1. Introduction
Dilute magnetic semiconductors (DMSs) have attracted much attention due to their unique potential usage of both charge and spin of freedom of carriers in magneto-optical, magneto-electrical, and magneto-transport devices [
1,
2,
3,
4], Especially, oxide-diluted magnetic semiconductors (such as ZnO) have shown excellent piezoelectric and photoelectric properties, and great potential applications in spintronic devices [
5,
6,
7]. Moreover, theoretical and experimental research has predicted Curie temperatures (
TC) above room temperature, high solubility of magnetic ions, and transparency for visible light in ZnO-based DMSs [
8,
9]. In recent years, great progress has been achieved in one-dimensional (1D) ZnO-based DMSs. Chang
et al. [
10] prepared Zn
1−xMn
xO nanowires (NWs) at 500 °C with a high doped content of 13%. Philipose
et al. [
11] also successfully prepared Zn
1−xMn
xO NWs, and the samples showed stable ferromagnetic (FM) and ultraviolet emission properties. Wang
et al. [
12] prepared Zn
1−xMn
xO NWs and no second phase was observed in the Mn-doped ZnO NWs. Theoreticians have found ZnO to be an excellent candidate host semiconductor for high-
TC ferromagnetism. Dietl
et al. [
2] made theoretical predictions about the higher Curie temperature of Mn-doped ZnO, Sato
et al. [
13] revealed the magnetic properties of
3d TMs-doped ZnO by using the local density approximation (LDA). Marcel
et al. [
14] investigated the ferromagnetic (FM) and antiferromagnetic (AFM) properties of
3d TMs-doped ZnO. Our previous studies have shown that potential FM ground states are more stable in V-doped and Fe-doped ZnO NWs [
15,
16], and have obvious half-metallic properties in Fe-doped ZnO NWs [
16]. In addition, He
et al. [
17] have reported on the electronic and magnetic properties of Mn-doped ZnO nanotubes using density functional theory (DFT) with the generalized gradient approximation (GGA). However, many problems in the research of ZnO-based DMSs still remain both scarce and controversial regarding the influence of impurities on the electronic, optical, and magnetic properties. The magnetic coupling mechanism and origin of the ferromagnetism is still not clear, and experimental observations demonstrate controversial results about optical phenomenon in the optical absorption with regards to Mn-doped ZnO NWs. In order to clarify the effect of Mn doping on the electronic, optical, and magnetic properties of ZnO NW, it is necessary to perform first-principles methods based on DFT.
In this paper, the geometric structures, and electronic and magnetic properties of Mn-doped ZnO NWs were systematically investigated by first-principles methods, based on DFT. The applicable methods to adjust and control oxide DMSs were obtained by analysis of the magnetic coupling mechanism of Mn-doped ZnO NWs. We systematically study mechanisms of FM and AFM coupling on the electronic, optical, and magnetic properties. The main results provide theoretical guidance for preparing ZnO-based DMS materials of high quality and high-TC.
2. Theoretical Models and Calculated Methods
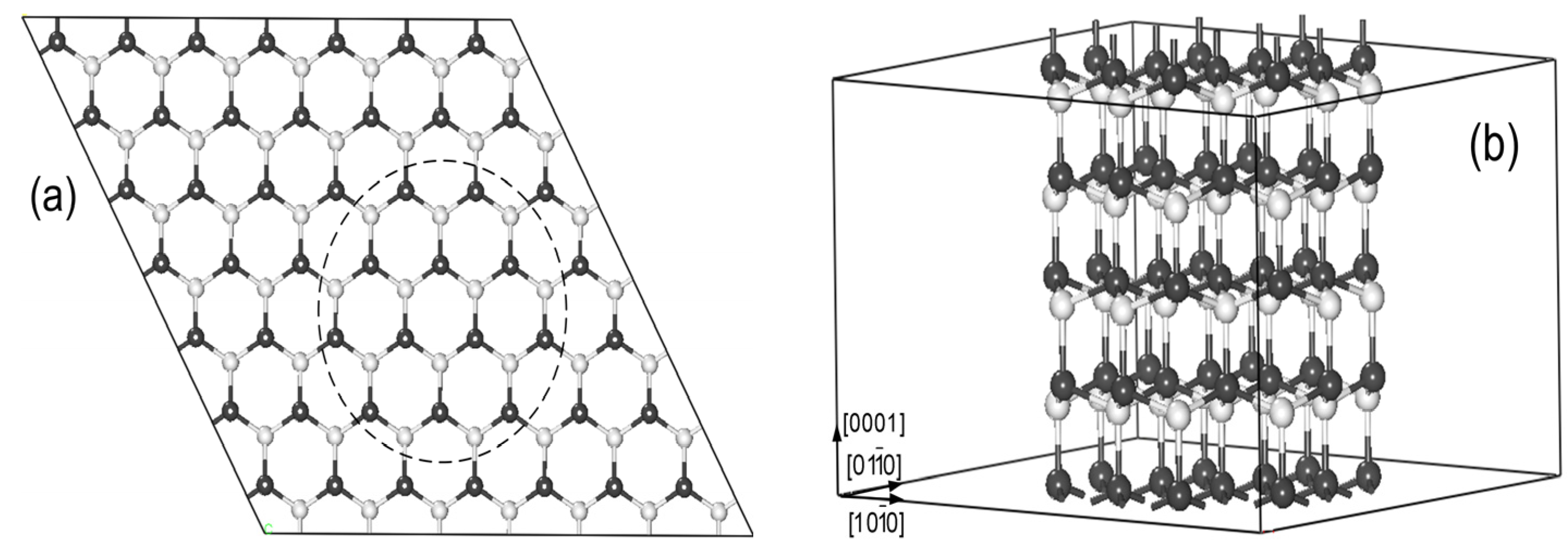
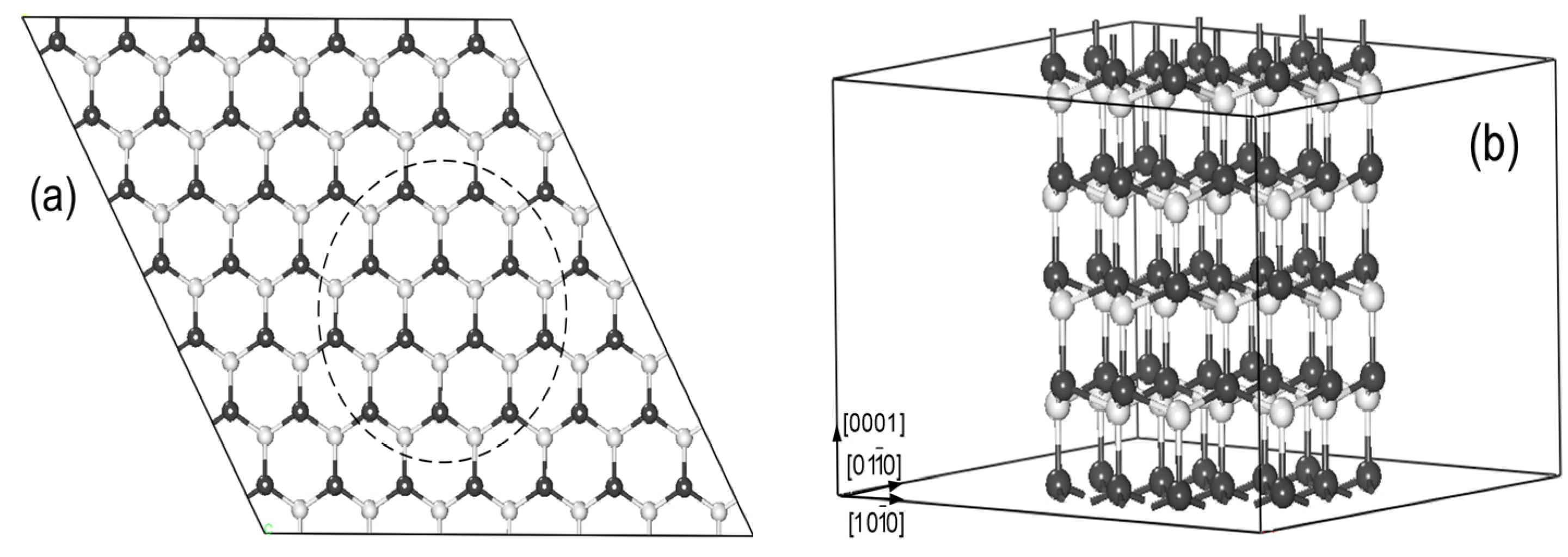
The models for Mn-doped ZnO NWs are generated from a wurtzite ZnO (7 × 7 × 2) supercell structure along the [0001] direction (see
Figure 1a). All atoms within the dashed line remained to build up the ZnO NW structure in
Figure 1b. Our NW model contains 96 atoms (Zn46Mn2O48). The vacuum region of ZnO NWs along [
] and [
] is 15 Å to avoid the effects of the interaction between Mn-doped ZnO NWs on the results, and we select the periodical structure along [0001] direction, as shown in
Figure 1b. To investigate the magnetic coupling mechanism between the two Mn atoms, we replace two of the Zn atoms with Mn in ZnO NWs. The six possible magnetic coupling models are as shown in
Figure 2, and Mn doping concentration is at 4.2%.
The calculations were carried out by using the first principles methods based on DFT, additionally, the Vienna
Ab initio Simulation Package (VASP) was applied in our work [
18]. The detailed parameters were set as follows, the exchange-correlation function was calculated based on GGA in the form of the Perdew-Burke-Ernzher (PBE) [
19], the valence electronic configuration of O, Zn and Mn atoms were 2
s22
p4, 4
s23
d10 and 4
s23
d5, respectively, the cut-off energy of plane wave was set at 420 eV, and the convergence in energy and force was less than 1 × 10
−5 eV and 10
−3 eV/Å, respectively, and the stress on the cell was less than 0.01 GPa. The Brillouin zone was sampled with a mesh of 1 × 1 × 16 k points, generated by the Monkhorst-Pack scheme.
Figure 1.
(a) The top view of the 7 × 7 × 2 ZnO supercell structure; (b) Zn48O48 nanowires (NWs) supercell along the [0001] direction (The white and black spheres represent O and Zn atoms).
Figure 1.
(a) The top view of the 7 × 7 × 2 ZnO supercell structure; (b) Zn48O48 nanowires (NWs) supercell along the [0001] direction (The white and black spheres represent O and Zn atoms).
Figure 2.
The structures of Mn-doped ZnO NWs. (a) Configuration I; (b) Configuration II; (c) Configuration III; (d) Configuration IV; (e) Configuration V; (f) Configuration VI. (The white, gray, and black spheres represent O, Zn, and Mn atoms, respectively).
Figure 2.
The structures of Mn-doped ZnO NWs. (a) Configuration I; (b) Configuration II; (c) Configuration III; (d) Configuration IV; (e) Configuration V; (f) Configuration VI. (The white, gray, and black spheres represent O, Zn, and Mn atoms, respectively).
3. Results and Discussion
To verify the geometric structure, and electronic and magnetic properties of the Mn-doped ZnO NWs, the geometric structures and electronic properties of pure Zn
48O
48 NW were calculated under full geometry relaxation. The total energy of the optimized ZnO NW was found to be 6.371 eV lower than that of the unoptimized one. The optimized Zn–O bond length along the [0001] direction was 1.899 Å on the outermost surface layer, which was 4.67% shorter than that of bulk ZnO. The optimized Zn–O bond lengths were 1.969 Å and 1.978 Å on the inner-layer NWs, and 1.15% and 0.7% shorter than that of bulk ZnO, respectively. The optimized Zn–O bond lengths along the [
] direction were between 1.955 Å and 1.962 Å on the outermost surface layer, their corresponding bond angles of ∠O–Zn–O (108.04°) and ∠Zn–O–Zn (108.04°) changed into ∠O–Zn–O (113.76°) and ∠Zn–O–Zn (105.81°). In addition, it was observed from
Figure 3a that the total density of states (DOS) for the spin-up and spin-down were identical, namely, pure ZnO NW is a nonmagnetic material. The calculated band gap was 1.86 eV, which was significantly larger than that of our calculated bulk ZnO (0.97 eV).
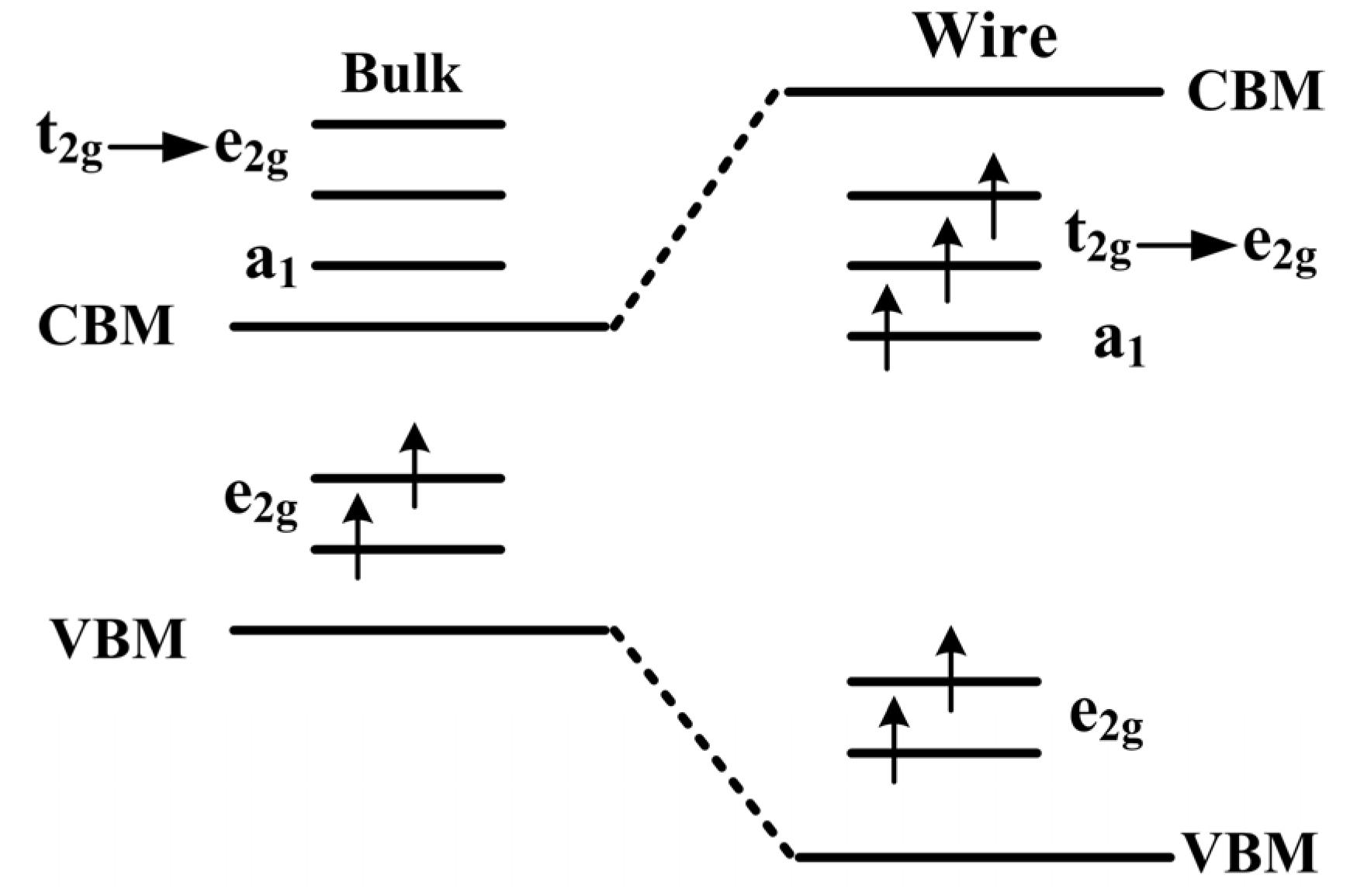
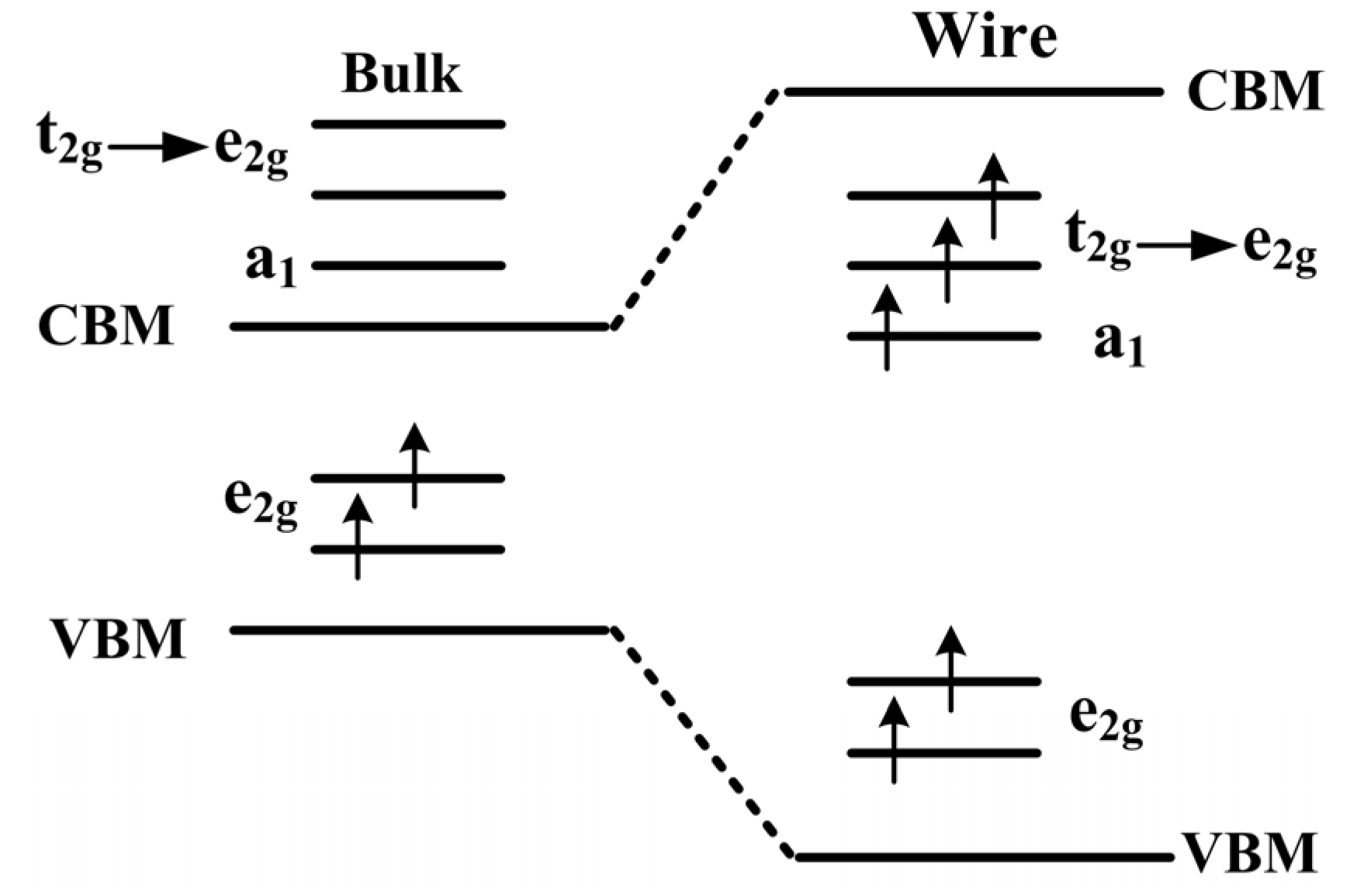
Figure 3.
The schematic plot of 3d level splitting in the ZnO bulk and NW.
Figure 3.
The schematic plot of 3d level splitting in the ZnO bulk and NW.
Table 1 illustrates the magnetic coupling modes, geometric parameters, energy, bond lengths, and magnetic moments of Mn-doped ZnO NW, in which Δ
E (Δ
E = EAFM −
EFM) denotes energy difference after optimized treatments, Δ
E0 (Δ
E = EAFM −
EFM) denotes the unoptimized energy difference, and the relative energy Δε denotes the energy difference corresponding to the ground state configuration. As shown in
Table 1, the Δ
E of Mn-doped ZnO NWs are all negative, which indicates that the AFM states are more stable than FM states. The configuration V corresponds to the ground state of Mn-doped ZnO nanowires, and its energy is lower than FM state energy by 0.229 eV. In this configuration, the two Mn atoms reside in the inner layer sites along the [0001] direction. As for configuration VI, the energy difference Δ
E is only −0.007 eV, which indicates that AFM coupling is slightly more stable than FM coupling. In order to further investigate AFM interactions, we note that the corresponding energies of FM and AFM states are almost same as shown in
Table 1 (
ΔE=-0.007 eV), and the AFM coupling is weak with the increase of Mn–Mn bond length for configuration VI (
dMn–Mn = 5.205 Å). Hence, it is prone to forming a paramagnetic state and spin-glass state. It is indicated that the Mn–O bond length may play a more important role than the Mn–Mn bond length in predicting the magnetic coupling, which suggests that the range of magnetic interactions between the two Mn atoms is rather short and independent of the Mn–Mn distance. While configurations II, III, and IV are in an AFM state for Mn-doped ZnO nanowires along other directions. Our calculated results are consistent with previously reported results [
11,
20,
21,
22]. Moreover, all magnetic moments of Mn atoms are 3.947–4.193 μ
B and mainly originate from
3d electrons of Mn atoms. Oxygen atoms also show AFM spin polarization in nearest-neighbor Mn atoms, which produce a weak magnetic moments of −0.061–−0.089 μ
B. As for the relaxed geometric structure, the calculated results show that the Mn–O and Mn–Mn bond lengths are 1%–3% shorter than that of an unrelaxed one. The outer layer of ZnO NWs shows more intense relaxation than the inner one, therefore presenting high surface effects and quantum size effects.
Table 1.
Magnetic coupling modes, energy, bond lengths, and magnetic moments.
Table 1.
Magnetic coupling modes, energy, bond lengths, and magnetic moments.
| Modes | ΔE (eV) | ΔE0 (eV) | Δε (eV) | Coupling | dMn–O (Å) | dMn–Mn (Å) | Mn1 (μB) | Mn2 (μB) | O (μB) |
|---|
| I | −0.302 | −0.052 | 0.721 | AFM | 1.914 | 3.134 | 4.171 | −4.176 | −0.061 |
| II | −0.219 | −0.053 | 0.208 | AFM | 1.819 | 3.111 | 4.134 | −4.070 | −0.069 |
| I | −0.144 | −0.047 | 0.405 | AFM | 1.916 | 3.060 | 3.947 | −4.113 | −0.074 |
| IV | −0.139 | −0.095 | 0.561 | AFM | 1.816 | 3.264 | 4.021 | −4.104 | −0.071 |
| V | −0.229 | −0.061 | 0.000 | AFM | 1.839 | 2.694 | 4.078 | −4.067 | −0.070 |
| VI | −0.007 | −0.003 | 0.117 | AFM | 1.823 | 5.205 | 4.193 | −4.021 | −0.089 |
To further study the origin of magnetic coupling between two Mn atoms,
Figure 3 displays the schematic of splitting 3
d level occupation in bulk ZnO and NW. For Mn-doped ZnO NW, the positions of Zn
2+ ions are replaced by Mn
2+ ions, and an Mn
2+ (
d5 configuration) ion has an tetrahedral structure in a crystalline field. The 3
d states of an Mn atom split into one upper, triply degenerate
state (
dxy,
dyz,
dxz) and one lower, doubly degenerate
state (
,
). However, the space group of ZnO with a wurtzite structure degrades from a high symmetric
Td to a
C3v group in the NW, the
states are no longer the original triply degenerate, but further divide into one doubly degenerate
state and one singly
a1 state. In particular, O 2
p states also splits into an
state and an
a1 state, due to the high symmetry of
T2 in O 2
p orbital. Magnetic coupling between O 2
p and Mn 3
d states cause localization characteristics of the
states owing to the same symmetry.
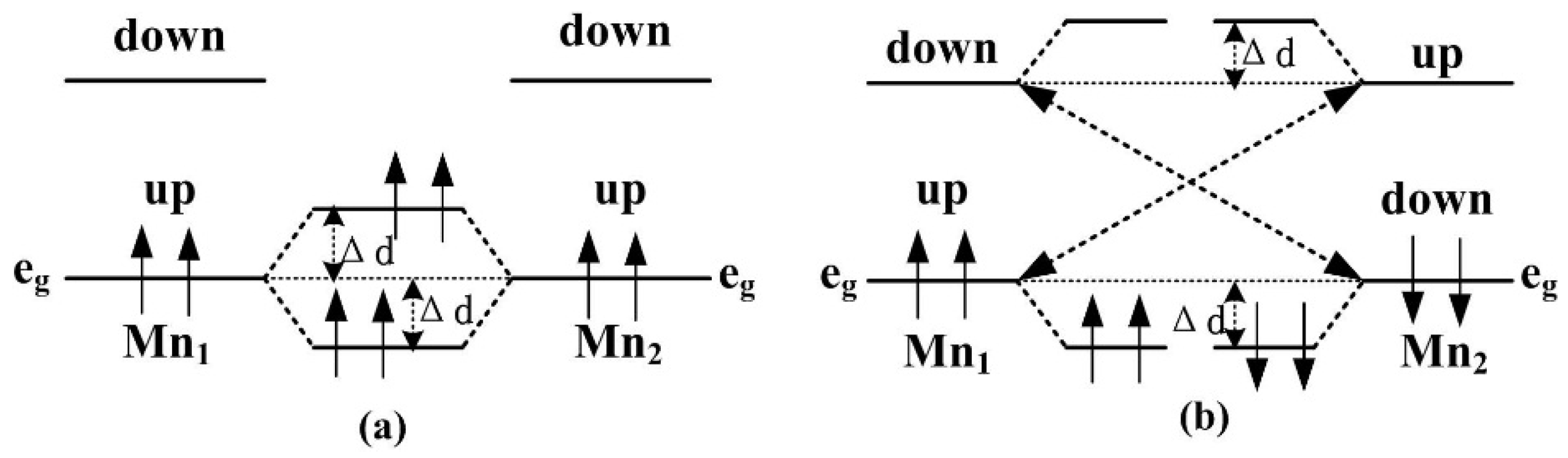
Figure 4 shows the FM and AFM mechanisms of the Mn-doped ZnO NW. According to the Hund rules and the Pauli exclusion principle, the two lower
levels located in the gap are fully occupied in the upper-spin bands, and pushed upward and downward by the same Δ
d to form an FM coupling when there is no energy input, as shown in
Figure 3 and
Figure 4a. For the AFM coupling in
Figure 4a, the upper-spin
level of Mn ion couples with that of another Mn ion with the same level, the occupied
levels are pushed up by Δ
d energy, and the unoccupied ones are pushed down by Δ
d energy due to the half-occupation of the
levels, as shown in
Figure 4b, thus, the super-exchange interaction is smaller than the double exchange interaction. By increasing distance between the two Mn
2+ ions, the AFM coupling of d-d state becomes weaker, and the doped system can form paramagnetic or spin-glass states, which is consistent with our analysis about geometric structure and stability in previous section. The Mn-doped ZnO nanowires show an AFM behavior, and are prone to form a zigzag chain of Mn–O–Mn atoms along the [0001] direction, which indicate that the doped systems have a good effect on the [0001] preferred orientation to prepare ZnO-based dilute magnetic semiconductors.
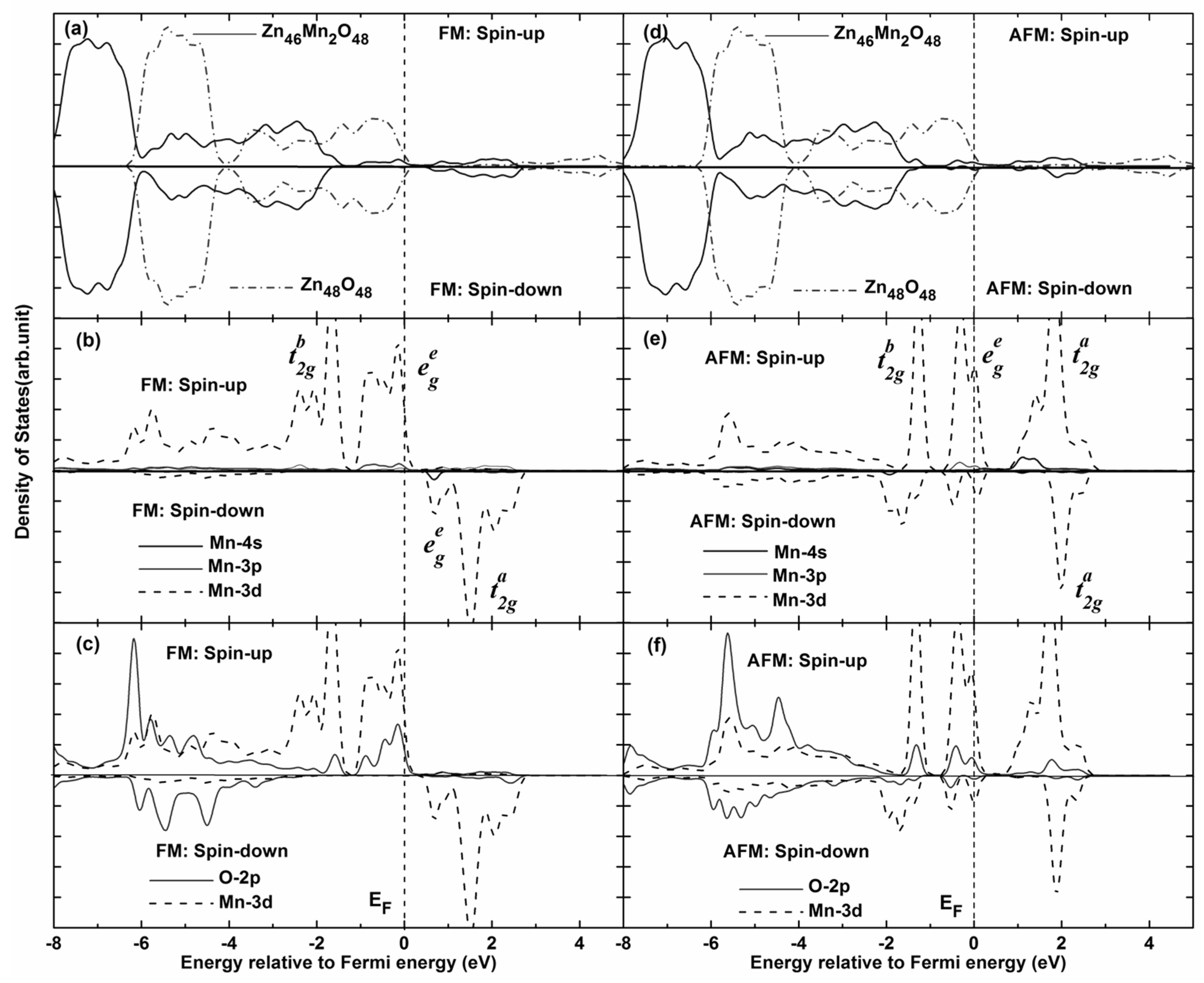
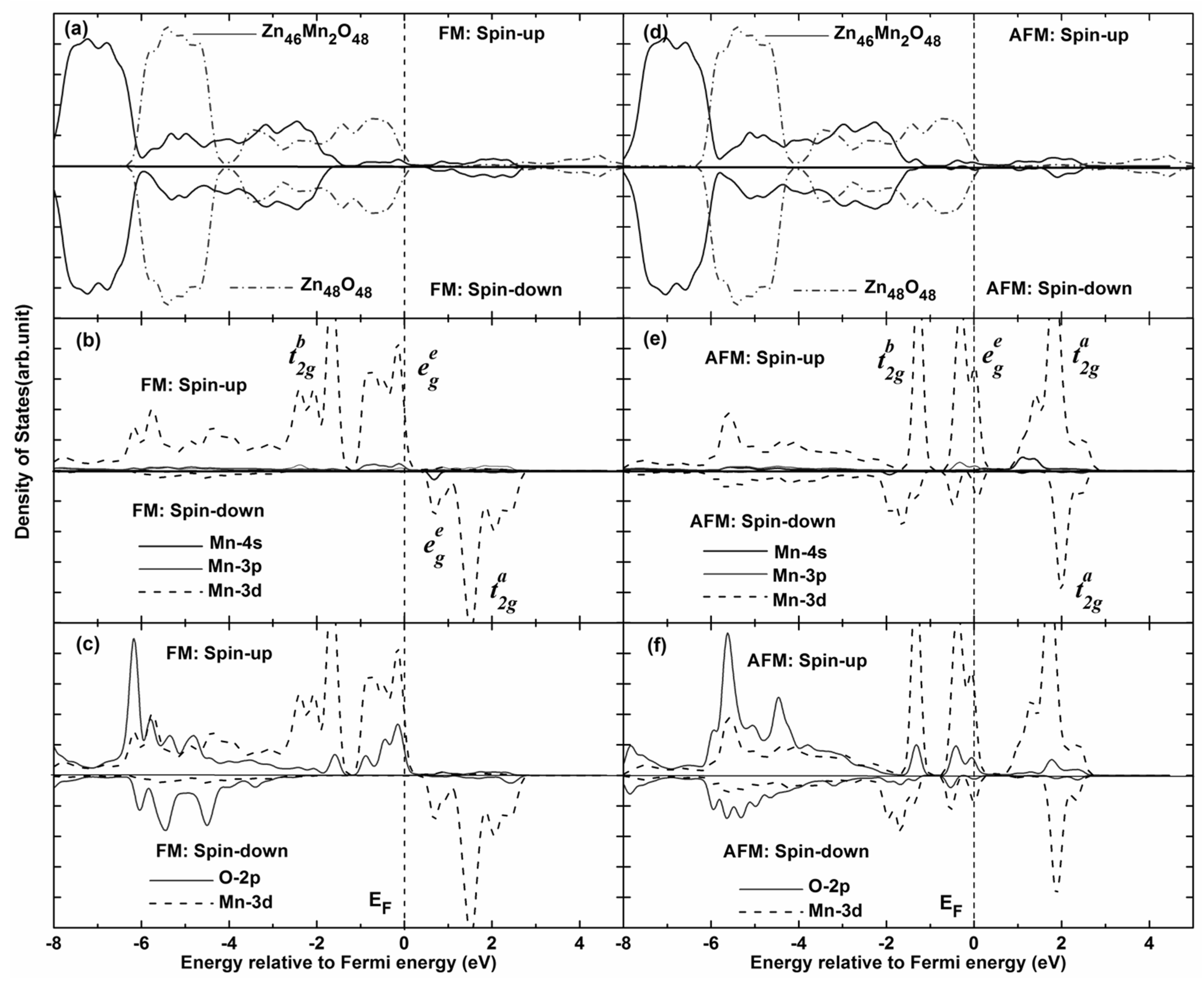
In order to further explain the magnetic coupling of Mn-doped ZnO NW,
Figure 5 shows the total density of states (TDOS) and partial density of states (PDOS) of FM and AFM states. It can be seen that the TDOS of Mn-doped ZnO NW moves towards low energy (0.95 eV), and one can see that the spin-up Mn
3d states pass through the Femi level for FM states, which confirms that the doped system is a half-metallic ferromagnet and exhibits 100% spin polarization. The strong FM coupling between the Mn
3d and O
2p states occurs at the top valence band (−1.0–0 eV) and the bottom valence bands (−4.0–−7.0 eV), the dispersive bonding
state and the local nonbonding
state is formed at the valence band (−2–−6 eV) and near the Femi level, respectively. The antibonding
state is formed at the conduction band (1.6–2.1 eV). In particular, one can see from
Figure 5c that the role of the O atom mediates the magnetic coupling between Mn atoms. The magnetic coupling chains of Mn
2+–O
2−–Mn
2+ would reveal a double exchange mechanism owing to impurity states in the gap region. On the other hand, the magnetic moments of Mn atoms are 4.012–4.193 μ
B and mainly come from the Mn 3
d orbitals, which are less than the theoretical value of 5 μ
B. However, small contributions to the magnetic moments come from O 2
p orbitals. As for AFM state, we find that the spin-up and spin-down DOS are asymmetrical, the spin-up electrons are more than the spin-down electrons. As the DOS of spin-up and spin-down states all pass through the Femi level, the AFM state is a possible metal ferromagnet. Spin-exchange splitting is also observed between Mn 3
d and O 2
p states, as shown in
Figure 5e,f. Thus, the local bonding
states are fully occupied between −1.5 eV and −1.0 eV. Whereas the nonbonding
states are 4/5 occupied between −1.0 eV and the Femi level, the anti-bonding
states are full empty between 0.6 eV and 2.5 eV, which correspond to the Mn
3d states. We note that the bandwidth of the bonding and antibonding band is greatly narrowed for AFM states, and the spin-orbital coupling and hybridization effects are significantly higher than FM states. Therefore, stable AFM states can be easily formed in Mn-doped ZnO NWs. This is mainly because the Mn
2+ (3
d5) ion shows a half-full
3d electronic configuration, the spin-up
3d orbitals of Mn
2+ atom are all occupied, and there are no excessive unoccupied orbitals for hybridization coupling with neighboring Mn
2+ ions. As a result,
3d orbital electrons of the neighboring Mn
2+ are prone to filling the levels along the reverse parallel direction, which is in accordance with the mechanisms shown in
Figure 3 and
Figure 4. In addition, the up-spin or down-spin orbitals are fully empty for the higher
state and AFM coupling is favored. Namely, the AFM coupling can be formed more easily in Mn-doped ZnO NWs according to theoretical analysis, which is similar to the reported results [
23,
24].
Figure 4.
The ferromagnetic (FM) and antiferromagnetic (AFM) mechanisms of the Mn-doped ZnO NW. (a) FM coupling; (b) AFM coupling.
Figure 4.
The ferromagnetic (FM) and antiferromagnetic (AFM) mechanisms of the Mn-doped ZnO NW. (a) FM coupling; (b) AFM coupling.
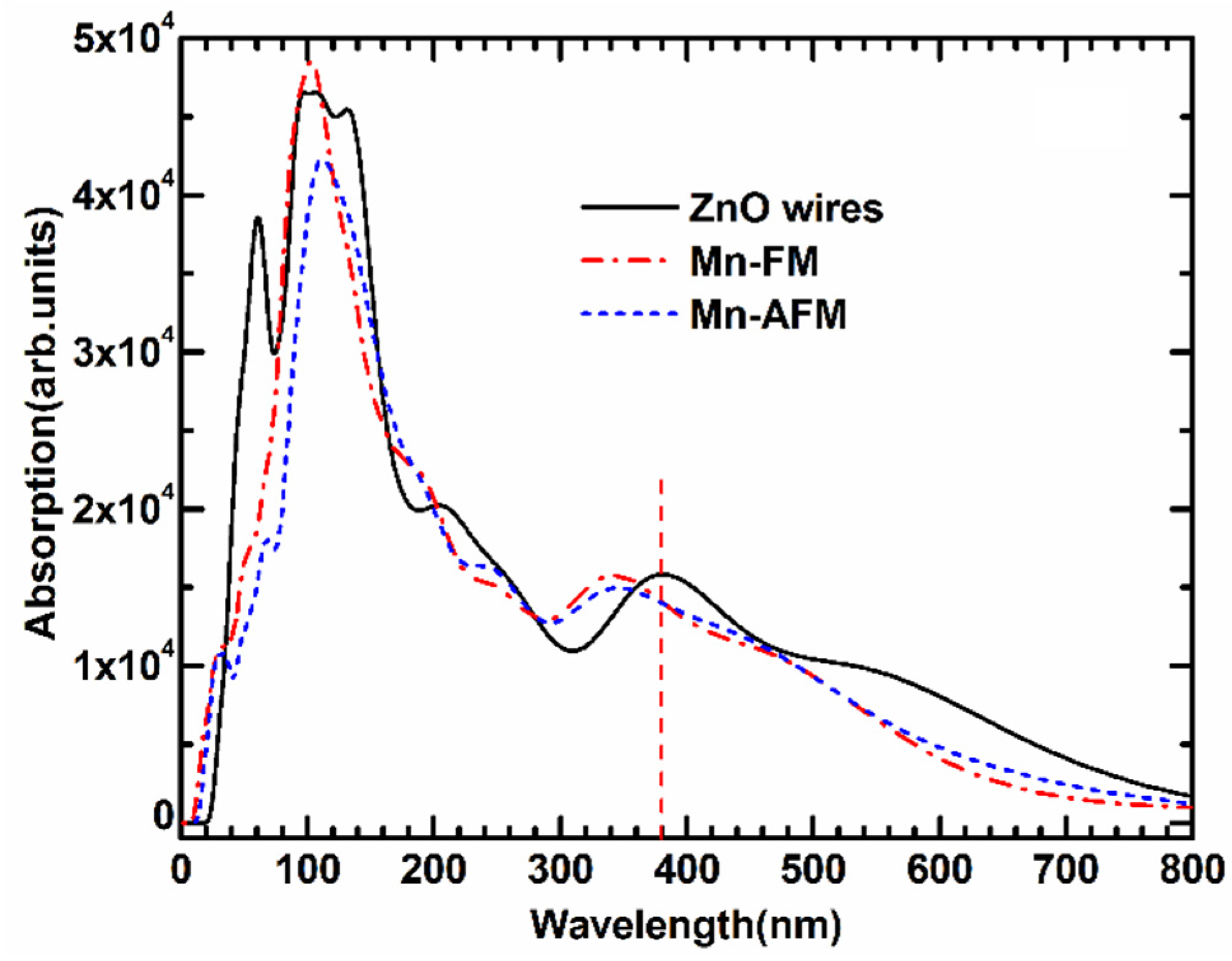
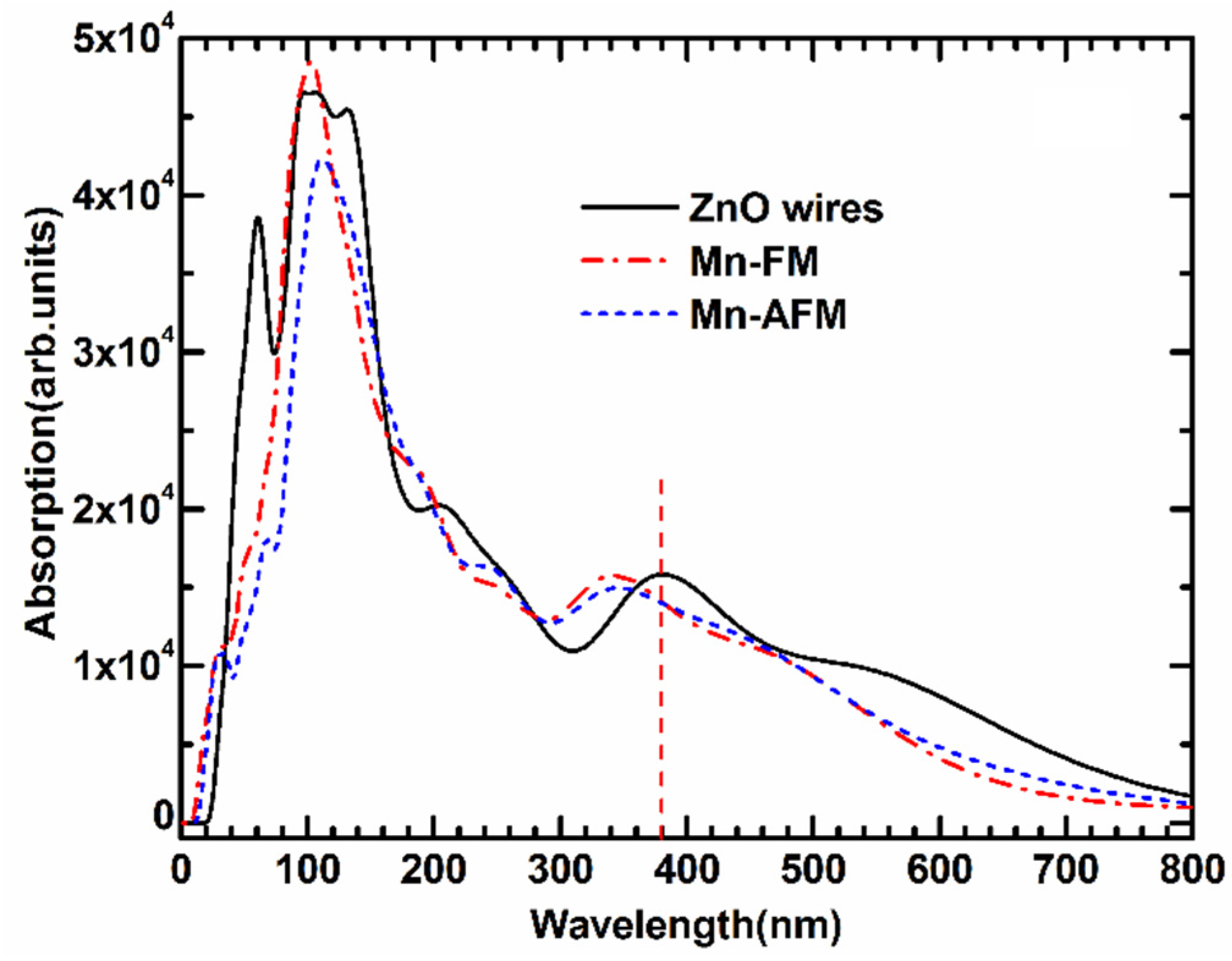
The optical absorption spectrum of Mn-doped ZnO NWs is illustrated in
Figure 6. We applied the scissors operator (
Escissor = 1.51 eV) to adjust the experimental and calculated band gap based on the experimental result (3.37 eV). To study the effect of Mn-doped on the optical properties, the absorption spectrum of pure ZnO NWs is also calculated. Some previous experimental studies [
25,
26,
27] found that pure ZnO NW shows a strong absorption peak in the ultraviolet region as a result of near-band edge absorption. A wide green-yellow luminous bands form within the visible region of 550–600 nm. As shown in
Figure 6, the calculated optical properties show that the absorption spectrum of the Mn-doped ZnO NWs are blue-shifted in the ultraviolet region, and the broad ultraviolet emission from 320 nm to 340 nm is observed in a doped system, which is related to electron transitions from the top valence band to the bottom conduction band. The calculated result is similar to the reported experimental optical absorption spectra of Mn-doped ZnO nanoparticles [
28,
29] and Mn-doped ZnO nanowires [
30,
31]. Additionally, one “sharp” absorption peak appears in the far-ultraviolet region near 90 nm, which are transitions from the
s–
d and
p–
d coupling of localized Mn 3
d orbitals. The increase in the intensity of the UV peak is considered to be due to the participation of the 3
d electrons of Mn [
10,
29,
30], and is very similar to Co-doped ZnO nanowires [
32]. The above-calculated results indicate a strong photo-response for ultraviolet-light luminescence. Namely, the Mn-doped ZnO NWs may be a new type of magneto-optical material with great promise.
Figure 5.
Total density of states (TDOS) and partial density of states (PDOS) of Mn-doped ZnO NW. (a) TDOS of pure Zn46O48 and Mn-doped ZnO nanowire for FM states; (b) PDOS of Mn atom for FM states; (c) PDOS of Mn 3d and O 2p for FM states; (d) TDOS of pure Zn46O48 and Mn-doped ZnO nanowire for FM states; (e) PDOS of Mn atom for AFM states; (f) PDOS of Mn 3d and O 2p for AFM states.
Figure 5.
Total density of states (TDOS) and partial density of states (PDOS) of Mn-doped ZnO NW. (a) TDOS of pure Zn46O48 and Mn-doped ZnO nanowire for FM states; (b) PDOS of Mn atom for FM states; (c) PDOS of Mn 3d and O 2p for FM states; (d) TDOS of pure Zn46O48 and Mn-doped ZnO nanowire for FM states; (e) PDOS of Mn atom for AFM states; (f) PDOS of Mn 3d and O 2p for AFM states.
Figure 6.
The optical absorption curves of FM and AFM for Mn-doped ZnO NW.
Figure 6.
The optical absorption curves of FM and AFM for Mn-doped ZnO NW.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}