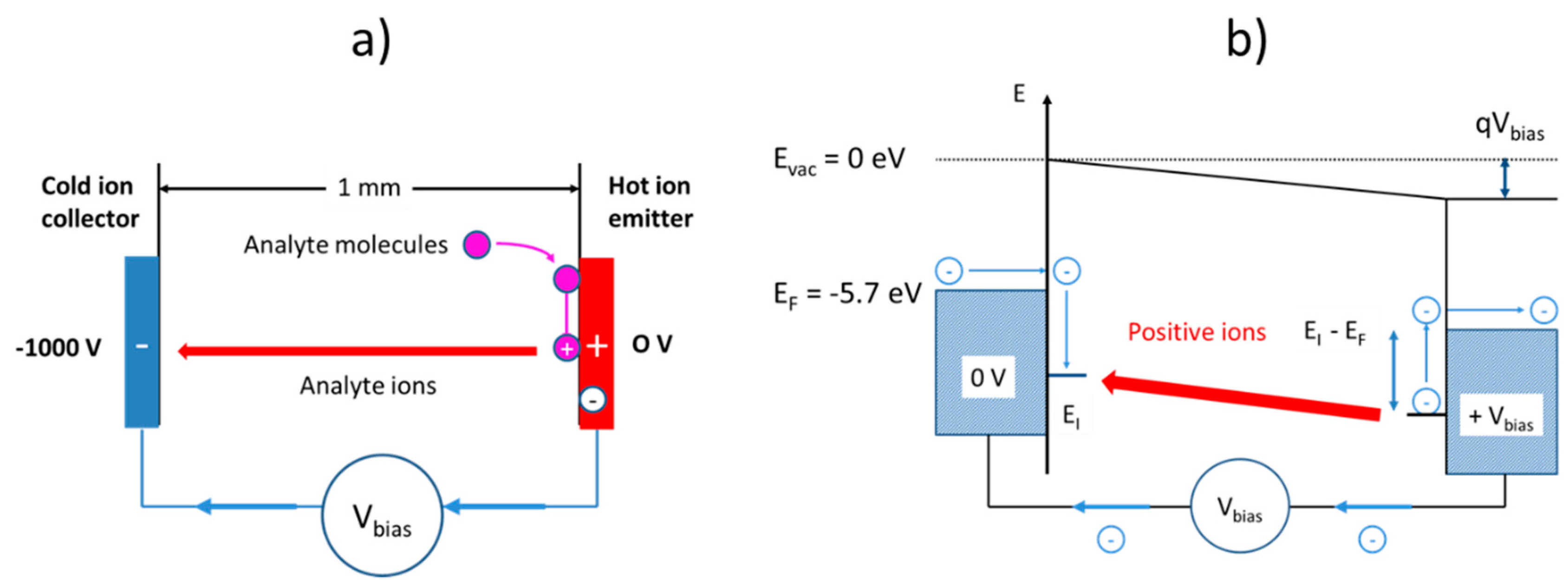
Although architecturally very similar, surface ionization (SI) gas sensors follow a very different kind of operation principle as illustrated in
Figure 1b. As described in more detail in our previous papers [
5,
6,
7,
8,
9,
10,
11,
12,
13], surface ionization can be observed when molecules become absorbed at heated solid surfaces, and when valence electrons are raised from the highest occupied molecular orbitals (HOMO) of the adsorbates to one of the lowest unoccupied electron orbitals (LUMO) inside the adsorbent solid. In this way, positive ion adsorbates are formed, which remain bound to the adsorbent surface with some energy,
, until these become thermally released from the heated emitter electrode. Once liberated from the surface, the positive ions start to move along the electrical field lines stretching across the air gap between emitter and collector electrodes and rapidly attain a constant drift velocity, which results in an ion current,
, that can be detected in an external circuit. Another important difference relative to conventional MOX gas sensors is that SI gas detectors can, in principle, be realized using simple metal electrodes as ion emitters. In the early work on SI gas detection, both noble-metal and refractory-metal electrodes were investigated [
1,
2,
3,
4]. As refractory metals tend to get oxidized upon high-temperature operation in air, the actual ion emission in most cases is likely to have occurred from some sort of MOX material, which moves SI gas detectors somewhat closer to conventional MOX gas sensors.
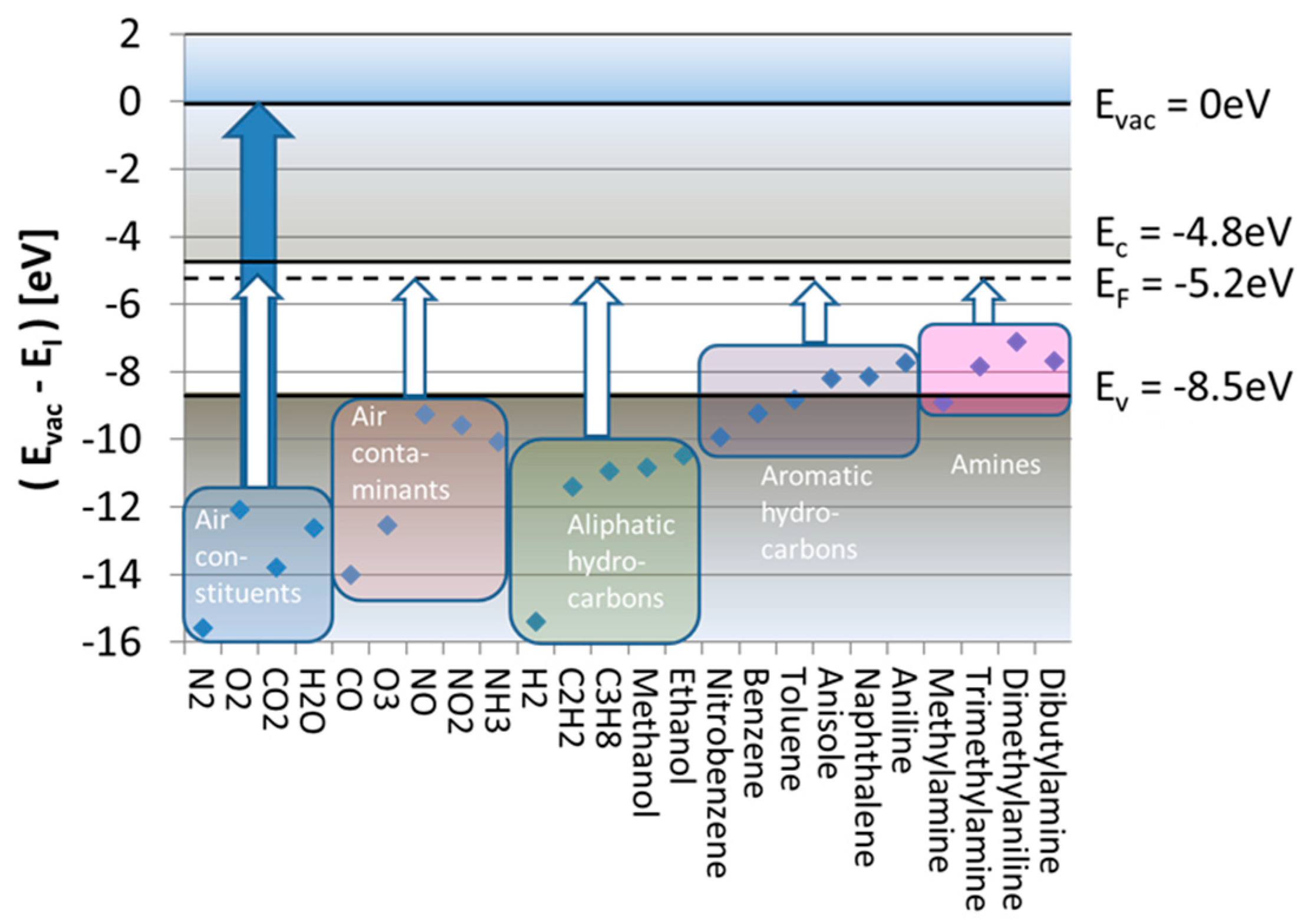
Figure 2 considers the subject of SI gas detection from its energetic point of view. Drawn there are the free-space ionization energies,
, of a range of different molecules relative to the vacuum energy level at which valence electrons become unbound, having zero kinetic energy (
). This plot reveals that free-space ionization energies range between
, which means that, if direct ionization with photons is desired, ionization requires illumination with deep ultraviolet light.
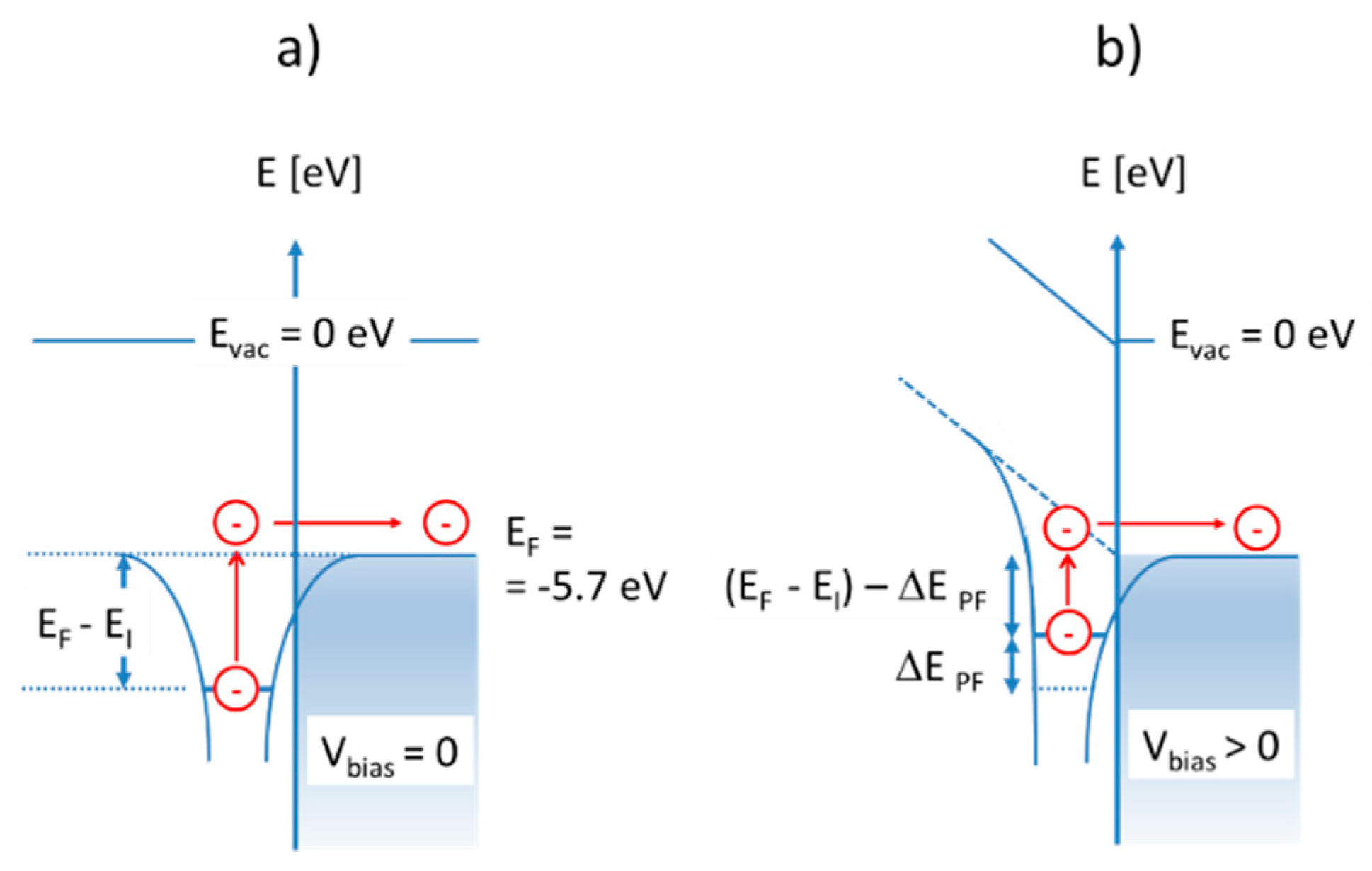
Much less energy is required in case these same molecules become adsorbed at heated solid surfaces. In case the emitter surface is a noble metal, such as, for instance, platinum (Pt), the LUMO energy is the metal Fermi energy, which lies roughly
below the vacuum energy [
19]:
Ionization of adsorbed molecules, therefore, is energetically much more favorable than the ionization of free molecules. In case the emitter material is an n-type metal oxide, the majority of accessible LUMO levels are those empty electron states immediately above the conduction band edge (). Alternatively, in p-type oxides, the accessible LUMO states will be those empty band states below the valence band edge (). Considering the realistic case that nanocrystalline MOX materials will contain a multitude of localized defect states in the bandgap, which will pin the Fermi energy, the relevant LUMO energy levels will again coincide with the Fermi energy of the respective MOX materials. From these considerations, it is evident that surface ionization energies, , can be controlled to some extent by a proper choice of emitter materials, emitter morphologies, and doping conditions.
To see how the energy expense for surface ionization,
, varies with the kind of analyte gas species, we consider in
Figure 2 the special case of a SnO
2 emitter electrode. Drawn there on the energy scale are horizontal lines representing the values of
, and
relevant to SnO
2. The white arrows ending at
indicate those energy inputs,
, that are necessary to ionize five relevant groups of analyte molecules. These arrows indicate that, even after adsorption, the ionization of all major background air constituents (N
2, O
2, CO
2, H
2O) still requires huge energy inputs of more than
, which are virtually impossible to supply out of the thermal reservoir of a heated emitter electrode. A similar situation arises in the case of H
2 and common aliphatic hydrocarbons. Much better chances for thermal ionization exist if the adsorbates come from the groups of aromatic hydrocarbons or amines. In these latter cases, the remaining energy inputs drop to
, only.
A key advantage of SI processes is that their probability scales in an Arrhenius-type manner with the ionization energy,
. In quantitative terms, the ionization current density,
, is expected to scale with the absolute temperature,
, and the analyte partial pressure,
, as [
6,
7]:
In this equation,
is the Fermi energy inside the adsorbent solid,
the free-space ionization energy of the adsorbate, and
its binding energy on the emitter surface. Due to this exponential cut-off criterion, all stable air constituents, such as N
2, O
2, CO
2, and H
2O, should remain completely undetectable. Analytically interesting molecules, such as aromatic hydrocarbons and amines, however, can potentially be detected with a high degree of selectivity. Concerning these latter groups of analytes, we mention that many toxic industrial compounds (TIC) feature aromatic rings [
20], while many organic decay products as well as medical and illicit drugs feature amine functional side groups on their hydrocarbon backbones [
8,
9,
10]. SI gas sensors therefore hold the promise of forming simple, sensitive, and selective sensors for interesting groups of analytes.
Fuji et al. [
3,
4] have studied surface ionization processes by applying mass spectroscopic analysis to the emitted ions. In this work, they found that surface ionization mainly proceeds through three different reaction channels:
In the first process, already discussed above, an electron is simply transferred from the adsorbate
across the HOMO-LUMO gap to the emitter surface, leaving a positive ion adsorbate
and an electron transferred to the emitter surface (
):
In the other two processes, hydrogen is involved in the ionization process. To enable ionization processes in which analyte ion adsorbates either contain smaller or larger numbers of covalently bonded hydrogen atoms than the neutral analytes themselves, sources and sinks for the transferred hydrogen need to exist on the emitter surface. Keeping this in mind, an example for a dissociative ionization process is:
Here, (
) stands for an analyte molecule with at least one covalently bonded hydrogen atom;
is a surface oxygen ion, and
a surface hydroxyl group. Similarly, an associative ionization process can be represented by:
In this latter process, the analyte molecule has become associated with a proton that has been withdrawn from an
adsorbate. Process (4c) is particularly advantageous in case analyte molecules with exceptionally high proton affinity are involved. Proton affinity (PA) is hereby defined as the energy gained in the reaction:
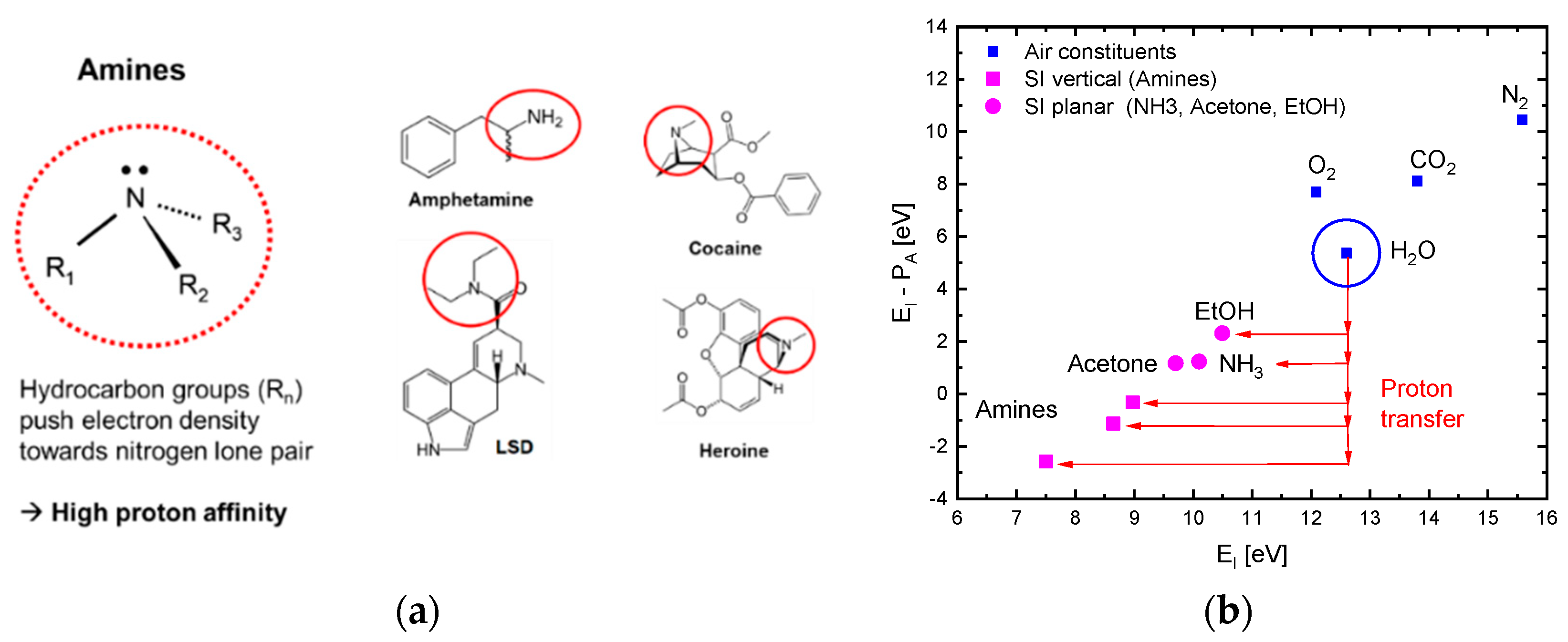
Figure 3a illustrates why high levels of proton affinity arise in the case of molecules with amine functional groups.
Figure 3b further shows that molecules, M, with low free-space ionization energy,
, normally also feature large values of proton affinity [
19]. This latter figure shows that, in the case of amines, the energy gain upon attaching a proton to a molecule
can exceed the energy input required for enabling free-space ionization of the compound ion
. This latter effect, in principle, turns the ionization of amines exothermic. The effect that ultimately turns the surface ionization of amines endothermic is that analytes first need to become chemisorbed before these can undergo surface ionization (Equation (2) and references [
6,
7]). Overall, the involvement of
groups in reactions (4b) and (4c) shows that despite the large ionization energy of water molecules (
), the presence of water vapor in the ambient air is likely to impact the energetics of dissociative and associative ionization processes. Evidence into this latter direction is presented in
Section 3.2.
4. Impact of Device Architecture on Sensitivity/Selectivity Characteristics
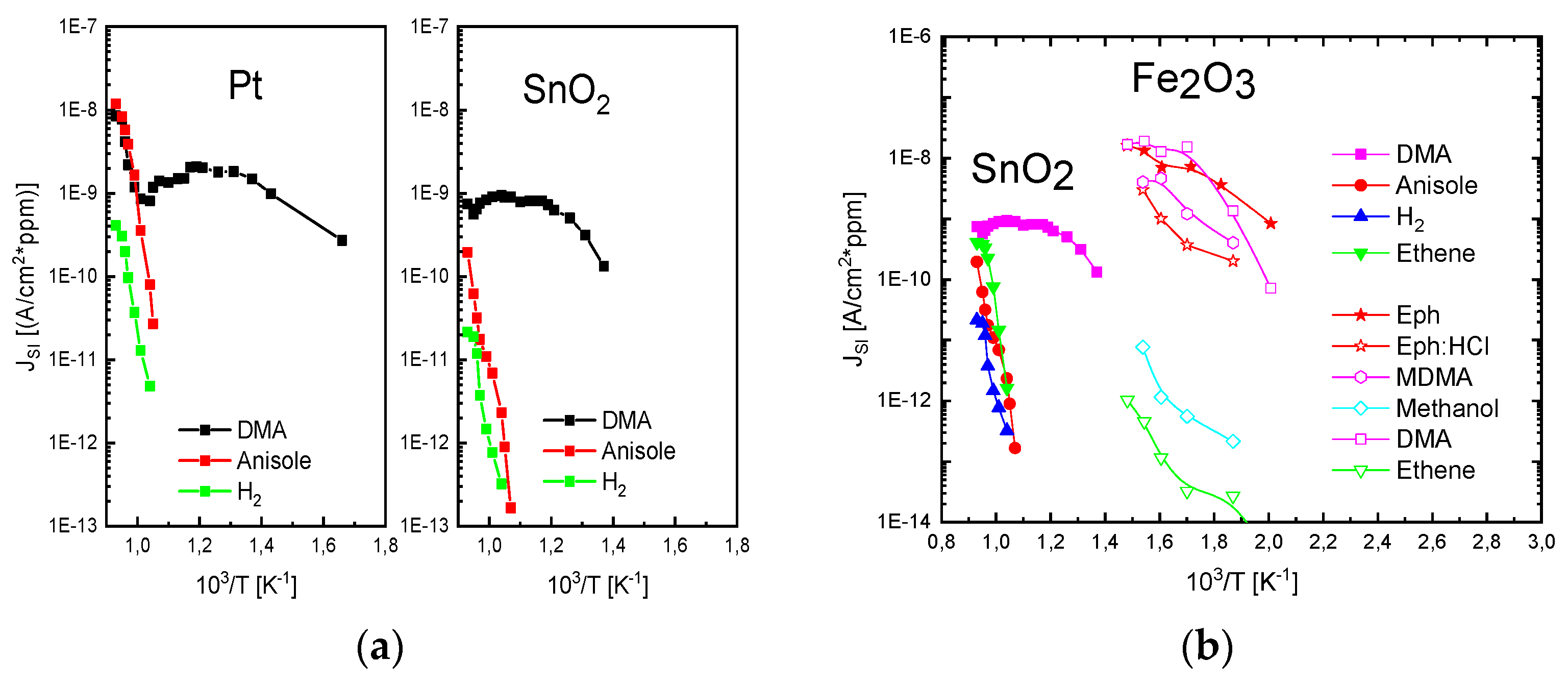
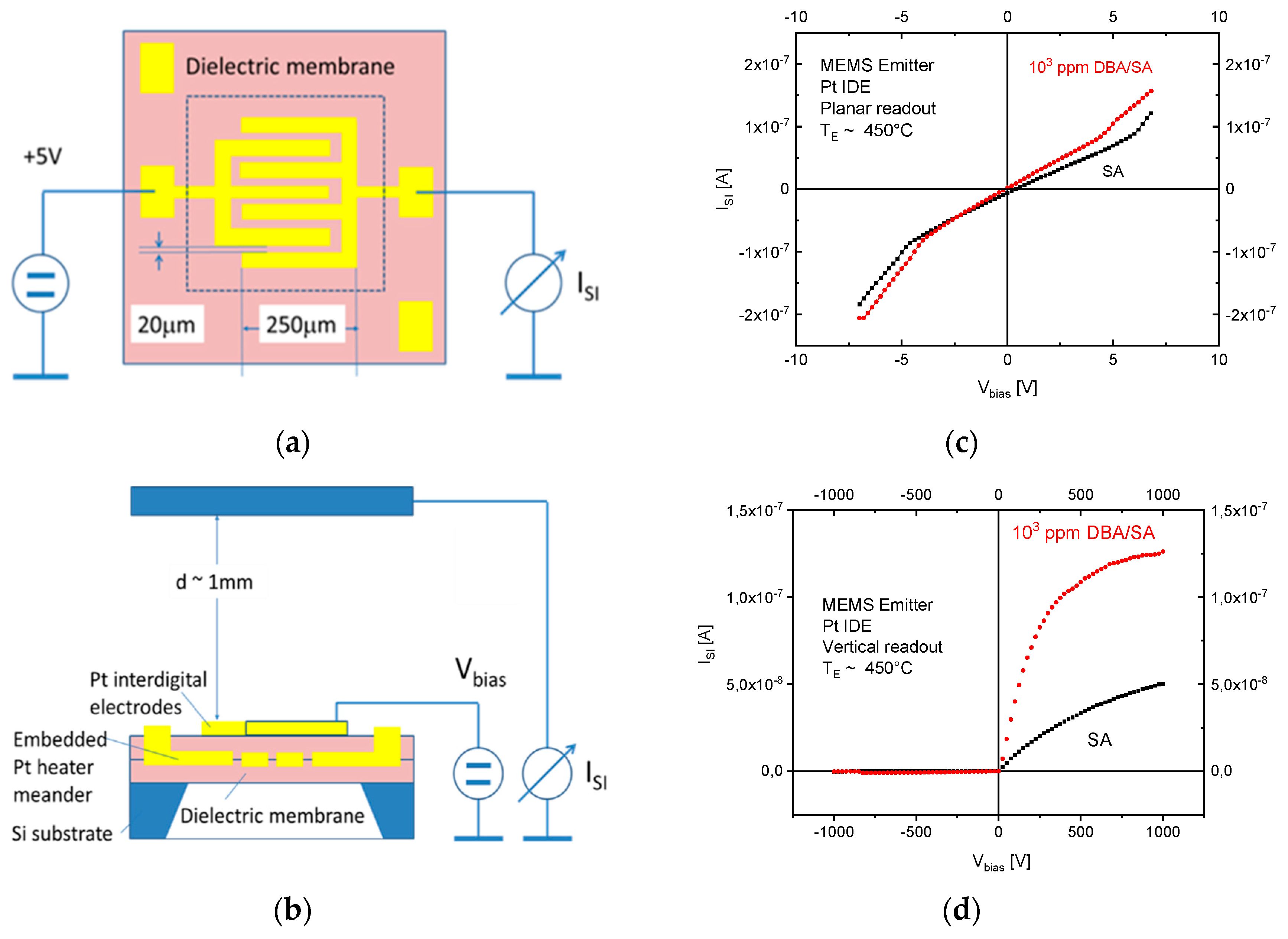
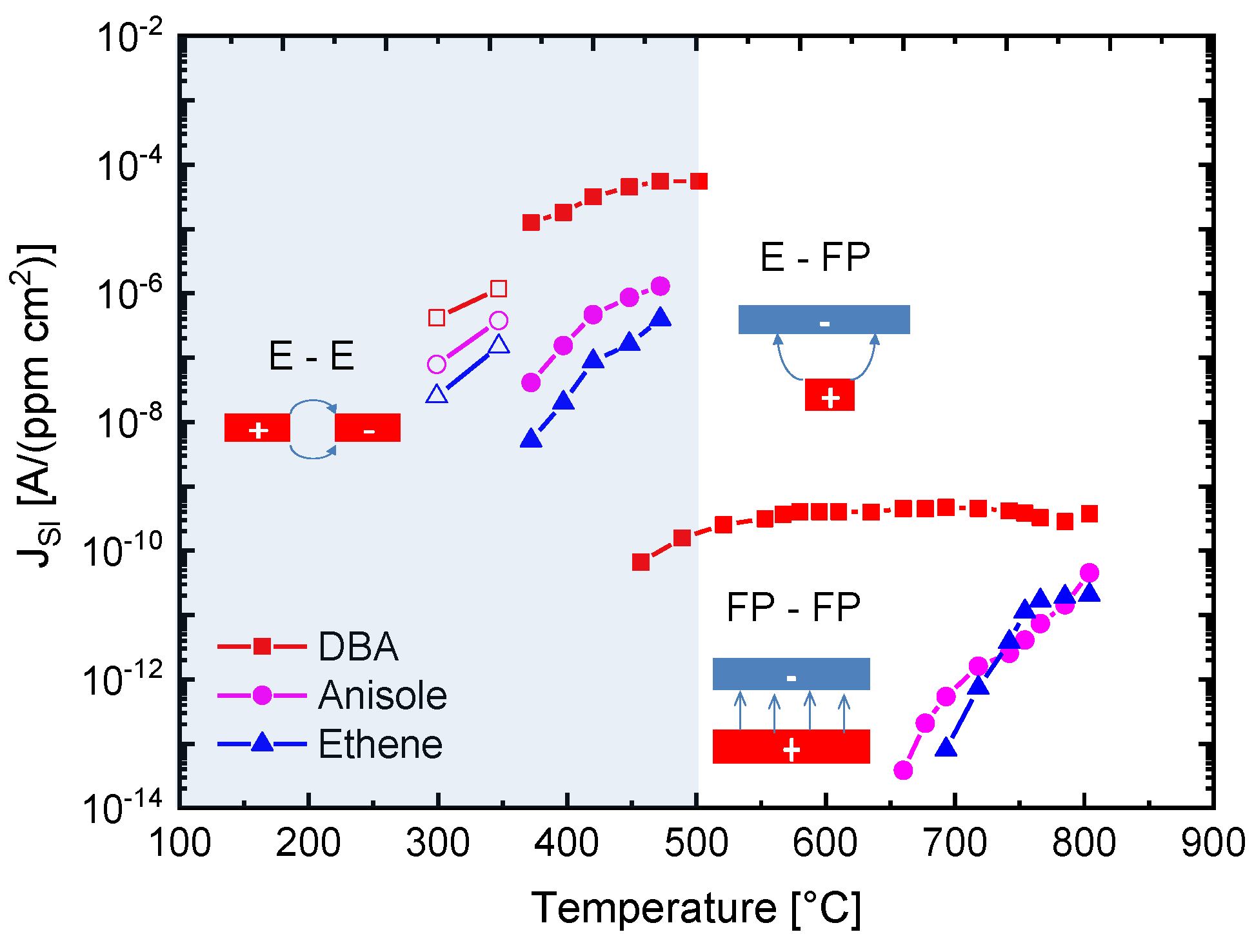
The results in
Figure 10 demonstrate with clarity that very different kinds of IV characteristics can be observed when one and the same kind of emitter material is shaped into different readout geometries. Whereas in these MEMS devices, the readout structures take the form of edge-to-edge (E-E) or edge-to-flat plate (E-FP) geometries, the initially investigated vertical read-out structures, shown in
Figure 4 and fitted with thin-film Pt electrodes, take the form of flat plate-to-flat plate (FP-FP) capacitor structures. In
Figure 11, we compare the SI current densities generated by these three different kinds of device structures as functions of the emitter temperature and in response to three gases with different free-space ionization energies: Dibutyl-amine,
; anisole,
; and ethene,
. The device configurations, on which the individual data have been obtained, are sketched in the form of insets in
Figure 11. In comparing the different data sets, the most striking observation is that the obtainable ion current densities and the level of amine selectivity of the SI response are both critically dependent on the form of the ion-emitting and ion-collecting electrodes [
6,
7,
12,
13].
By far, the lowest ion current densities are produced by FP-FP configurations, in which both emitter and collector electrodes take the form of flat-plates [
5,
6,
7]. In these parallel-plate capacitor-type devices, emitters and collectors are separated by an air gap of 1mm, and a DC potential of 1000 V is applied between both electrodes to allow ion currents to flow through the air gap. In this first vertical readout configuration, sizeable ion current densities could only be observed at moderate emitter temperatures, in the case of dibutyl-amine (DBA), which features a particularly low ionization energy (
) and a concomitantly high proton affinity of
. Analytes with slightly higher ionization energies (
,
) hardly yielded any observable ion currents at emitter temperatures lower than 600 °C. FP-FP configurations, therefore, are almost selective to low-ionization-energy amines with a concomitantly high proton affinity. From an application point of view, it is important to note that a whole range of illicit drugs does fall into this category. FP-FP types of sensors therefore can make very simple, sensitive, and selective sensors for illicit drugs [
8,
9,
10].
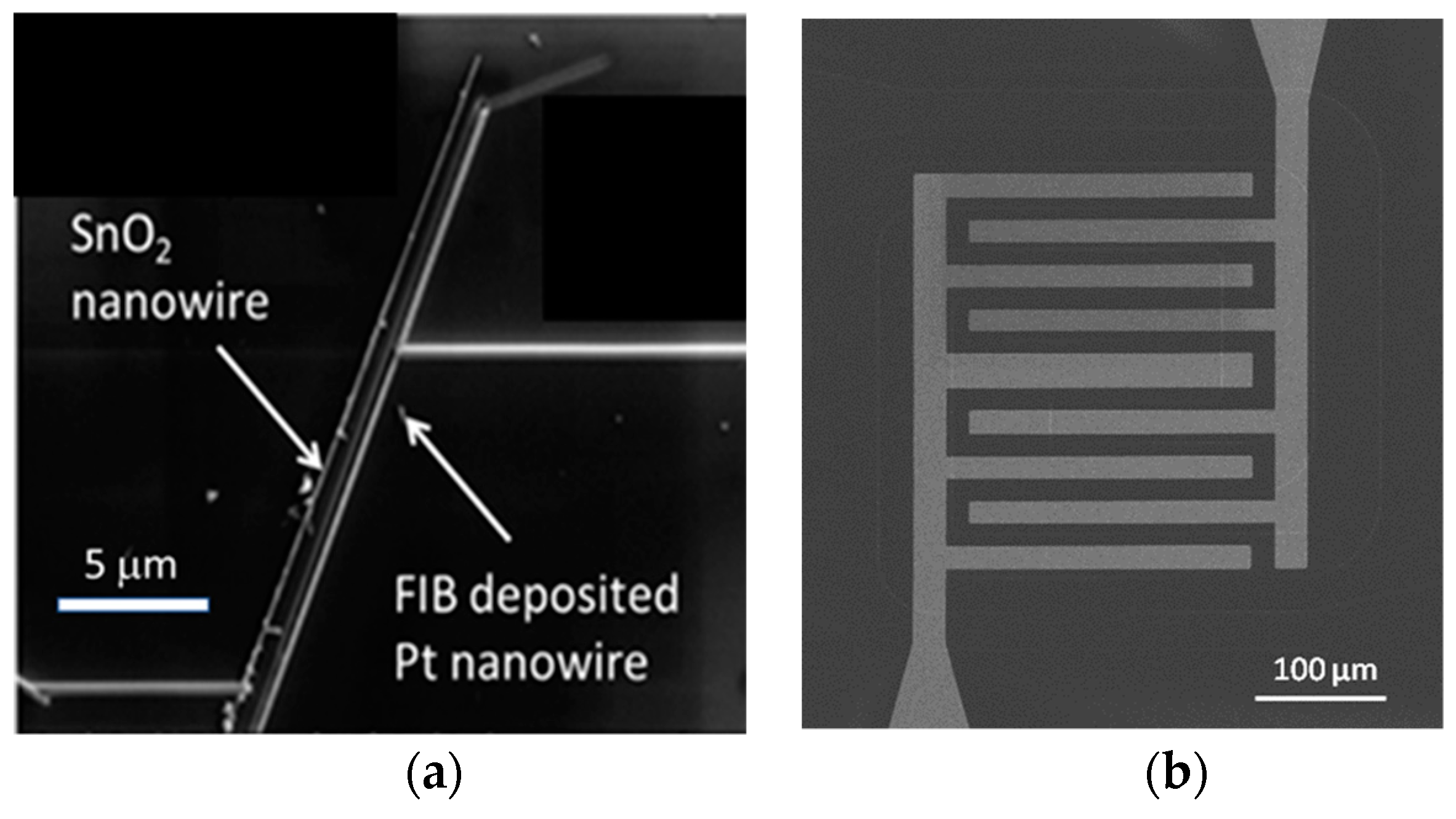
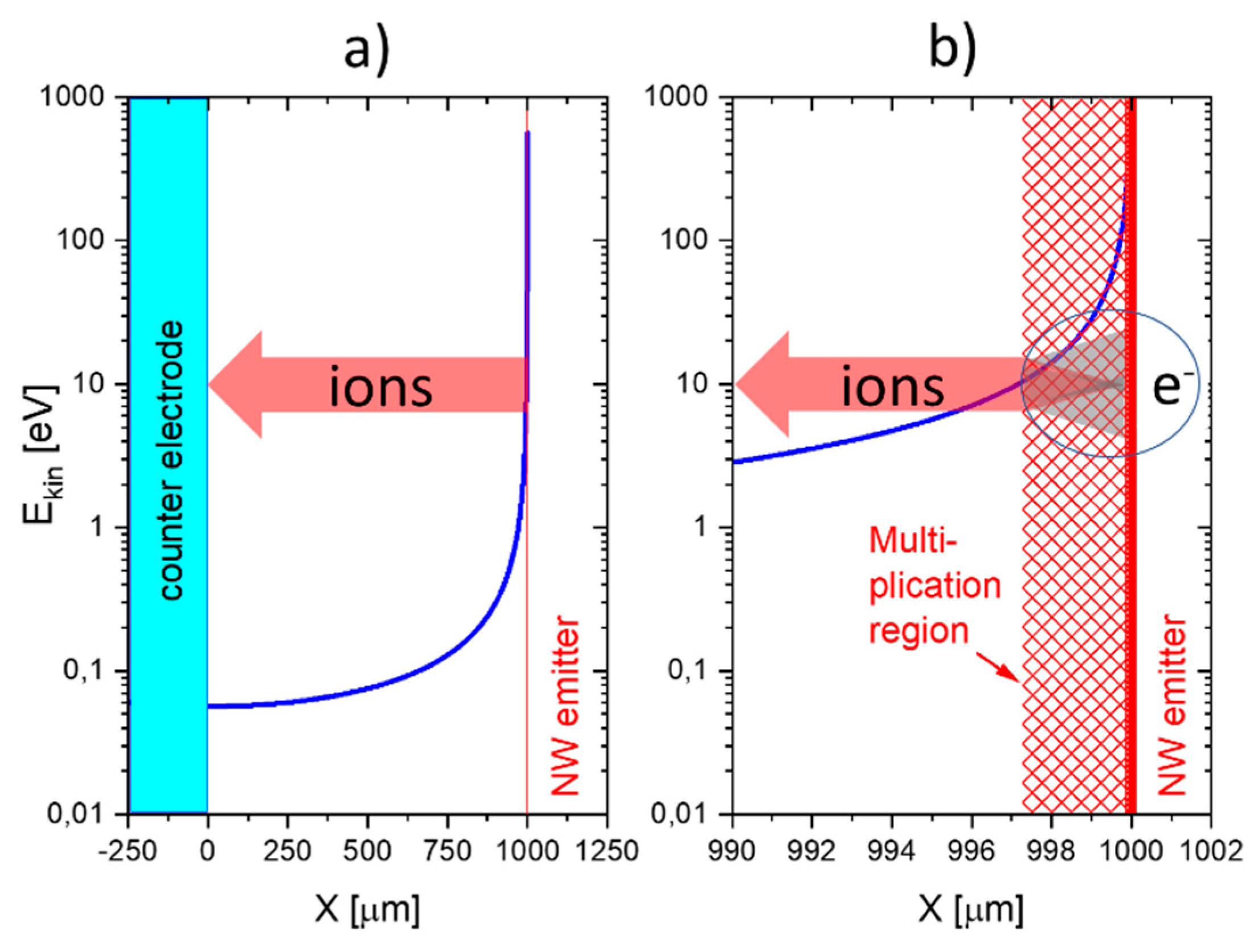
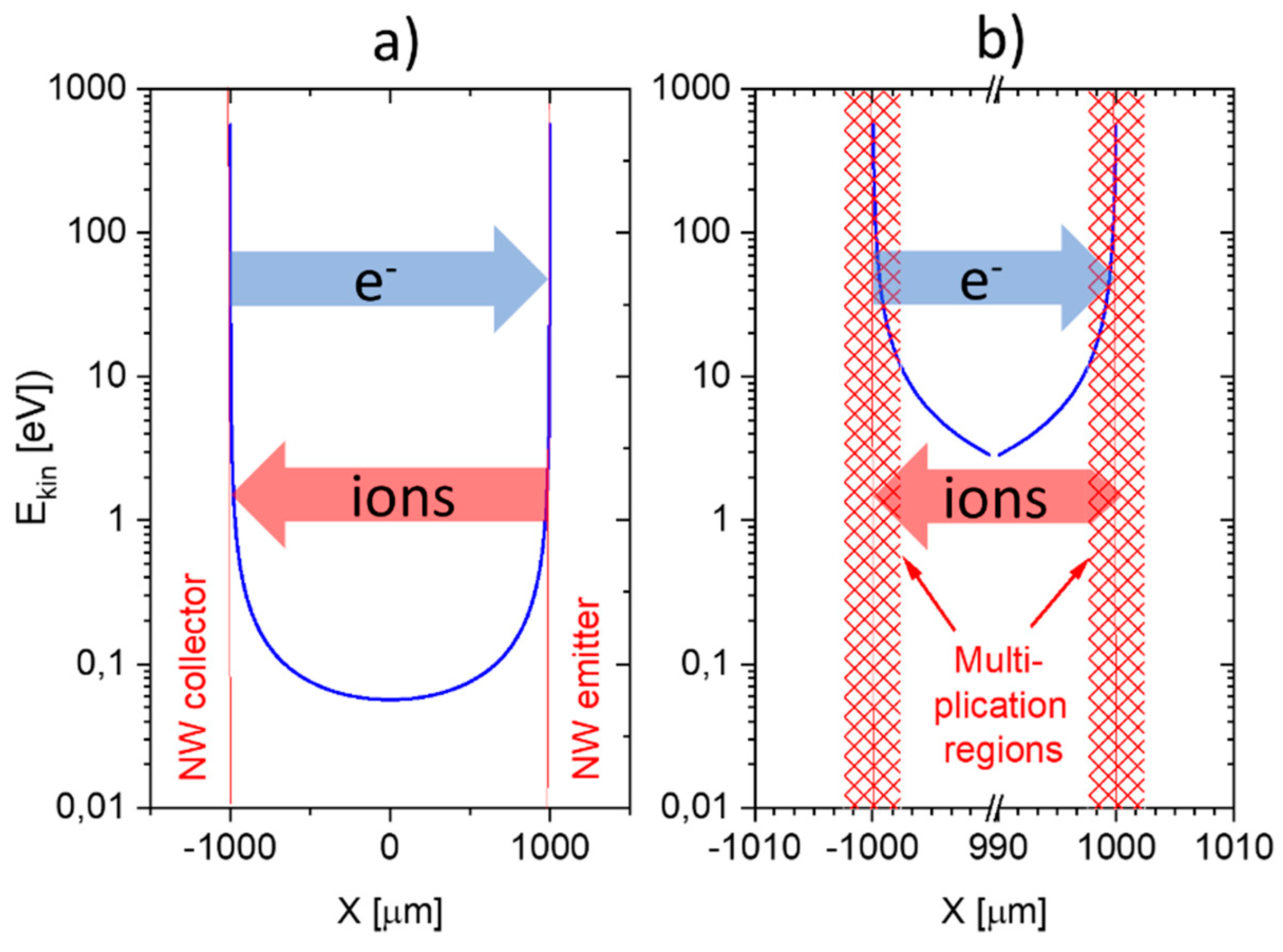
This situation changes completely when emitter electrodes with radii of curvature in the order of several tens of nanometers are employed. As described above, such emitters can be realized either by employing single nanowires [
11,
13] or by using the sharp, photo-lithographically defined edges of Pt interdigital capacitors as line emitters [
12]. In case these line emitters are read out with the help of flat-plate collector electrodes as before, orders of magnitude larger ion current densities can be obtained at significantly lower emission temperatures (
). As evidenced in
Figure 11, an obvious downside of the higher ionization current densities and the lower emission temperatures is a significantly lower amine selectivity of the SI response.
A further lowering of the emitter temperatures can be attained by shaping both emitter and collector electrodes into the forms of line emitters and line-collectors, respectively [
11,
12,
13]. In these kinds of devices, both emitter and collector electrodes sit on a common insulating substrate while being heated to a common emitter and collector temperature,
T. The data in
Figure 11 show that with such pairs of line-emitting and line-collecting devices, emission temperatures as low as 200 °C can be attained; however, at the expense of an almost complete loss of amine selectivity.
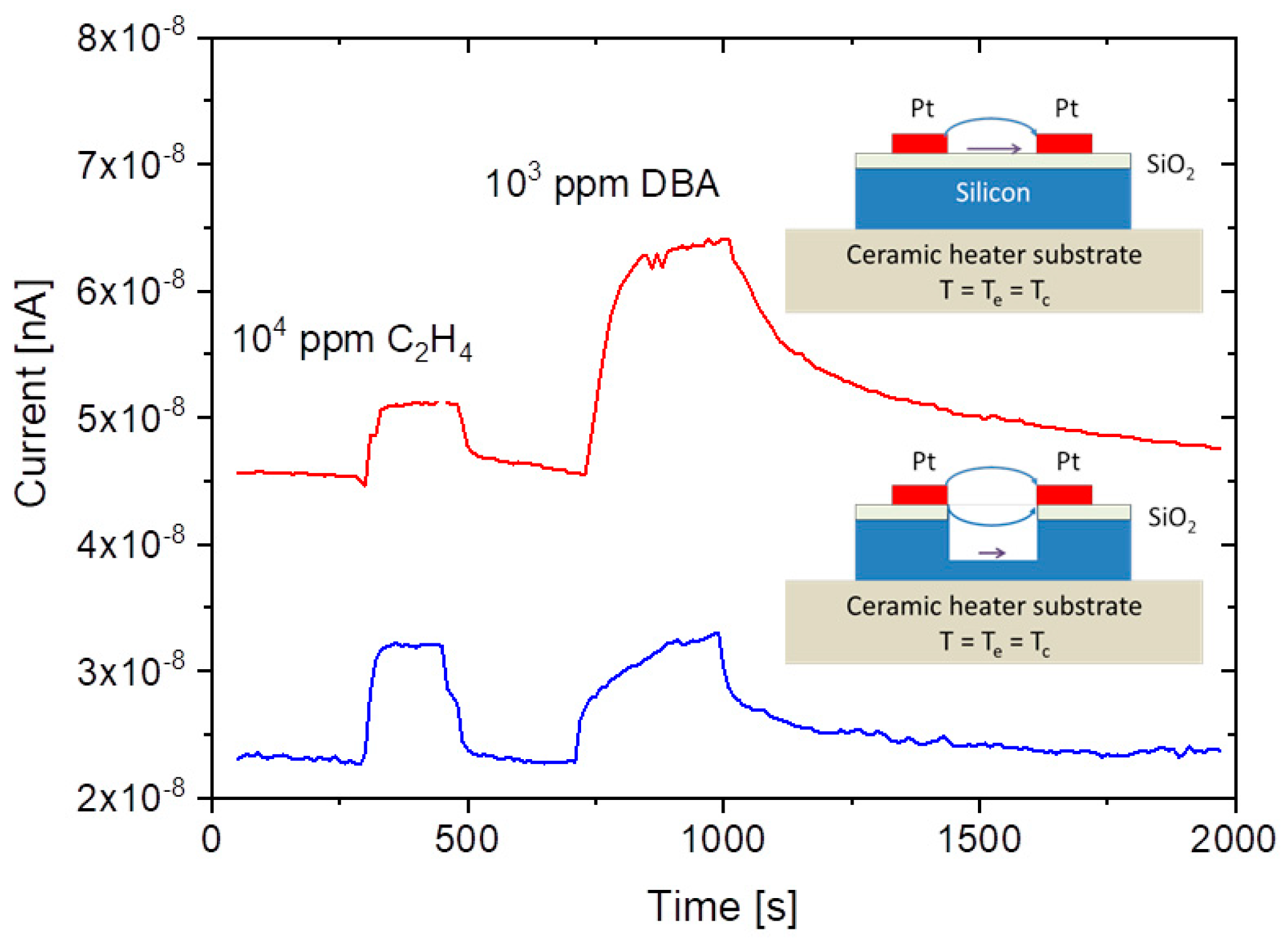
A common problem of all presently investigated E-E readout structures is that these have been realized on common heater substrates. Consequently, these devices contain heated collector electrodes, which make them somewhat difficult to compare to the FP-FP and E-FP devices discussed above. A first problem is that heated collector electrodes, in principle, could emit negative ions, which would raise the positive ion currents emerging from the hot, positive emitter electrodes. A second problem is that the presently available E-E devices do not contain a fully developed air gap in between the emitter and collector electrode. Parasitic surface currents creeping across the insulator gap between emitter and collector electrodes could therefore superimpose on the ion currents flowing through the air gap in the upper half-sphere of the device. Attempting to assess the magnitude of such parasitic current flows,
Figure 12 shows data obtained on E-E devices both with and without a deep saw-cut trench in between emitter and collector electrodes to suppress any parasitic surface currents. This comparison shows that a slightly better DBA/ethene selectivity is obtained when a deep trench cut had been formed. This enhanced selectivity, however, is orders of magnitude smaller than in the other device configurations considered above. In view of this situation, it is unlikely that the selectivity characteristics of E-E devices are seriously limited by parasitic surface currents. Rather, the different electric field profiles across the readout gap seem to play a dominating role.
5. Electrostatic Potential and Field Distributions
As the huge enhancements of the ionization efficiency in nanowire or edge-emitting SI devices are undoubtedly related to the enhanced electrical field strengths that exist at the surfaces of nanowire or MEMS edge emitters, estimates for the acting field strengths are required. Detailed potential and electrical field distributions can be obtained from finite-element modelling (FEM). As, in this paper, we are more concerned with the gross features of the different electrode configurations, we prefer here to use analytical approximations for the acting field strengths. The advantage of analytical solutions is that these provide a much clearer view on those geometrical and electrical parameters that determine the actual device behavior.
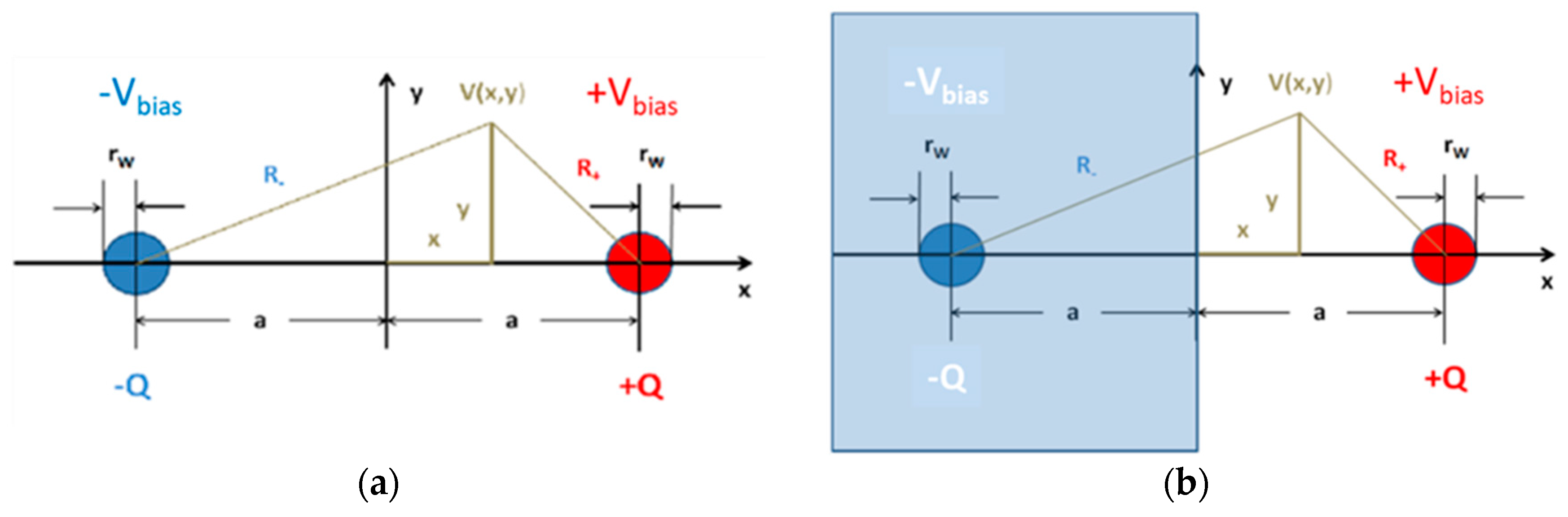
The idealized device configurations, which we should like to analyze in more detail, are shown in
Figure 13.
Figure 13a presents a cross section through a parallel-nanowire arrangement with both nanowires being separated by a distance,
. The radius of curvature of both nanowires is assumed to be
.
Figure 13b shows this same double-wire arrangement. This time, however, the negatively charged nanowire represents the image charge that is induced, when a positively charged nanowire is placed opposite to an electrically conducting medium filling the half-space,
. The induced image charge yields the same solution for the potential,
, as before, however, with the validity of
being limited to the half-space,
. In this latter case, the potential,
, yields the potential distribution on an electrically conducting flat counter electrode placed in the plane,
. In summary, the two idealized double-line arrangements can provide analytical approximations to the electrical potential and field distributions both in a parallel nanowire arrangement [
11] and in a nanowire/flat-plate arrangement [
13]. Both arrangements also provide reasonable estimates for the acting field strengths when the nanowire emitters and collectors are replaced by pairs of interdigitated electrodes with sharp, photo-lithographically defined edges [
12].
The potential distribution,
, generated by both nanowires at the position,
, can be obtained by superimposing the textbook solutions for the potential of a single electrically charged infinite wire [
38,
39], yielding:
While vanishes in the symmetry plane, i.e., , attains the values, and , at the nanowire positions.
Considering the symmetries of the parallel and vertical nanowire arrangements, it is sufficient to concentrate on the line of sight between the nanowire emitter and the real or virtual collector wires:
and:
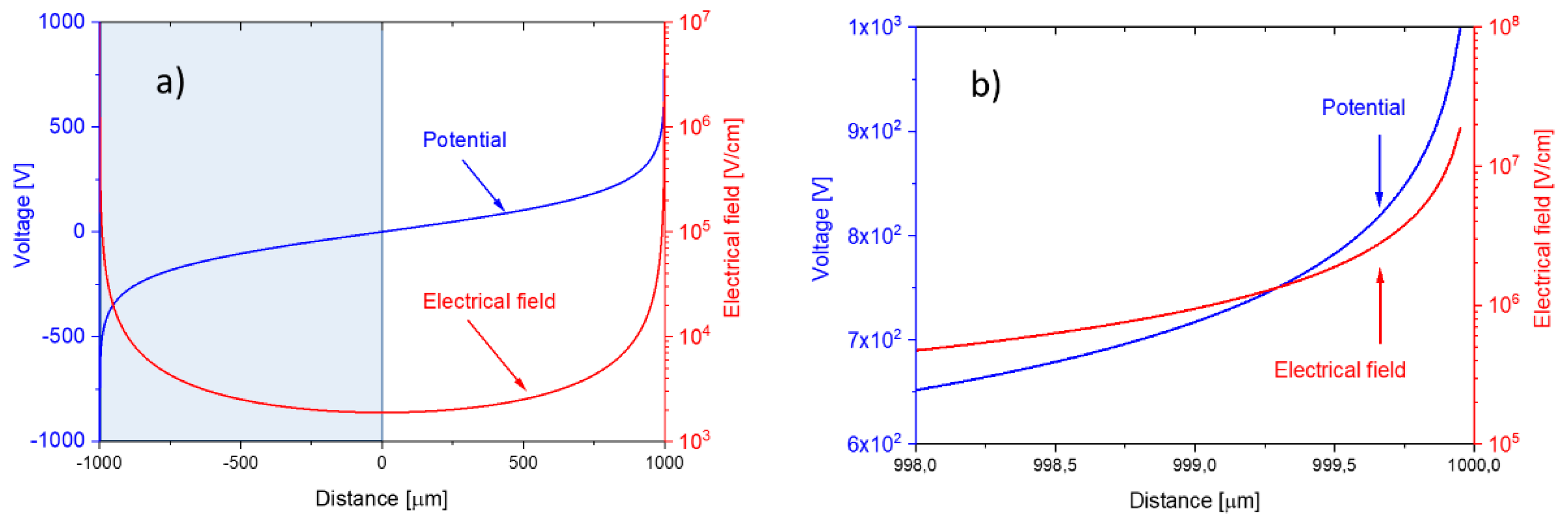
Both distributions are drawn up in
Figure 14, assuming an external bias voltage of
and a nanowire-to-nanowire distance of
. The nanowire diameter,
, in this case was assumed to be
. Concentrating on the right-hand side of both distributions, i.e., at the half-space,
, the truncated distributions also represent the case of a positively charged nanowire facing a flat-plate collector electrode, placed at a distance of
.
The data in
Figure 14a,b show that field strengths in the order of
are generated directly at the nanowire surface. Fields higher than 10
5 V/cm reach out to distances of about
from the nanowire surface. In the middle, in between both nanowires, the field strength drops below the average parallel flat-plate capacitor value of
. Evaluation of Equation (7) at the positions
(Equation (9a)) and at the nanowire surface itself (Equation (9b)) shows that the electrical field at a flat-plate collector electrode is sensitive to the nanowire-collector distance,
, whereas the field at the nanowire surface itself is largely determined by the nanowire radius of curvature,
:
7. Concluding Remarks
The efficiency with which adsorbed analyte gas species can become ionized at hot emitter surfaces depends both on adsorbate as well as on adsorbent parameters. Key adsorbate parameters are the free-space ionization energy, , and the proton affinity, . These two quantities are constants of nature, which make the surface ionization of amines and aromatic compounds much more probable than the ionization of most other analytes. Key adsorbent characteristics are the electron work function of the emitter materials, , as well as the binding energies of the adsorbates, , on the respective surfaces. These latter two parameters can clearly be influenced by the sensor developer. The considerations in this paper have shown that the magnitudes of SI current densities as well as the temperature ranges, in which these can be observed, depend to a large extent on a proper choice of the emitter material. In comparison to MOX materials, which are widely employed in resistively read out gas sensors, much less is known about those processes that control the efficiency of surface ionization processes. On the present kinds of emitter materials, the SI detection of aromatic compounds appears to be severely hampered by large adsorbate binding energies, . In case such energies could be reduced by appropriate material design, sensitive and selective SI detectors for an additional interesting class of analytes would become possible.
Perhaps the most striking result of our investigations was that, besides the materials’ properties of the emitters themselves, the geometry of the ion emitting and ion collecting electrodes can have a huge impact on the sensitivity-selectivity characteristics of SI gas sensors. A key result in this direction was that the intrinsic amine-selectivity of SI emitters only shines up when both the ion emitting and ion collecting electrodes take the form of 2d-geometries with lateral extensions much larger than the nanomorphology of the emitter materials and the lengths of the mean-free paths in the adjacent air ambient (FP-FP devices). By shaping one and the same kind of material into the form of 1d-line emitters or line collectors, huge gains in sensitivity can be attained at the expense of worsening selectivity (E-FP and E-E devices). Our considerations suggest that such massive sensitivity-selectivity trade-offs occur due to the interference of unselective physical ionization processes with the initiating and chemically selective surface ionization processes. As such processes have received little attention in the field of gas sensors yet, significant progress appears to be possible in this area as well.
Our final remark concerns micro-miniaturized SI gas sensors, capable of working at low heating-power-consumption levels and at voltages compatible with battery-operated equipment. Here, our experiments on MEMS and NEMS versions of SI gas sensors have revealed that those micro-miniaturization technologies, which have already proven to be successful for arriving at resistive MOX gas sensors with very low heating power consumption, also appear to be applicable to arrive at low-power-consumption SI gas sensors. To fully arrive at the final goal of low-voltage operation, additional developments in the field of multi-wafer micro-assembly techniques are necessary. With the help of such assembly technologies it appears possible to produce small and rugged devices with air gaps in the range of micrometers, which will reduce the extraction voltages of vertically readout SI devices into the range of normal battery voltages.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}