
Human Serum Albumin Nanoparticles for Use in Cancer Drug Delivery: Process Optimization and In Vitro Characterization

 ,
, 
Abstract
:1. Introduction
2. Results
2.1. Nanoparticles Optimization and Preparation
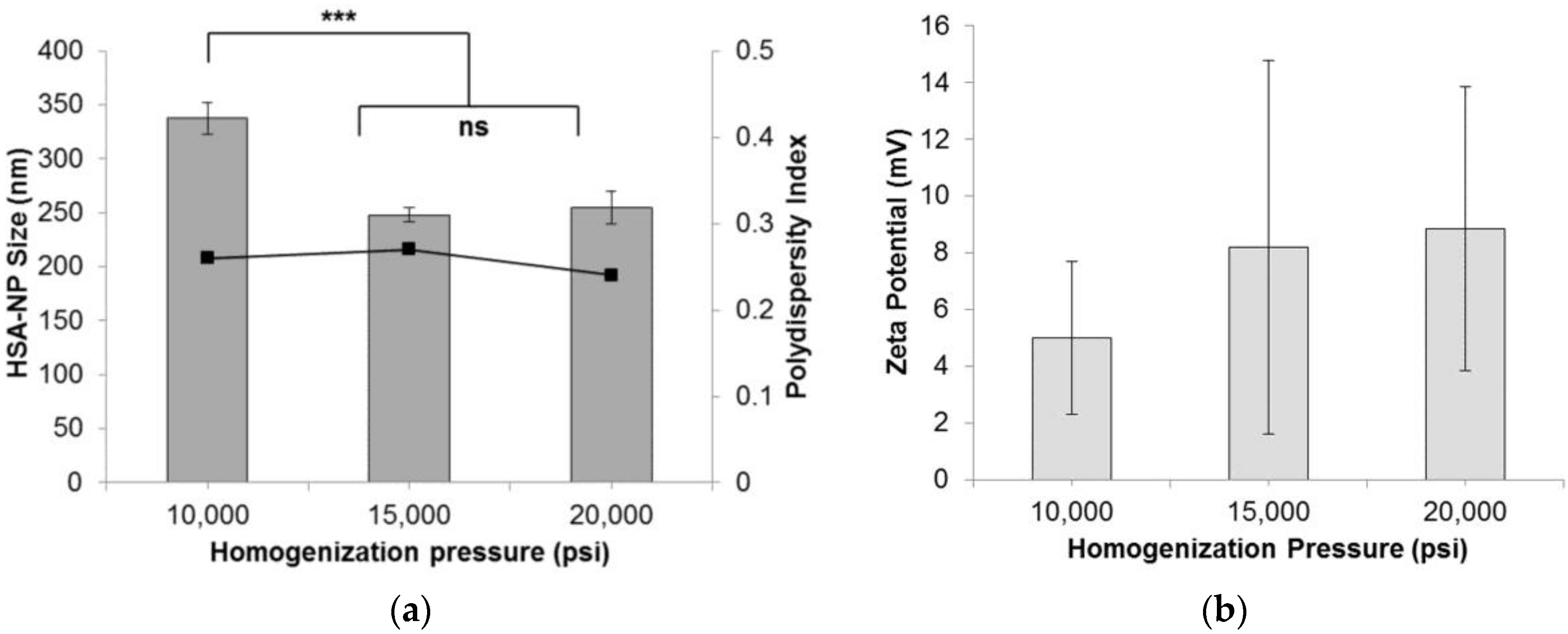
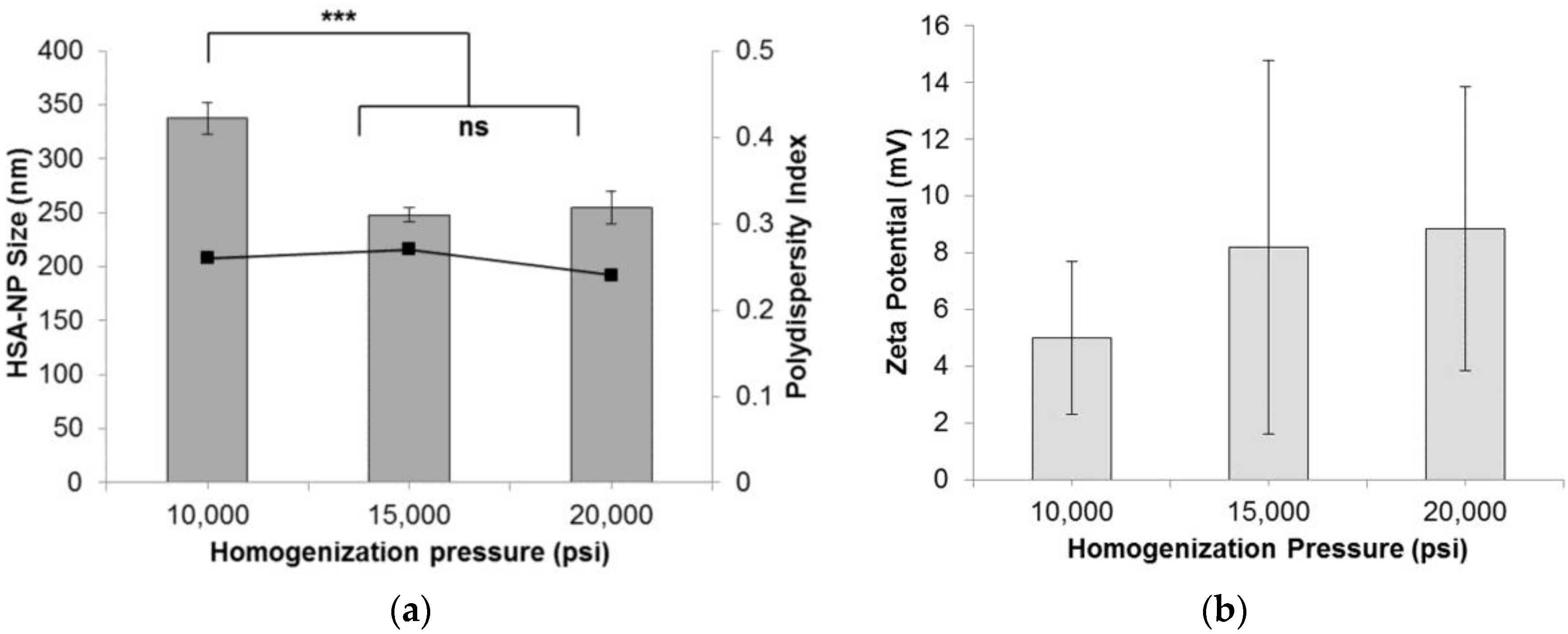
2.1.1. Effect of Homogenization Pressure on Nanoparticles
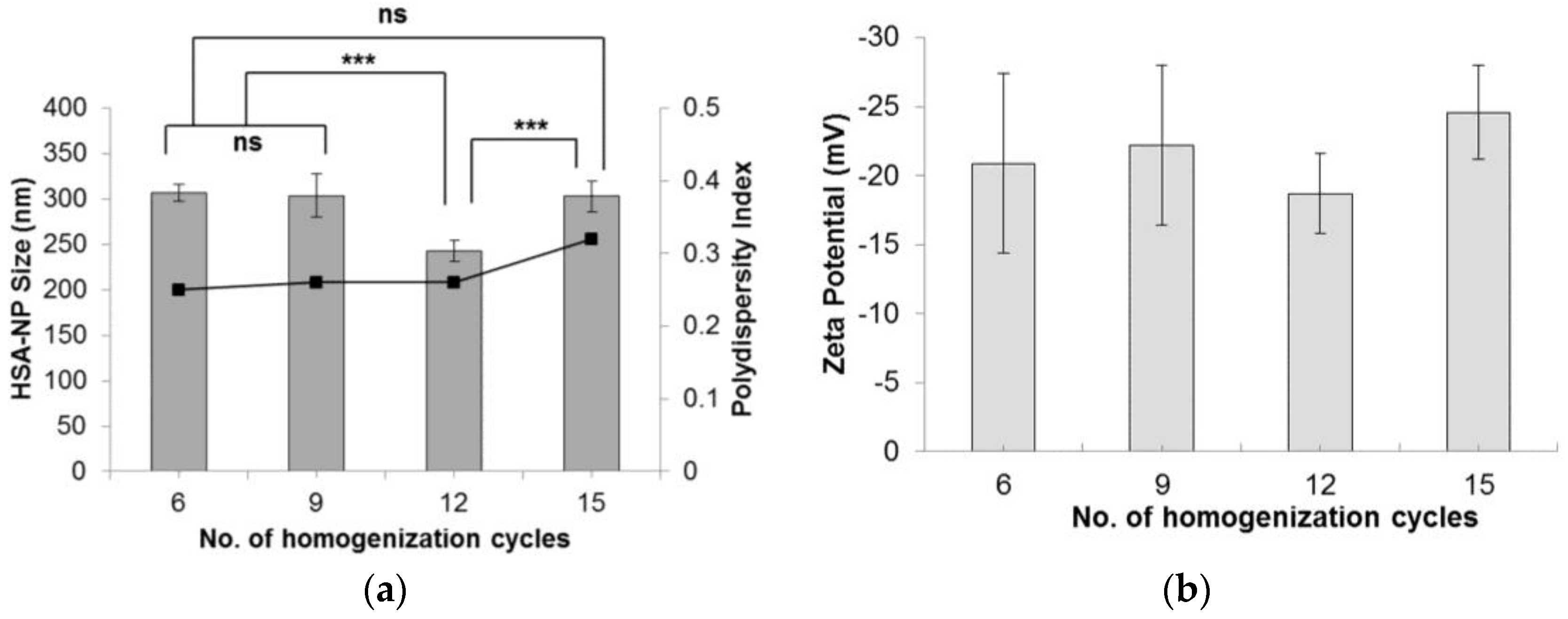
2.1.2. Effect of Homogenization Cycles on Nanoparticles
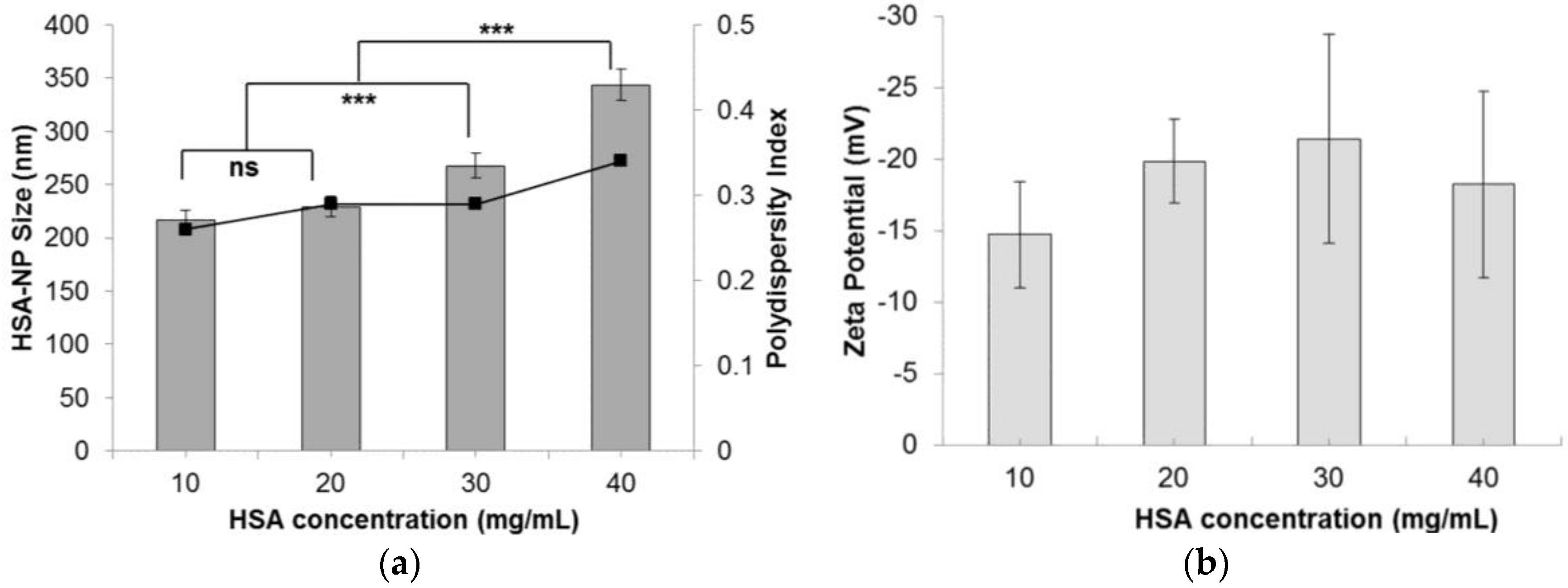
2.1.3. Effect of HSA Concentration on Nanoparticle Size
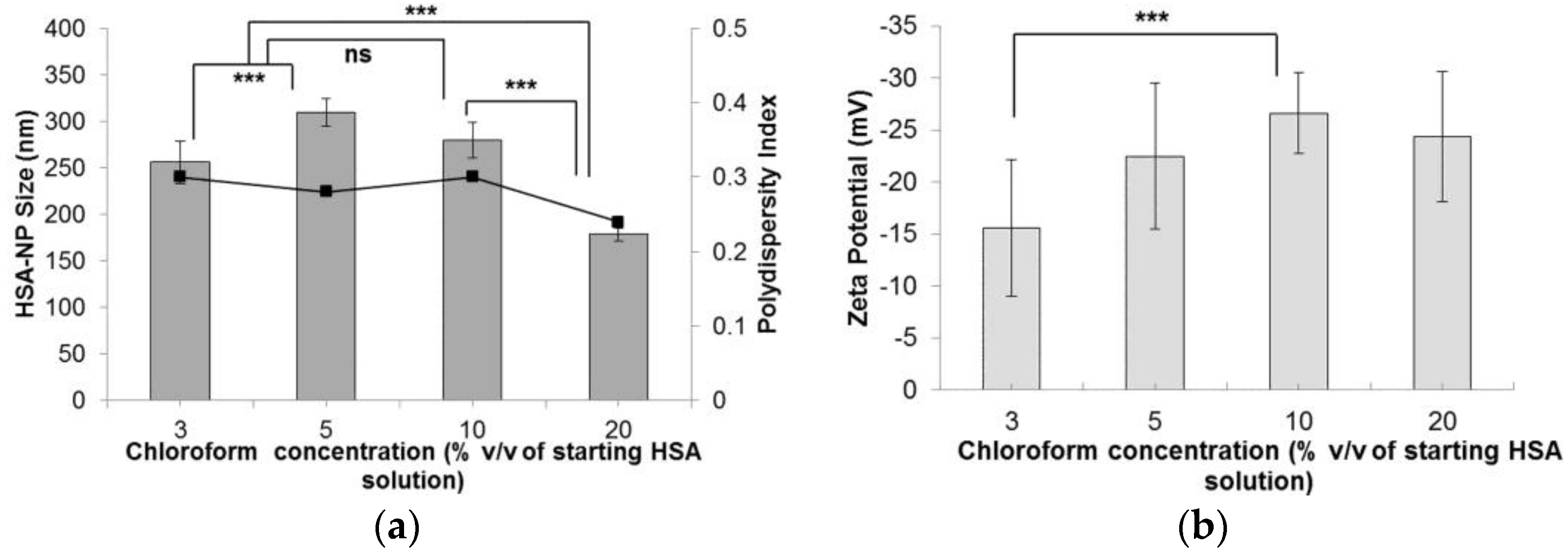
2.1.4. Effect of Chloroform on Nanoparticles
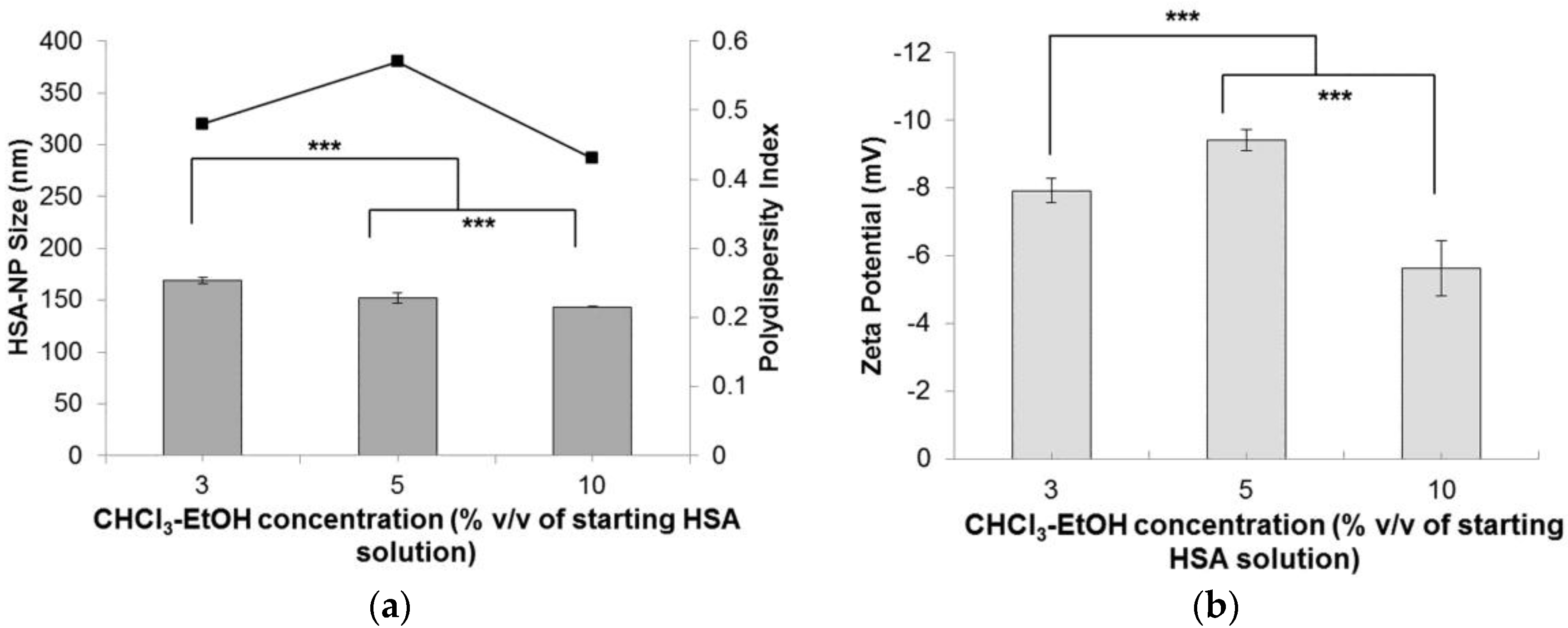
2.1.5. Effect of Chloroform-Ethanol Concentration on Nanoparticles
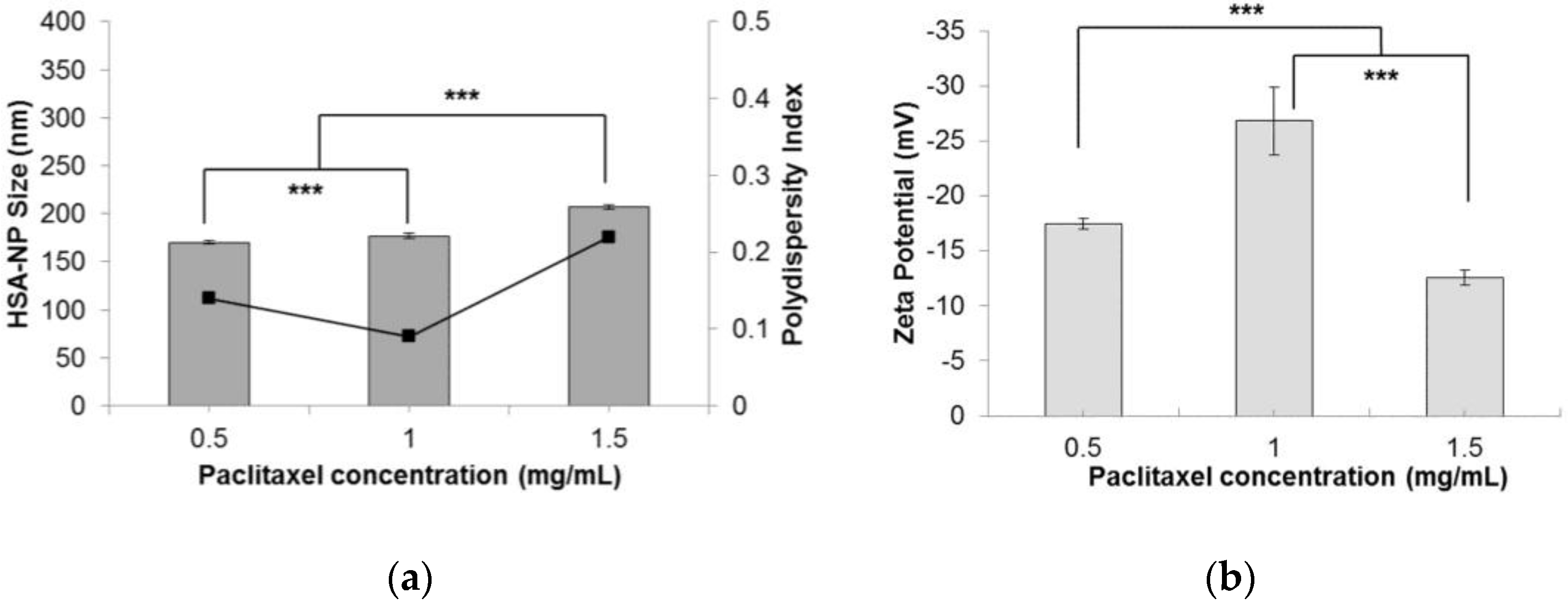
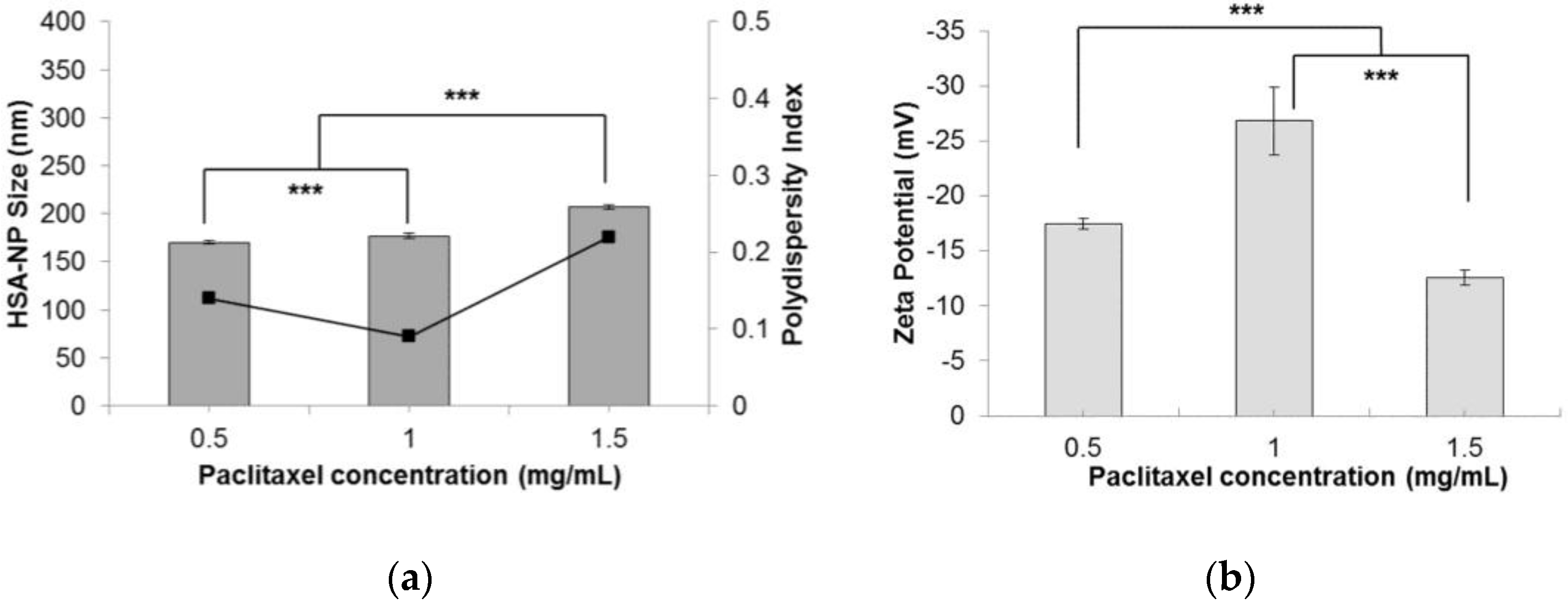
2.1.6. Effect of Paclitaxel Addition in Nanoparticles
2.2. Size, Stability, Morphology, Yield and Encapsulation Efficiency of HSA-NPs
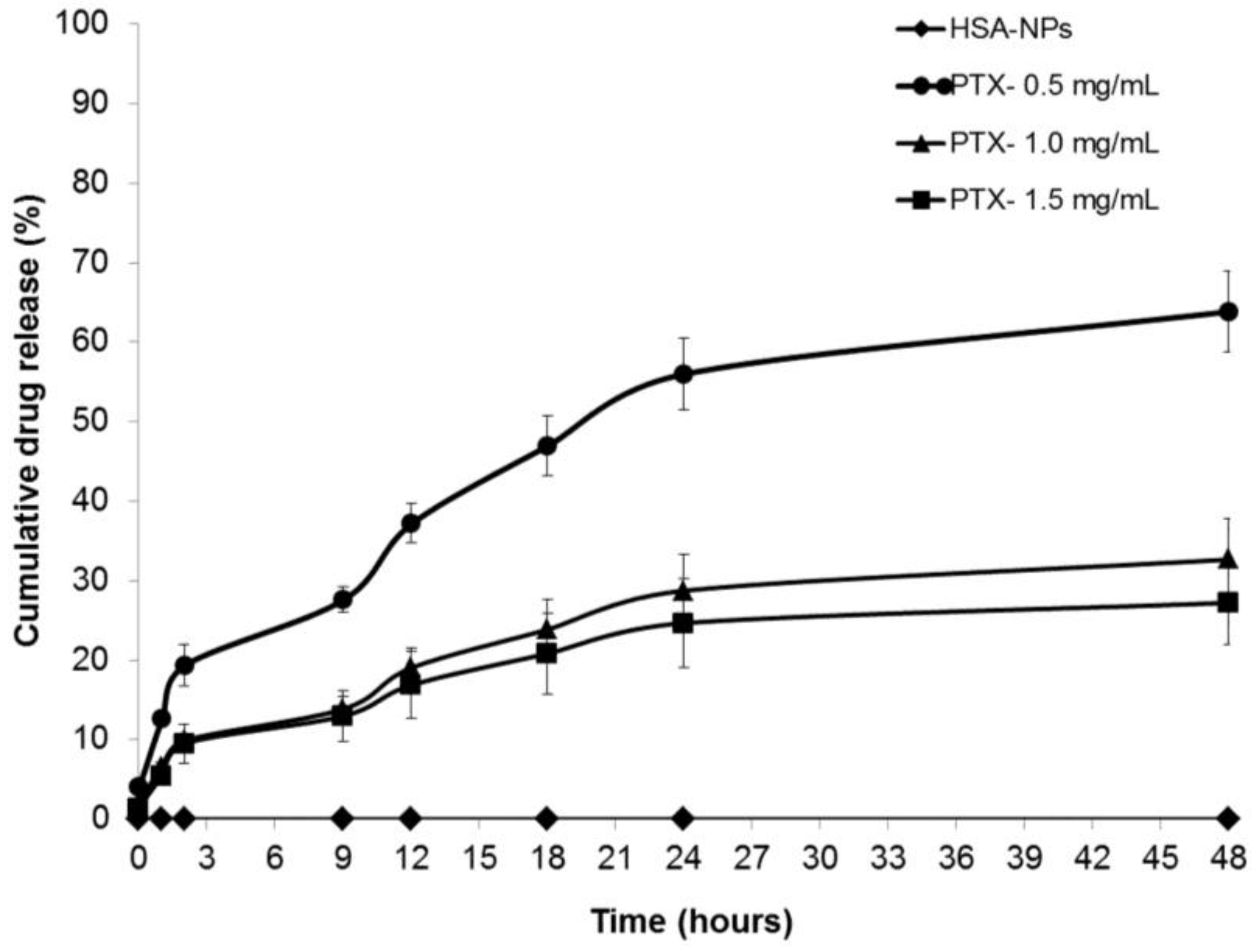
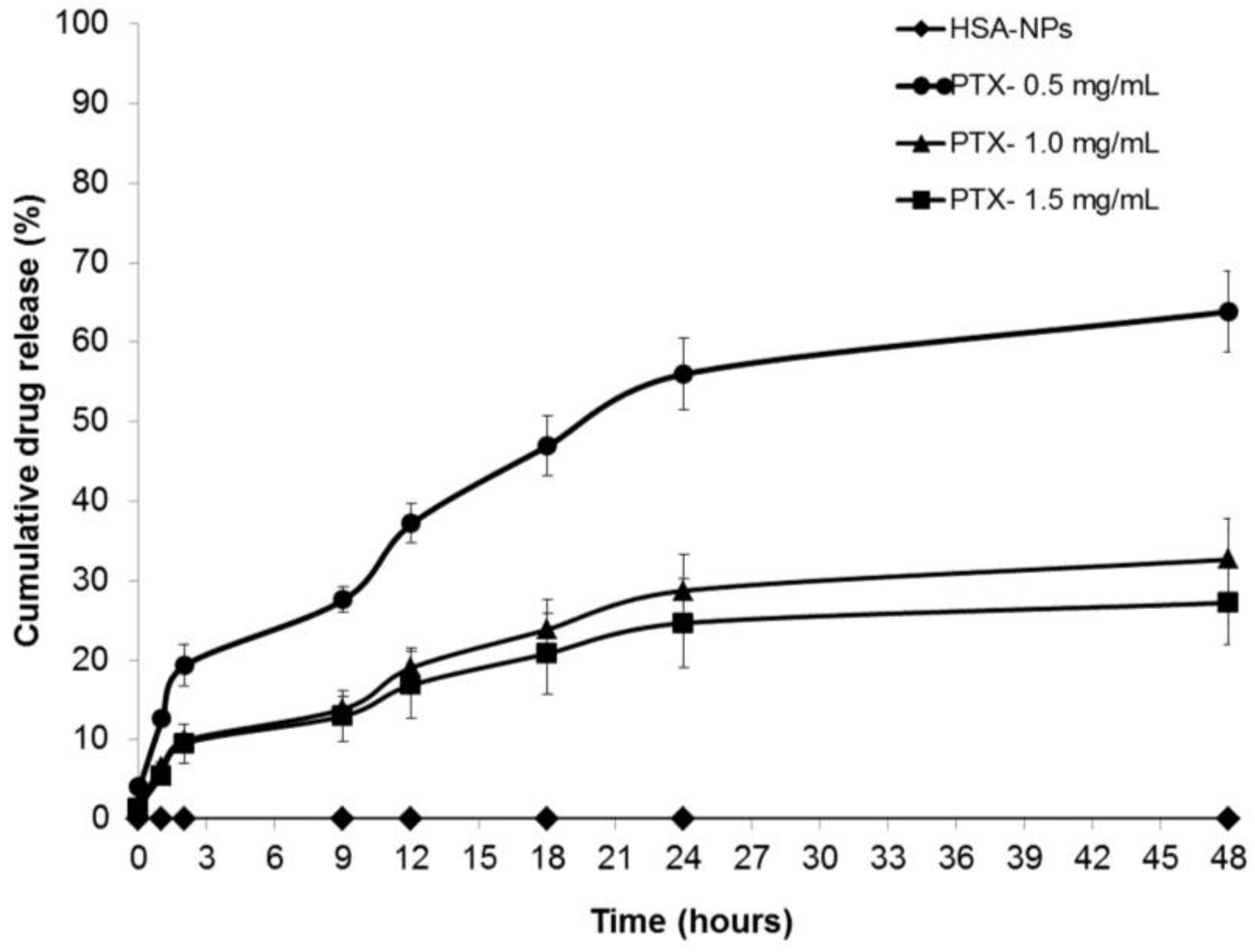
2.3. In Vitro Drug Release Study
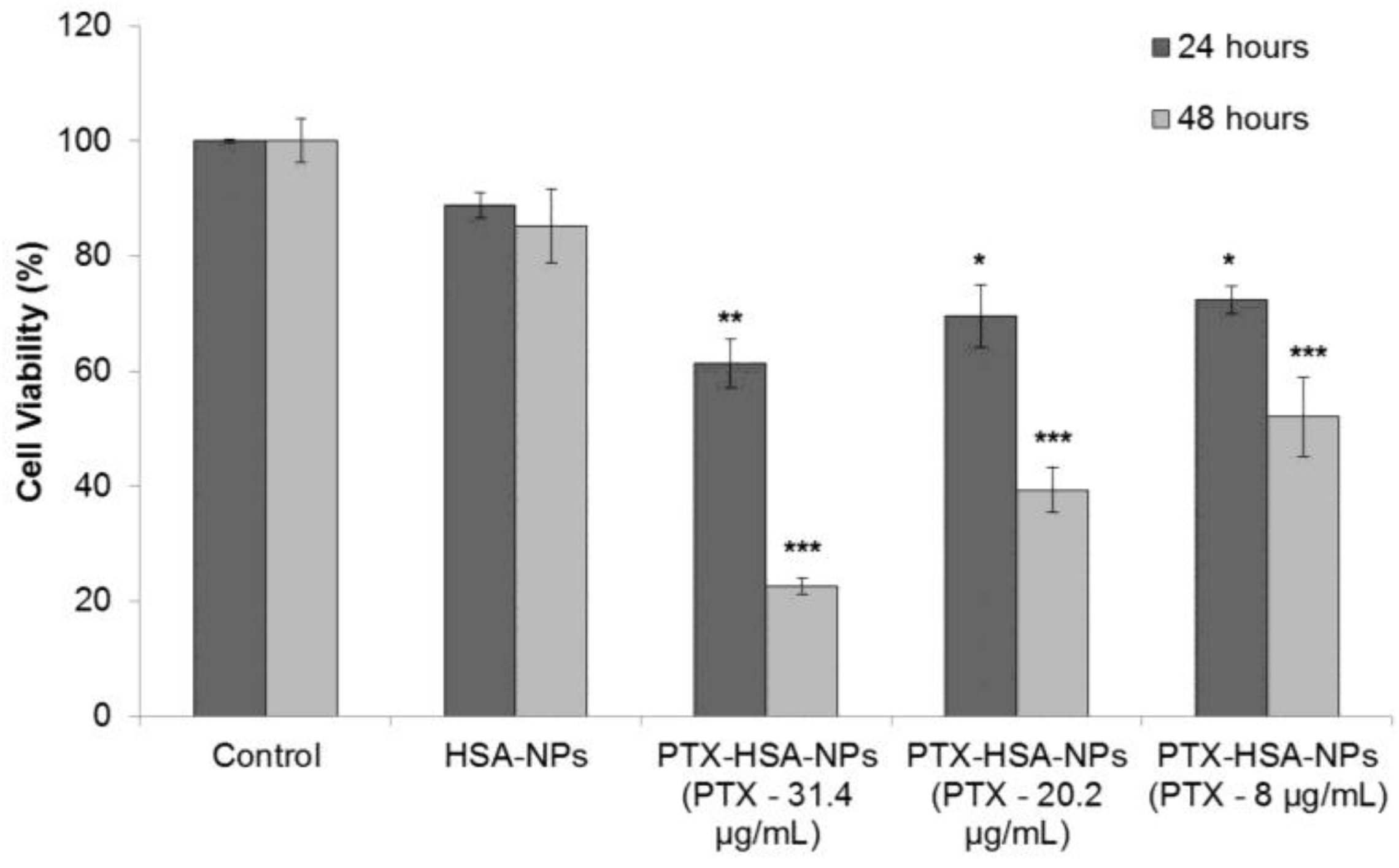
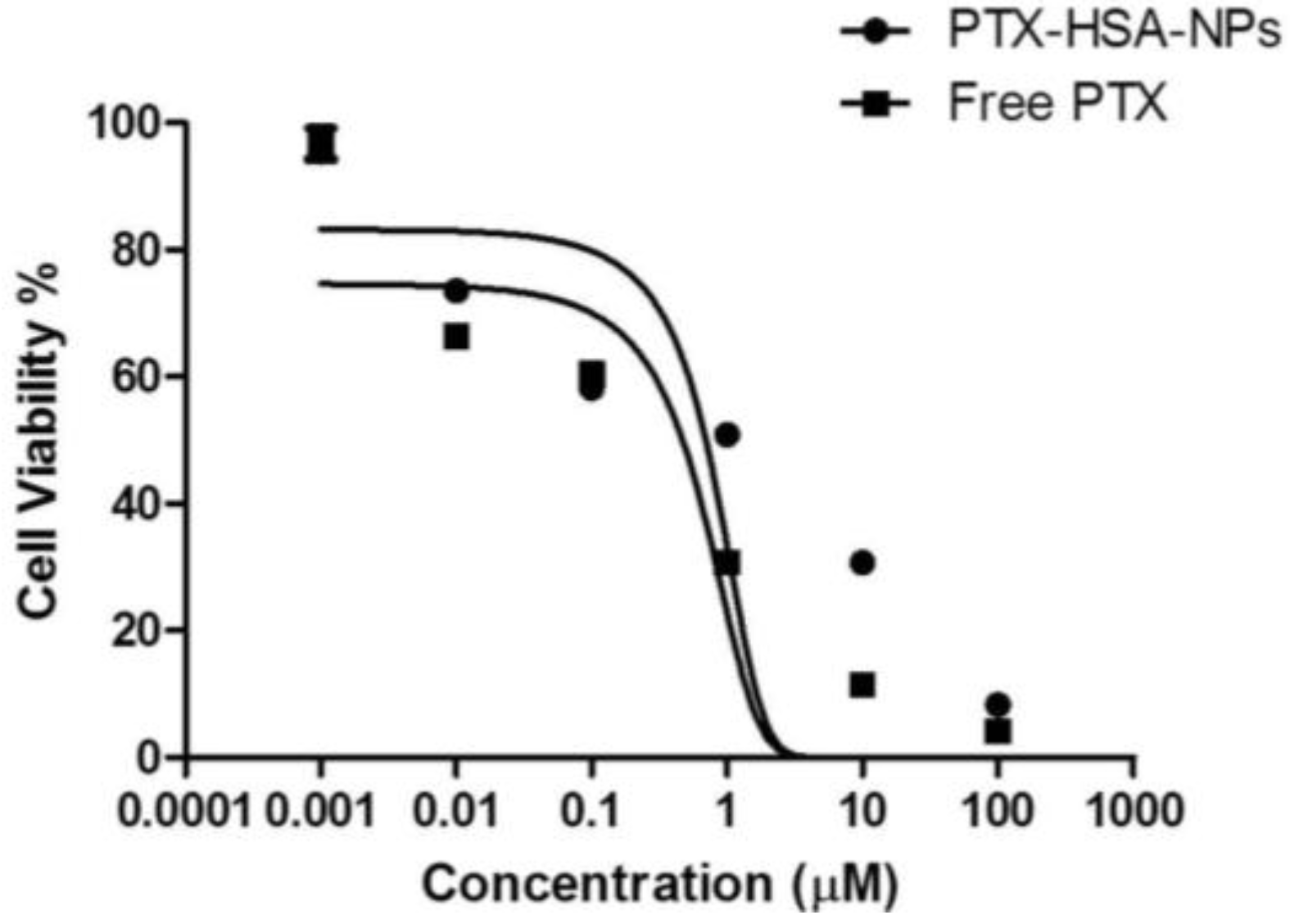
2.4. Cell Culture and In Vitro Cell Viability
3. Discussion
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. High Pressure Homogenizer
4.3. Process Optimization and Preparation of PTX-HSA-NPs
4.4. Nanoparticles Size Measurement, Zeta Potential Analysis and Surface Morphology
4.5. Yield and Encapsulation Efficiency of HSA-NPs
4.6. Measuring In Vitro Paclitaxel Drug Release
4.7. Cell Viability Due to PTX-HSA-NPs
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Urruticoechea, A.; Alemany, R.; Balart, J.; Villanueva, A.; Viñals, F.; Capellá, G. Recent advances in cancer therapy: An overview. Curr. Pharm. Des. 2010, 16, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Lordick, F.; Hacker, U. Chemotherapy and Targeted Therapy, in Imaging of Complications and Toxicity following Tumor Therapy; Kauczor, H.-U., Bäuerle, T., Eds.; Springer International Publishing: Cham (ZG, Switzerland), 2015; pp. 3–15. [Google Scholar]
- Jeong, K.; Kang, C.S.; Kim, Y.; Lee, Y.-D.; Kwon, I.C.; Kim, S. Development of highly efficient nanocarrier-mediated delivery approaches for cancer therapy. Cancer Lett. 2016, 374, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Sebak, S.; Mirzaei, M.; Malhotra, M.; Kulamarva, A.; Prakash, S. Human serum albumin nanoparticles as an efficient noscapine drug delivery system for potential use in breast cancer: Preparation and in vitro analysis. Int. J. Nanomed. 2010, 5, 525–532. [Google Scholar]
- Abbasi, S.; Paul, A.; Shao, W.; Prakash, S. Cationic albumin nanoparticles for enhanced drug delivery to treat breast cancer: Preparation and in vitro assessment. J. Drug Deliv. 2011, 2012. [Google Scholar] [CrossRef]
- Kratz, F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J. Control. Release 2008, 132, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Dockal, M.; Carter, D.C.; Rüker, F. The three recombinant domains of human serum albumin structural characterization and ligand binding properties. J. Biol. Chem. 1999, 274, 29303–29310. [Google Scholar] [CrossRef] [PubMed]
- Elzoghby, A.O.; Samy, W.M.; Elgindy, N.A. Albumin-based nanoparticles as potential controlled release drug delivery systems. J. Control. Release 2012, 157, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Haley, B.; Frenkel, E. Nanoparticles for drug delivery in cancer treatment. Urol. Oncol. Semin. Orig. Investig. 2008, 26, 57–64. [Google Scholar]
- Nateghian, N.; Goodarzi, N.; Amini, M.; Atyabi, F.; Khorramizadeh, M.R.; Dinarvand, R. Biotin/Folate-decorated Human Serum Albumin Nanoparticles of Docetaxel: Comparison of Chemically Conjugated Nanostructures and Physically Loaded Nanoparticles for Targeting of Breast Cancer. Chem. Biol. Drug Des. 2016, 87, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wang, W.; Huang, Y.; Fu, Y.; Cheng, Y. Paclitaxel loaded human serum albumin nanoparticles stabilized with intermolecular disulfide bonds. MedChemComm 2014, 5, 1658–1663. [Google Scholar] [CrossRef]
- Dranitsaris, G.; Yu, B.; Wang, L.; Sun, W.; Zhou, Y.; King, J.; Kaura, S.; Zhang, A.; Yuan, P. Abraxane® versus Taxol® for patients with advanced breast cancer: A prospective time and motion analysis from a Chinese health care perspective. J. Oncol. Pharm. Pract. 2014, 22, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, S.; Arghya, P.; Afshan, K.; Prakash, S. siRNA Delivery Using Biodegradable Nanoparticles for Breast Cancer Therapy. Nanotechnology 2016, 13, 74–76. [Google Scholar]
- Desai, N.P.; Tao, C.; Yang, A.; Louie, L.; Zheng, T.; Yao, Z.; Soon-Shiong, P.; Magdassi, S. Protein Stabilized Pharmacologically Active Agents, Methods for the Preparation Thereof and Methods for the use Thereof. U.S. Patent 6749868 B1, 15 January 2004. [Google Scholar]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Langer, K.; Baltasar, S.; Voqel, V.; Dinauer, N.; von Briesen, H.; Schubert, D. Optimization of the preparation process for human serum albumin (HSA) nanoparticles. Int. J. Pharm. 2003, 257, 169–180. [Google Scholar] [CrossRef]
- Longmire, M.; Choyke, P.L.; Kobayashi, H. Clearance properties of nano-sized particles and molecules as imaging agents: Considerations and caveats. Nanomedicine (Lond.) 2008, 3, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Harashima, H.; Sakata, K.; Funato, K.; Kiwada, H. Enhanced hepatic uptake of liposomes through complement activation depending on the size of liposomes. Pharm. Res. 1994, 11, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.E.; Peppas, N.A. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, A.P.; Mukerjee, A.; Helson, L.; Vishwanatha, J.K. Scale up, optimization and stability analysis of Curcumin C3 complex-loaded nanoparticles for cancer therapy. J. Nanobiotechnol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Mumper, R.J. Paclitaxel nano-delivery systems: A comprehensive review. J. Nanomed. Nanotechnol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Jiang, H.H.; Youn, Y.S.; Park, C.W.; Tak, K.K.; Lee, S.; Kim, H.; Jon, S.; Chen, X.; Lee, K.C. Preparation and characterization of water-soluble albumin-bound curcumin nanoparticles with improved antitumor activity. Int. J. Pharm. 2011, 403, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Floury, J.; Desrumaux, A.; Lardieres, J. Effect of high-pressure homogenization on droplet size distributions and rheological properties of model oil-in-water emulsions. Innov. Food Sci. Emerg. Technol. 2000, 1, 127–134. [Google Scholar] [CrossRef]
- Schultz, S.; Wagner, G.; Urban, K.; Ulrich, J. High-pressure homogenization as a process for emulsion formation. Chem. Eng. Technol. 2004, 27, 361–368. [Google Scholar] [CrossRef]
- Qian, C.; McClements, D.J. Formation of nanoemulsions stabilized by model food-grade emulsifiers using high-pressure homogenization: Factors affecting particle size. Food Hydrocoll. 2011, 25, 1000–1008. [Google Scholar] [CrossRef]
- Mu, L.; Feng, S. A novel controlled release formulation for the anticancer drug paclitaxel (Taxol®): PLGA nanoparticles containing vitamin E TPGS. J. Control. Release 2003, 86, 33–48. [Google Scholar] [CrossRef]
- Danhier, F.; Lecouturier, N.; Vroman, B.; Jérôme, C.; Marchand-Brynaert, J.; Feron, O.; Préat, V. Paclitaxel-loaded PEGylated PLGA-based nanoparticles: In vitro and in vivo evaluation. J Control. Release 2009, 133, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S.; Malhotra, M.; Shao, W.; Tomaro-Duchesneau, C.; Abbasi, S. Polymeric nanohybrids and functionalized carbon nanotubes as drug delivery carriers for cancer therapy. Adv. Drug Deliv. Rev. 2011, 63, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.-B.; Fu, D.-H.; Nie, L.; Zou, P.; Suo, J.-P. A detailed view of PLGA-mPEG microsphere formation by double emulsion solvent evaporation method. Chin J. Polym. Sci. 2015, 33, 955–963. [Google Scholar] [CrossRef]
- Yadav, K.; Yadav, D.; Yadav, M.; Kumar, S. Noscapine Loaded PLGA Nanoparticles Prepared Using Oil-in-Water Emulsion Solvent Evaporation Method. J. Nanopharm. Drug Deliv. 2015, 3, 97–105. [Google Scholar] [CrossRef]
- Pooja, D.; Tunki, L.; Kulhari, H.; Reddy, B.B.; Sistla, R. Optimization of solid lipid nanoparticles prepared by a single emulsification-solvent evaporation method. Data Brief. 2016, 6, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Byeon, H.J.; Thao le, Q.; Lee, S.; Min, S.Y.; Lee, E.S.; Shin, B.S.; Choi, H.G.; Youn, Y.S. Doxorubicin-loaded nanoparticles consisted of cationic-and mannose-modified-albumins for dual-targeting in brain tumors. J. Control. Release 2016, 225, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Lian, X.; Dong, J.; Wan, Z.; Xia, C.; Song, X.; Fu, Y.; Gong, T.; Zhang, Z. Co-delivery of Pirarubicin and Paclitaxel by Human Serum Albumin Nanoparticles to Enhance Antitumor Effect and Reduce Systemic Toxicity in Breast Cancers. Mol. Pharm. 2015, 12, 4085–4098. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhang, Y.; Liang, H. Interactive association of drugs binding to human serum albumin. Int. J. Mol. Sci. 2014, 15, 3580–3595. [Google Scholar] [CrossRef] [PubMed]
- Fasano, M.; Curry, S.; Terreno, E.; Galliano, M.; Fanali, G.; Narciso, P.; Notari, S.; Ascenzi, P. The extraordinary ligand binding properties of human serum albumin. IUBMB Life 2005, 57, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Sahni, J.K.; Ali, J.; Baboota, S. Effect of high-pressure homogenization on formulation of TPGS loaded nanoemulsion of rutin–pharmacodynamic and antioxidant studies. Drug Deliv. 2015, 22, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.Y.; Nguyena, H.H.; Paika, S.-Y.-R.; Chunb, H.S.; Kangc, B.-C.; Koa, S. Preparation of size-controlled bovine serum albumin (BSA) nanoparticles by a modified desolvation method. Food Chem. 2011, 127, 1892–1898. [Google Scholar] [CrossRef]
- Song, K.C.; Leea, H.S.; Chounga, Y.; Choa, K.I.; Ahnb, Y.; Choic, E.J. The effect of type of organic phase solvents on the particle size of poly(d,l-lactide-co-glycolide) nanoparticles. Colloids Surf A 2006, 276, 162–167. [Google Scholar] [CrossRef]
- Kubo, M.T.K.; Augusto, P.E.; Cristianini, M. Effect of high pressure homogenization (HPH) on the physical stability of tomato juice. Food Res. Int. 2013, 51, 170–179. [Google Scholar] [CrossRef]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D. Preparation, characterization, and in vitro targeted delivery of folate-decorated paclitaxel-loaded bovine serum albumin nanoparticles. Int. J. Nanomed. 2010, 5, 669–677. [Google Scholar]
- Bober, B.G.; Shah, S.B. Paclitaxel alters sensory nerve biomechanical properties. J. Biomech. 2015, 48, 3559–3567. [Google Scholar] [CrossRef] [PubMed]
- Bernabeu, E.; Helgueraa, G.; Legaspia, M.J.; Gonzalezb, L.; Hochtd, C.; Tairab, C.; Chiappetta, D.A. Paclitaxel-loaded PCL-TPGS nanoparticles: In vitro and in vivo performance compared with Abraxane®. Colloids Surf. B 2014, 113, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, J.; Bellucci, J.J.; Weitzhaandler, I.; McDaniel, J.R.; Spasojevic, I.; Li, X.; Lin, C.-C.; Chi, J.-T.A.; Chilkoti, A. A paclitaxel-loaded recombinant polypeptide nanoparticle outperforms Abraxane in multiple murine cancer models. Nat. commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Lei, C.; Yang, Y.; Bu, X.; Ma, H.; Gong, H.; Liu, J.; Fang, X.; Hu, Z.; Fang, Q. Abraxane, the Nanoparticle Formulation of Paclitaxel Can Induce Drug Resistance by Up-Regulation of P-gp. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Kesarwani, P.; Tekade, R.K.; Jain, N. Spectrophotometric estimation of paclitaxel. Int. J. Adv. Pharm. Sci. 2011, 2. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Homogenization Pressure | |||
| Pressure (psi) | Size (nm) | Zeta Potential (mV) | PDI |
| 10,000 | 337.7 ± 14.8 | 5.0 ± 12.7 | 0.26 |
| 15,000 | 248.2 ± 6.6 | 8.2 ± 16.6 | 0.27 |
| 20,000 | 254.7 ± 15.5 | 8.8 ± 8.0 | 0.24 |
| Number of Homogenization Cycles | |||
| Cycles | Size (nm) | Zeta Potential (mV) | PDI |
| 6 | 307.1 ± 9.2 | −20.8 ± 6.5 | 0.25 |
| 9 | 303.7 ± 24.1 | −22.2 ± 5.8 | 0.26 |
| 12 | 242.8 ± 11.7 | −18.7 ± 2.9 | 0.26 |
| 15 | 302.8 ± 17.3 | −24.5 ± 3.4 | 0.32 |
| Human serum albumin (HSA) Concentration | |||
| HSA (mg/mL) | Size (nm) | Zeta Potential (mV) | PDI |
| 10 | 216.6 ± 9.8 | −14.7 ± 13.7 | 0.26 |
| 20 | 229.3 ± 9.5 | −19.8 ± 2.9 | 0.29 |
| 30 | 267.9 ± 11.7 | −21.4 ± 7.3 | 0.29 |
| 40 | 343.6 ± 14.4 | −18.2 ± 6.5 | 0.34 |
| Chloroform Concentration | |||
| Chloroform (% v/v) | Size (nm) | Zeta Potential (mV) | PDI |
| 3 | 256.1 ± 22.8 | −15.5 ± 6.6 | 0.30 |
| 5 | 309.4 ± 14.5 | −22.4 ± 7.0 | 0.28 |
| 10 | 280.1 ± 19.1 | −26.6 ± 3.9 | 0.30 |
| 20 | 178.4 ± 6.7 | −24.3 ± 6.3 | 0.24 |
| Chloroform-Ethanol Concentration (94:6) | |||
| CHCl3-EtOH (% v/v) | Size (nm) | Zeta Potential (mV) | PDI |
| 3 | 169.1 ± 2.6 | −7.9 ± 0.36 | 0.48 |
| 5 | 152.4 ± 5.1 | −9.4 ± 0.3 | 0.57 |
| 10 | 143.4 ± 0.7 | −5.62 ± 0.8 | 0.43 |
| Paclitaxel Concentration | |||
| PTX (mg/mL) | Size (nm) | Zeta Potential (mV) | PDI |
| 0.5 | 170.2 ± 1.4 | −17.4 ± 0.5 | 0.14 |
| 1 | 177.1 ± 2.5 | −26.8 ± 3.1 | 0.09 |
| 1.5 | 207.5 ± 2.4 | −12.6 ± 0.7 | 0.22 |
| Type of HSA-NPs with PTX | Yield | Encapsulation Efficiency |
|---|---|---|
| PTX-0.5 mg/mL | 93.40% | 81.80% |
| PTX-1.0 mg/mL | 93.70% | 93.94% |
| PTX-1.5 mg/mL | 92.50% | 97.96% |
| PTX-0 mg/mL | 93.90% | - |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lomis, N.; Westfall, S.; Farahdel, L.; Malhotra, M.; Shum-Tim, D.; Prakash, S. Human Serum Albumin Nanoparticles for Use in Cancer Drug Delivery: Process Optimization and In Vitro Characterization. Nanomaterials 2016, 6, 116. https://doi.org/10.3390/nano6060116
Lomis N, Westfall S, Farahdel L, Malhotra M, Shum-Tim D, Prakash S. Human Serum Albumin Nanoparticles for Use in Cancer Drug Delivery: Process Optimization and In Vitro Characterization. Nanomaterials. 2016; 6(6):116. https://doi.org/10.3390/nano6060116
Chicago/Turabian StyleLomis, Nikita, Susan Westfall, Leila Farahdel, Meenakshi Malhotra, Dominique Shum-Tim, and Satya Prakash. 2016. "Human Serum Albumin Nanoparticles for Use in Cancer Drug Delivery: Process Optimization and In Vitro Characterization" Nanomaterials 6, no. 6: 116. https://doi.org/10.3390/nano6060116
APA StyleLomis, N., Westfall, S., Farahdel, L., Malhotra, M., Shum-Tim, D., & Prakash, S. (2016). Human Serum Albumin Nanoparticles for Use in Cancer Drug Delivery: Process Optimization and In Vitro Characterization. Nanomaterials, 6(6), 116. https://doi.org/10.3390/nano6060116