
Single-Atom Catalysts Dispersed on Graphitic Carbon Nitride (g-CN): Eley–Rideal-Driven CO-to-Ethanol Conversion



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Computational Methods and Models
3. Results and Discussion
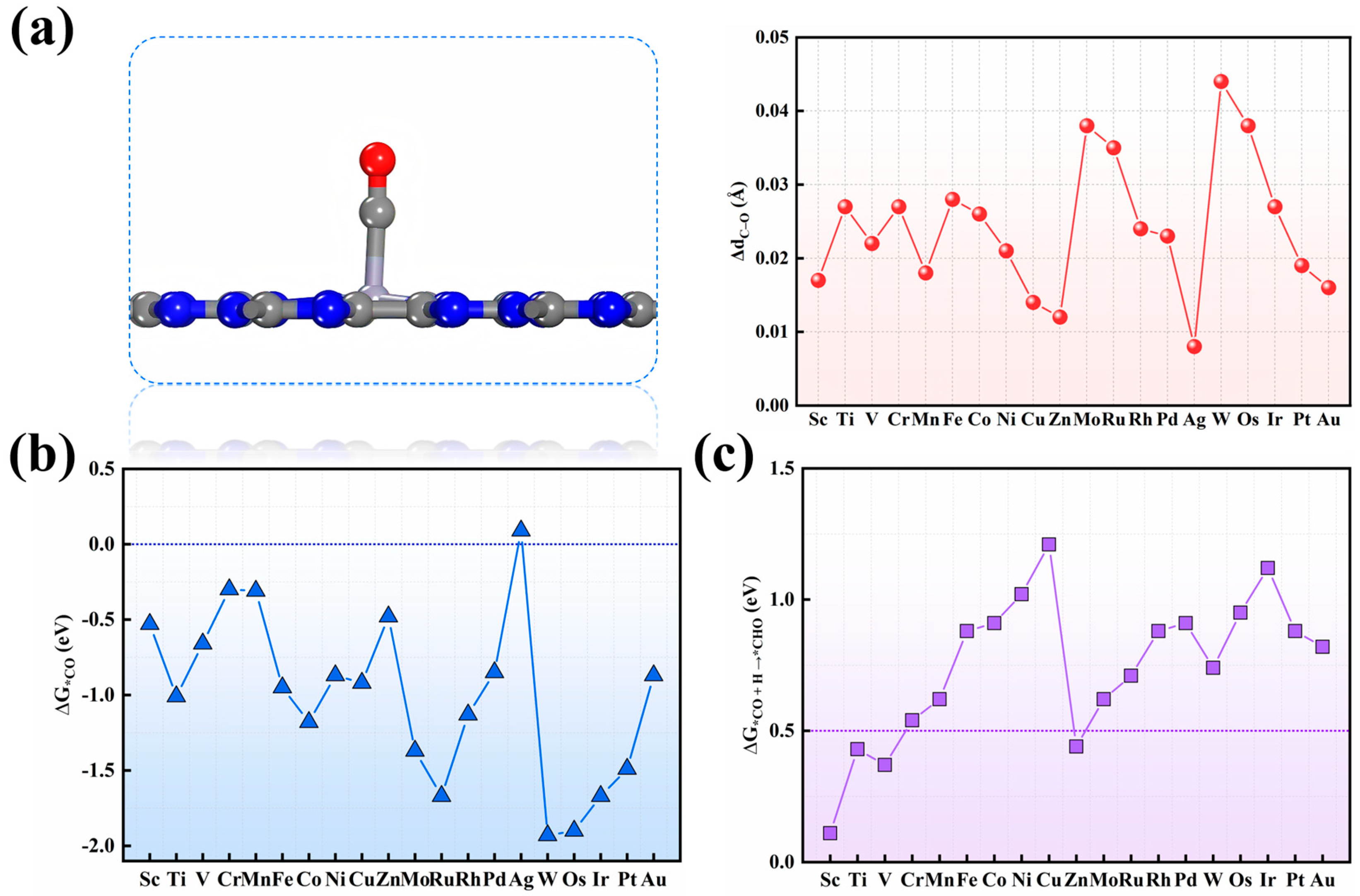
3.1. Screening of SACs Candidates
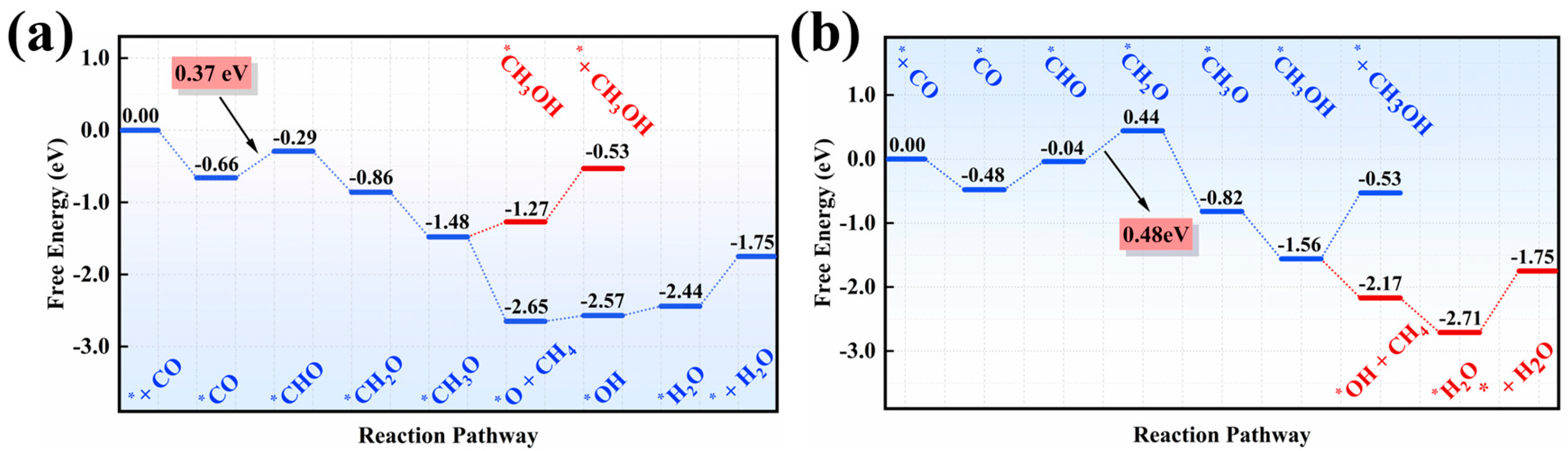
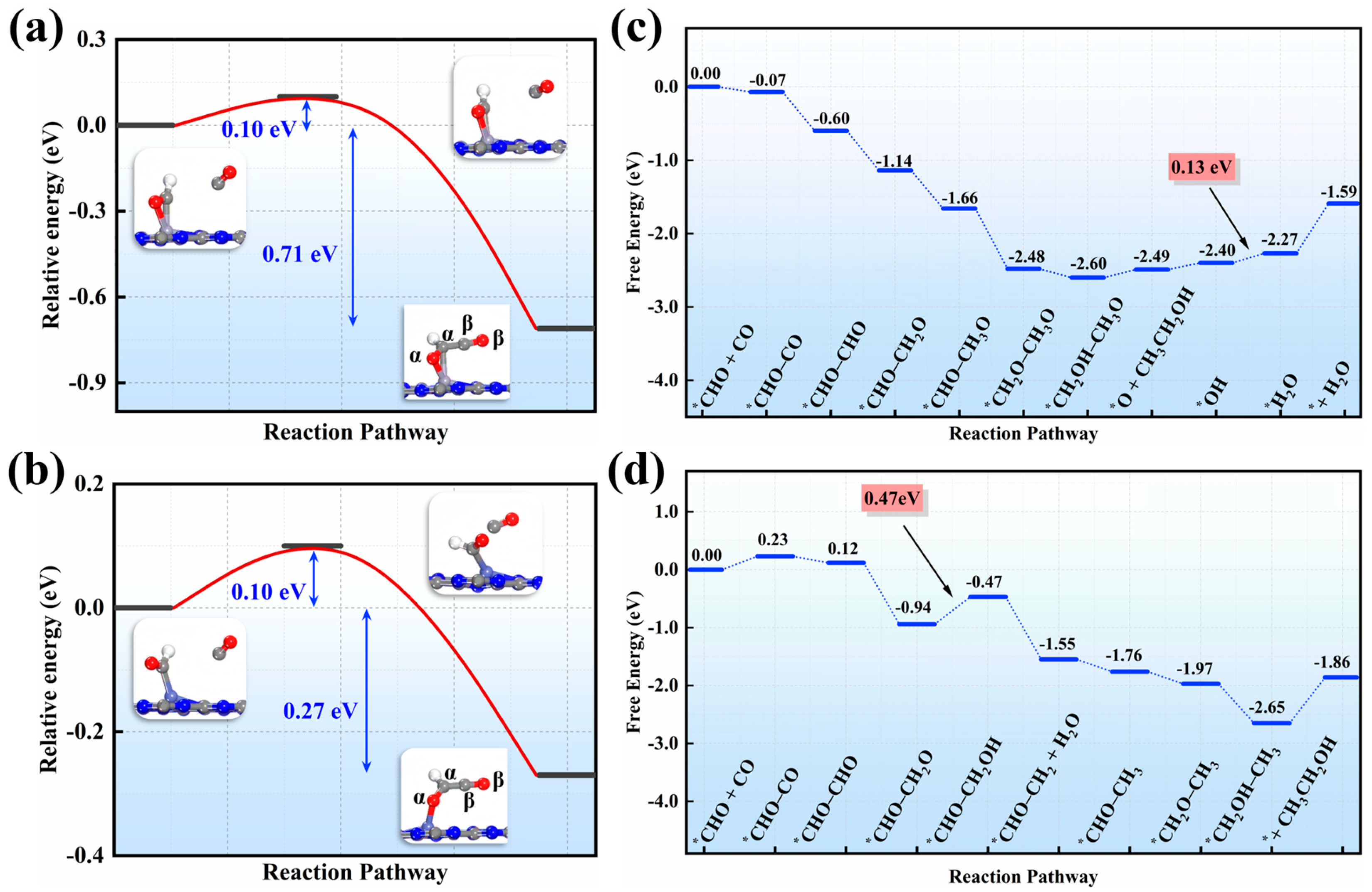
3.2. Product Distribution and Reaction Mechanism
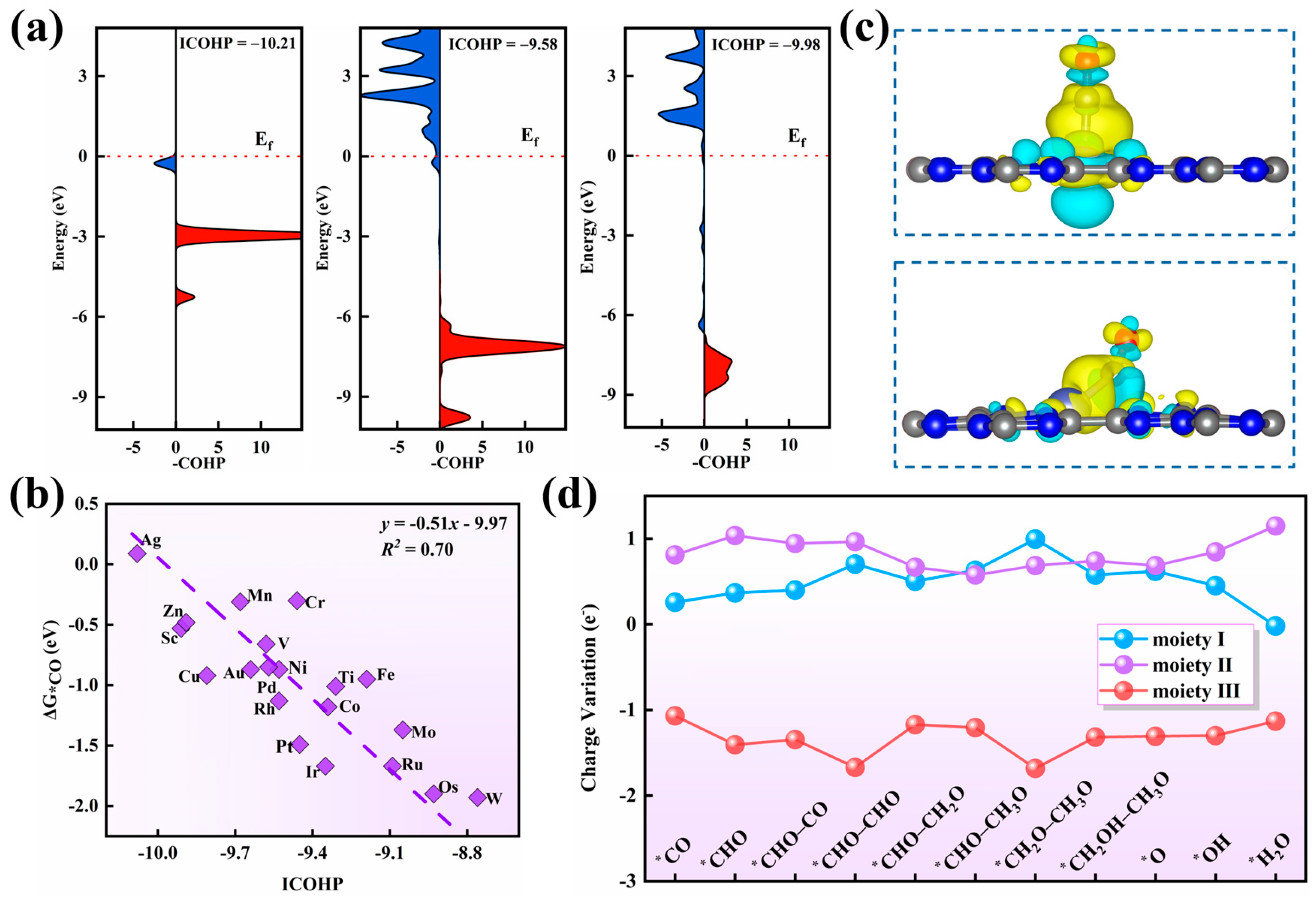
3.3. V/g–CN and Zn/g–CN of COER Activity Origin
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chang, B.; Pang, H.; Raziq, F.; Wang, S.; Huang, K.W.; Ye, J.; Zhang, H. Electrochemical reduction of carbon dioxide to multicarbon (C2+) products: Challenges and perspectives. Energy Environ. Sci. 2023, 16, 4714–4758. [Google Scholar] [CrossRef]
- Hu, J.; Chen, X.; Liu, J.; Guan, J. Advances in the CO2 electroreduction to multi-carbon products. Chem. Eng. J. 2025, 516, 164199. [Google Scholar] [CrossRef]
- Chen, J.; Wang, T.; Li, Z.; Yang, B.; Zhang, Q.; Lei, L.; Feng, P.; Hou, Y. Recent progress and perspective of electrochemical CO2 reduction towards C2-C5 products over non-precious metal heterogeneous electrocatalysts. Nano Res. 2021, 14, 3188–3207. [Google Scholar] [CrossRef]
- She, X.; Wang, Y.; Xu, H.; Tsang, S.C.E.; Lau, S.P. Challenges and opportunities in electrocatalytic CO2 reduction to chemicals and fuels. Angew. Chem. Int. Ed. 2022, 61, e202211396. [Google Scholar] [CrossRef]
- Overa, S.; Feric, T.G.; Park, A.H.A.; Jiao, F. Tandem and hybrid processes for carbon dioxide utilization. Joule 2021, 5, 8–13. [Google Scholar] [CrossRef]
- Qiu, R.; Cui, L.; Peng, L.; Syzgantseva, O.A.; Li, J.; Fang, N.; Syzgantseva, M.A.; Jiang, Y.; Zhang, J.; Zhang, B.; et al. Cooperative promotion of electroreduction of CO to n-propanol by *CO enrichment and proton regulation. Chem. Sci. 2025, 16, 8897–8909. [Google Scholar] [CrossRef]
- Tran, N.H.; Schreiber, M.W.; Fontecave, M. Catalysts for selective CO2/CO electroreduction to C3+ compounds. EES Catal. 2025, 3, 644–668. [Google Scholar] [CrossRef]
- Wang, X.; Ou, P.; Ozden, A.; Hung, S.F.; Tam, J.; Gabardo, C.M.; Howe, J.Y.; Sisler, J.; Bertens, K.; de Arquer, F.P.G.; et al. Efficient electrosynthesis of n-propanol from carbon monoxide using a Ag–Ru–Cu catalyst. Nat. Energy 2022, 7, 170–176. [Google Scholar] [CrossRef]
- Zhuang, T.T.; Pang, Y.; Liang, Z.Q.; Wang, J.; Li, Y.; Tan, C.S.; Li, J.; Thang, D.C.; De Luna, P.; Hsieh, P.L.; et al. Copper nanocavities confine intermediates for efficient electrosynthesis of C3 alcohol fuels from carbon monoxide. Nat. Catal. 2018, 1, 946–951. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, Y.; Liu, G.; Zhang, H.; Yan, Y.; Ke, Q.; Liu, M.; Liu, L.; Lin, Z. Turning the Selectivity of CO Electroreduction from Acetate to Ethanol by Alloying FCC-Phased Cu with Atomically Dispersed Mn Atoms. Nano Lett. 2025, 25, 6771–6779. [Google Scholar] [CrossRef]
- Phong, D.H.; Rivera de La Cruz, J.G.; Portehault, D.; Zitolo, A.; Louis, J.; Zanna, S.; Arnoux, Q.; Schreiber, M.W.; Menguy, N.; Tran, N.H.; et al. Incorporation of isolated Ag atoms and Au nanoparticles in copper nitride for selective CO electroreduction to multicarbon alcohols. Nat. Mater. 2025, 24, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Li, T.; Song, Y.; Li, R.; Wei, P.; Liao, Z.; Wu, Z.; Gao, D.; Fu, Q.; Wang, G.; et al. Selective CO Electroreduction to Multicarbon Oxygenates over Atomically Dispersed Cu-Ag Sites in Alkaline Membrane Electrode Assembly Electrolyzer. Angew. Chem. Int. Ed. 2025, 64, e202507062. [Google Scholar] [CrossRef] [PubMed]
- Niu, W.; Feng, J.; Chen, J.; Deng, L.; Guo, W.; Li, H.; Zhang, L.; Li, Y.; Zhang, B. High-efficiency C3 electrosynthesis on a lattice-strain-stabilized nitrogen-doped Cu surface. Nat. Commun. 2024, 15, 7070. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Tan, X.; Jia, C.; Rong, C.; Wang, S.; Han, C.; Xiao, Y.; Qi, H.; Smith, S.C.; Zhao, C. Molecule Dispersed of Atomically Dispersed Cu–Au Alloy for Enhancing Electroreduction of CO to C2+ Products. Adv. Funct. Mater. 2024, 34, 2406281. [Google Scholar] [CrossRef]
- Wu, Z.; Meng, N.; Yang, R.; Chen, M.; Pan, J.; Chi, S.; Wu, C.; Xi, S.; Liu, Y.; Ou, Y.; et al. Boosting C2+ Alcohols Selectivity and Activity in High-Current CO Electroreduction using Synergistic Cu/Zn Co-Catalysts. Angew. Chem. Int. Ed. 2025, 64, e202420283. [Google Scholar] [CrossRef]
- Zhuang, Z.; Wang, G.; Zhao, W.; Yang, R.; Zhou, Y.; Zhu, W. Silver-Doped Porous Copper Catalysts for Efficient Resource Utilization of CO-Containing Flue Gases. ACS Environ. Au 2025, 5, 287–297. [Google Scholar] [CrossRef]
- Hori, Y.; Takahashi, I.; Koga, O.; Hoshi, N. Electrochemical reduction of carbon dioxide at various series of copper single crystal electrodes. J. Mol. Catal. A Chem. 2003, 199, 39–47. [Google Scholar] [CrossRef]
- Zhao, T.T.; Yan, T.Y.; Sun, Y.T.; Wang, Z.X.; Cai, Q.H.; Zhao, J.X.; Chen, Z.F. Constructing a square-like copper cluster to boost C–C coupling for CO2 electroreduction to ethylene. J. Mater. Chem. A 2023, 11, 19444–19454. [Google Scholar] [CrossRef]
- Luc, W.; Fu, X.; Shi, J.; Lv, J.J.; Jouny, M.; Ko, B.H.; Xu, Y.; Tu, Q.; Hu, X.; Wu, J.; et al. Two-dimensional copper nanosheets for electrochemical reduction of carbon monoxide to acetate. Nat. Catal. 2019, 2, 423–430. [Google Scholar] [CrossRef]
- Yang, R.; Xia, L.; Jiang, W.; Cheng, Y.; Wang, K.; Chen, T.; Li, F.; Zhao, X.; Wang, B.; Zhou, Y.; et al. Cu-Based Tandem Architectures for CO2 Electrolysis to Multicarbon Products. Adv. Energy Mater. 2025, 15, 2405964. [Google Scholar] [CrossRef]
- Overa, S.; Crandall, B.S.; Shrimant, B.; Tian, D.; Ko, B.H.; Shin, H.; Bae, C.; Jiao, F. Enhancing acetate selectivity by coupling anodic oxidation to carbon monoxide electroreduction. Nat. Catal. 2022, 5, 738–745. [Google Scholar] [CrossRef]
- Lei, Y.; Wang, Y.; Liu, Y.; Song, C.; Li, Q.; Wang, D.; Li, Y. Designing atomic active centers for hydrogen evolution electrocatalysts. Angew. Chem. Int. Ed. 2020, 59, 20794–20812. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Lu, B.; Chen, S. Carbon-supported single atom catalysts for electrochemical energy conversion and storage. Adv. Mater. 2018, 30, 1801995. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, W.; Wang, D.; Li, Y. Electronic metal–support interaction of single-atom catalysts and applications in electrocatalysis. Adv. Mater. 2020, 32, 2003300. [Google Scholar] [CrossRef]
- Zhang, Q.; Guan, J. Single-atom catalysts for electrocatalytic applications. Adv. Funct. Mater. 2020, 30, 2000768. [Google Scholar] [CrossRef]
- Zhu, C.; Fu, S.; Shi, Q.; Du, D.; Lin, Y. Single-atom electrocatalysts. Angew. Chem. Int. Ed. 2017, 56, 13944–13960. [Google Scholar] [CrossRef]
- Wang, Y.; Li, B.; Xue, B.; Libretto, N.; Xie, Z.; Shen, H.; Wang, C.; Raciti, D.; Marinkovic, N.; Zong, H.; et al. CO electroreduction on single-atom copper. Sci. Adv. 2023, 9, eade3557. [Google Scholar] [CrossRef]
- Bao, H.; Qiu, Y.; Peng, X.; Wang, J.A.; Mi, Y.; Zhao, S.; Liu, X.; Liu, Y.; Cao, R.; Zhuo, L.; et al. Isolated copper single sites for high-performance electroreduction of carbon monoxide to multicarbon products. Nat. Commun. 2021, 12, 238. [Google Scholar] [CrossRef]
- Miao, K.; Wen, J.; Luo, M.; Xiang, D.; Jiang, Y.; Duan, D.; Jiang, Z.; Sun, W.; Mei, B.; Xiong, Y.; et al. Phosphorus Coordination in Second Shell of Single-Atom Cu Catalyst toward Acetate Production in CO Electroreduction. Nano Lett. 2024, 24, 12849–12856. [Google Scholar] [CrossRef]
- Ohashi, K.; Nagita, K.; Yamamoto, H.; Nakanishi, S.; Kamiya, K. C−C Coupling in CO2 Electroreduction on Single Cu-Modified Covalent Triazine Frameworks: A Static and Dynamic Density Functional Theory Study. ChemElectroChem 2024, 11, e202300693. [Google Scholar] [CrossRef]
- Yu, Z.; Li, Y.; Torres-Pinto, A.; LaGrow, A.P.; Diaconescu, V.M.; Simonelli, L.; Sampaio, M.J.; Bondarchuk, O.; Amorim, I.; Araujo, A. Single-atom Ir and Ru anchored on graphitic carbon nitride for efficient and stable electrocatalytic/photocatalytic hydrogen evolution. Appl. Catal. B Environ. 2022, 310, 121318. [Google Scholar] [CrossRef]
- Lv, X.; Wei, W.; Huang, B.; Dai, Y.; Frauenheim, T. High-throughput screening of synergistic transition metal dual-atom catalysts for efficient nitrogen fixation. Nano Lett. 2021, 21, 1871–1878. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yue, Y.; Yang, C.; Zhang, X.; Qin, J.; Liu, R. Transition metal single-atom anchored g-CN monolayer for constructing high-activity multifunctional electrocatalyst. Appl. Surf. Sci. 2021, 565, 150547. [Google Scholar] [CrossRef]
- Zhong, J.J.; Huang, S.P.; Gu, J.F.; Li, Y.; Ding, K.N.; Zhang, Y.F.; Lin, W.; Chen, W.K. Theoretical study of transition metal atom pairs anchored in g-CN monolayers for ammonia decomposition. Appl. Surf. Sci. 2023, 609, 155280. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jonsson, H. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Nørskov, J.K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 2010, 3, 1311–1315. [Google Scholar] [CrossRef]
- González-González, R.; Salas-Zepeda, M.G.; Tlahuice-Flores, A. New two-dimensional carbon nitride allotrope with 1 : 1 stoichiometry featuring spine-like structures: A structural and electronic DFT-D study. Phys. Chem. Chem. Phys. 2019, 21, 15282–15285. [Google Scholar] [CrossRef]
- Niu, H.; Zhang, Z.; Wang, X.; Wan, X.; Shao, C.; Guo, Y. Theoretical insights into the mechanism of selective nitrate-to-ammonia electroreduction on single-atom catalysts. Adv. Funct. Mater. 2021, 31, 2008533. [Google Scholar] [CrossRef]
- Niu, H.; Wang, X.; Shao, C.; Zhang, Z.; Guo, Y. Computational screening single-atom catalysts supported on g-CN for N2 reduction: High activity and selectivity. ACS Sustain. Chem. Eng. 2020, 8, 13749–13758. [Google Scholar] [CrossRef]
- Wang, S.; Wei, W.; Lv, X.; Huang, B.; Dai, Y. W supported on g-CN manifests high activity and selectivity for N2 electroreduction to NHJ. Mater. Chem. A 2020, 8, 1378–1385. [Google Scholar] [CrossRef]
- Li, X.; Cui, P.; Zhong, W.; Li, J.; Wang, X.; Wang, Z.; Jiang, J. Graphitic carbon nitride supported single-atom catalysts for efficient oxygen evolution reaction. Chem. Commun. 2016, 52, 13233–13236. [Google Scholar] [CrossRef]
- Ruqia, B.; Tomboc, G.M.; Kwon, T.; Kundu, J.; Kim, J.Y.; Lee, K.; Choi, S.I. Recent advances in the electrochemical CO reduction reaction towards highly selective formation of Cx products (X = 1–3). Chem. Catal. 2022, 2, 1961–1988. [Google Scholar] [CrossRef]
- Montoya, J.H.; Shi, C.; Chan, K.; Nørskov, J.K. Theoretical insights into a CO dimerization mechanism in CO2 electroreduction. J. Phys. Chem. Lett. 2015, 6, 2032–2037. [Google Scholar] [CrossRef]
- He, M.; Jiang, C.H.; Yan, H.M.; Wang, G.; Wang, Y.G. Unraveling the C–C Coupling Mechanism on Dual-Atom Catalysts for CO2/CO Reduction Reaction: The Critical Role of CO Hydrogenation. J. Phys. Chem. Lett. 2024, 16, 324–332. [Google Scholar] [CrossRef]
- Wang, S.; Li, F.; Zhao, J.; Zeng, Y.; Li, Y.; Lin, Z.Y.; Lee, T.J.; Liu, S.; Ren, X.; Wang, W.; et al. Manipulating CC coupling pathway in electrochemical CO2 reduction for selective ethylene and ethanol production over single-atom alloy catalyst. Nat. Commun. 2024, 15, 10247. [Google Scholar] [CrossRef]
- Goyal, A.; Marcandalli, G.; Mints, V.A.; Koper, M.T.M. Competition between CO2 reduction and hydrogen evolution on a gold electrode under well-defined mass transport conditions. J. Am. Chem. Soc. 2020, 142, 4154–4161. [Google Scholar] [CrossRef]
- Wang, X.; Chen, Y.; Li, F.; Miao, R.K.; Huang, J.E.; Zhao, Z.; Li, X.Y.; Dorakhan, R.; Chu, S.; Wu, J.; et al. Site-selective protonation enables efficient carbon monoxide electroreduction to acetate. Nat. Commun. 2024, 15, 616. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Song, Q.; Shang, Y.; Liu, Y.; Zhao, J. Single-Atom Catalysts Dispersed on Graphitic Carbon Nitride (g-CN): Eley–Rideal-Driven CO-to-Ethanol Conversion. Nanomaterials 2025, 15, 1111. https://doi.org/10.3390/nano15141111
Wang J, Song Q, Shang Y, Liu Y, Zhao J. Single-Atom Catalysts Dispersed on Graphitic Carbon Nitride (g-CN): Eley–Rideal-Driven CO-to-Ethanol Conversion. Nanomaterials. 2025; 15(14):1111. https://doi.org/10.3390/nano15141111
Chicago/Turabian StyleWang, Jing, Qiuli Song, Yongchen Shang, Yuejie Liu, and Jingxiang Zhao. 2025. "Single-Atom Catalysts Dispersed on Graphitic Carbon Nitride (g-CN): Eley–Rideal-Driven CO-to-Ethanol Conversion" Nanomaterials 15, no. 14: 1111. https://doi.org/10.3390/nano15141111
APA StyleWang, J., Song, Q., Shang, Y., Liu, Y., & Zhao, J. (2025). Single-Atom Catalysts Dispersed on Graphitic Carbon Nitride (g-CN): Eley–Rideal-Driven CO-to-Ethanol Conversion. Nanomaterials, 15(14), 1111. https://doi.org/10.3390/nano15141111