
DFT Prediction of Structural and Physical Properties of Cr3AlC2 Under Pressure


Abstract
1. Introduction
2. Theoretical Methods
3. Results and Discussion
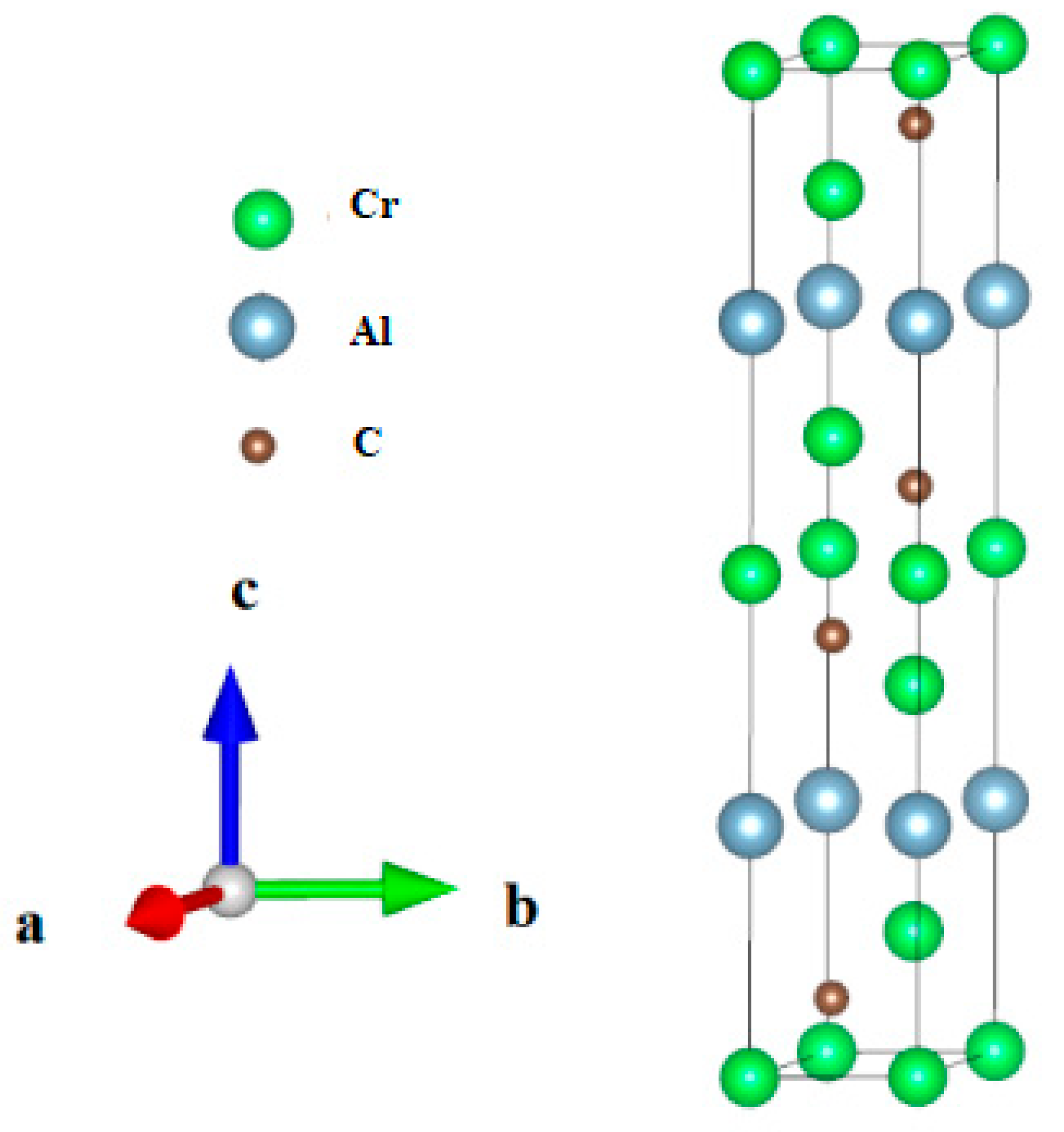
3.1. Structural Properties
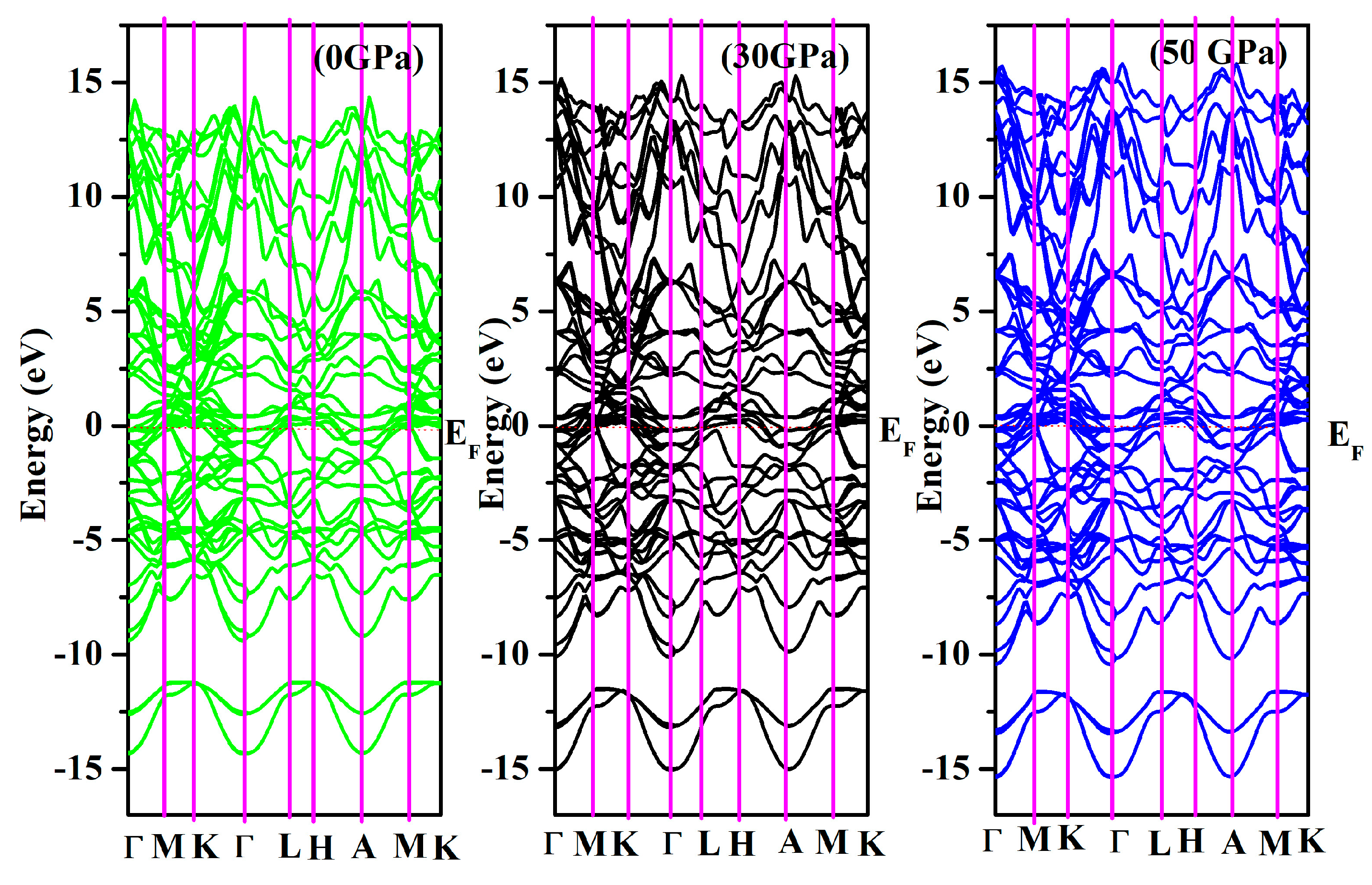
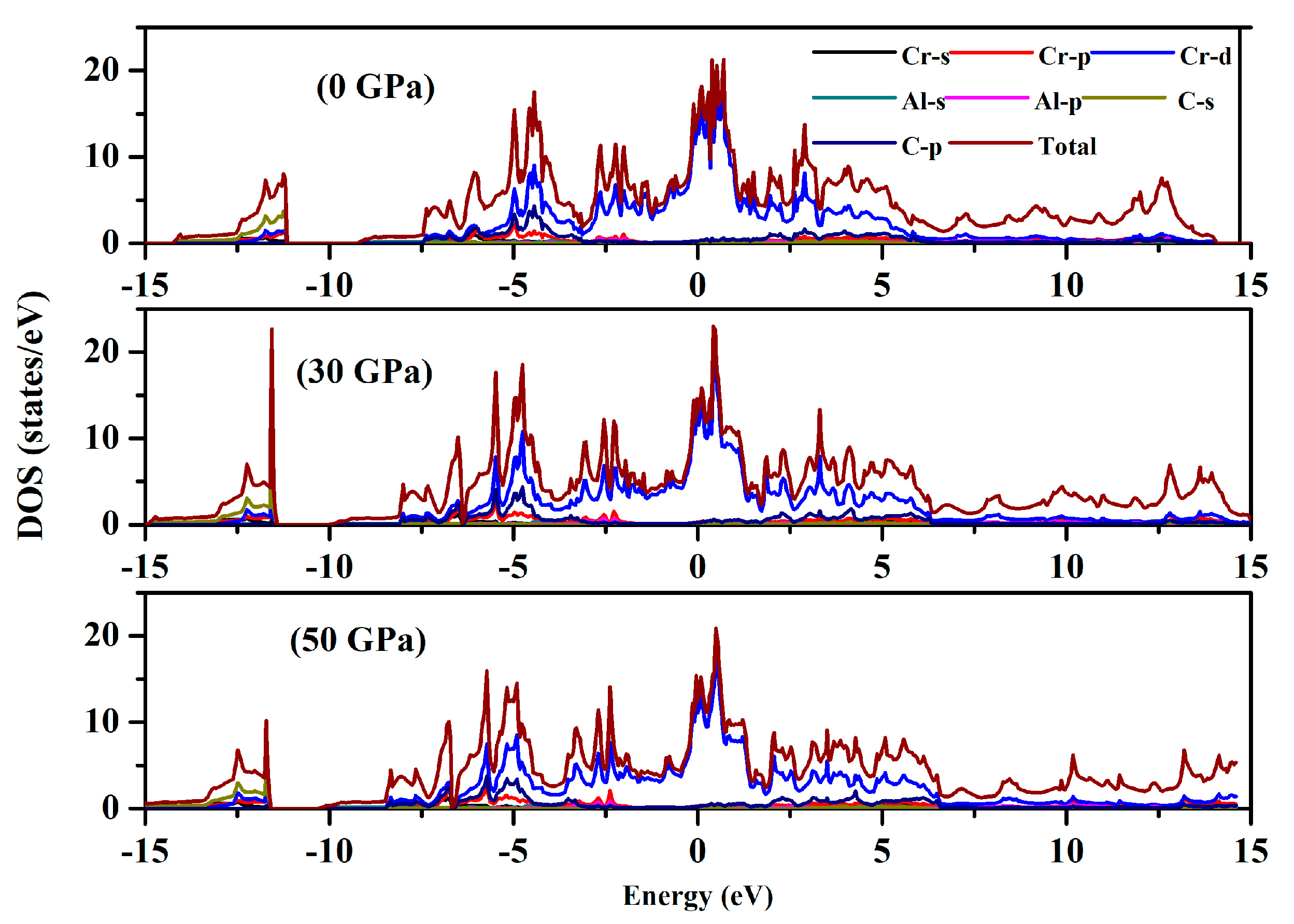
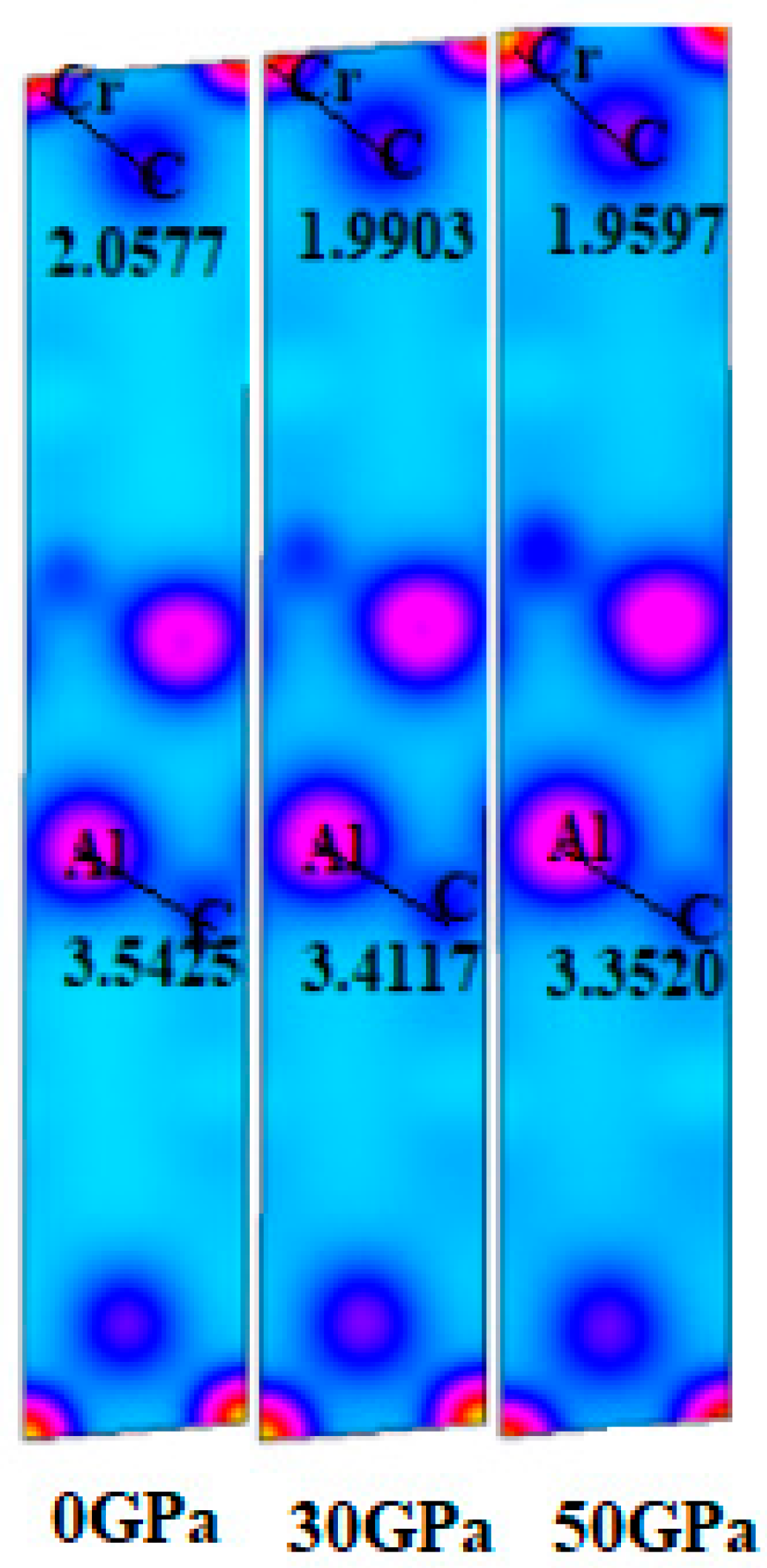
3.2. Electronic Properties
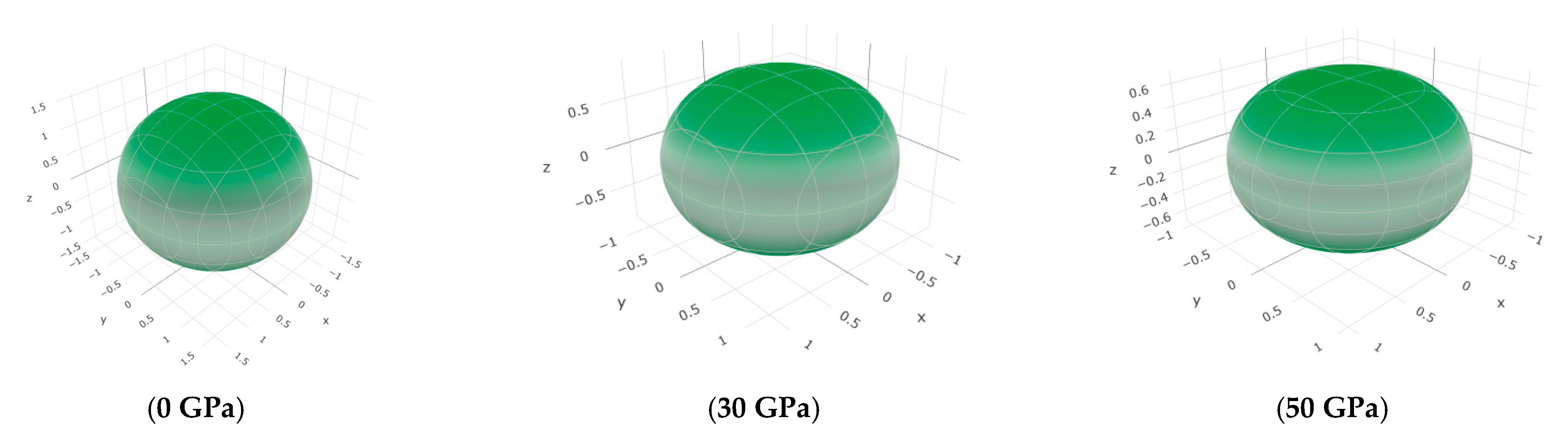
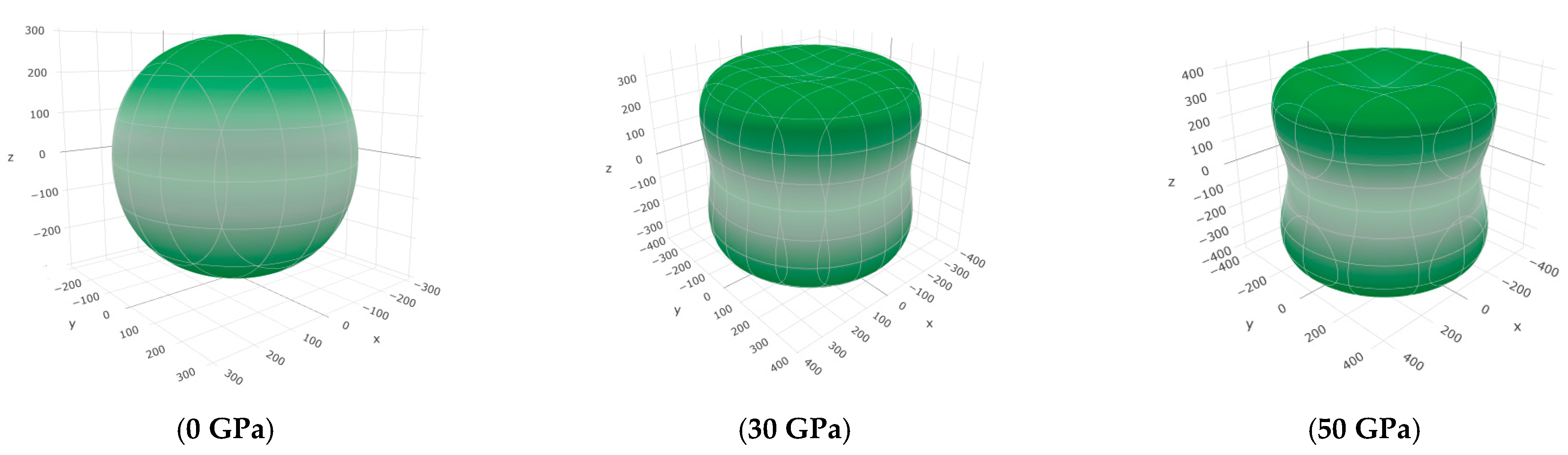
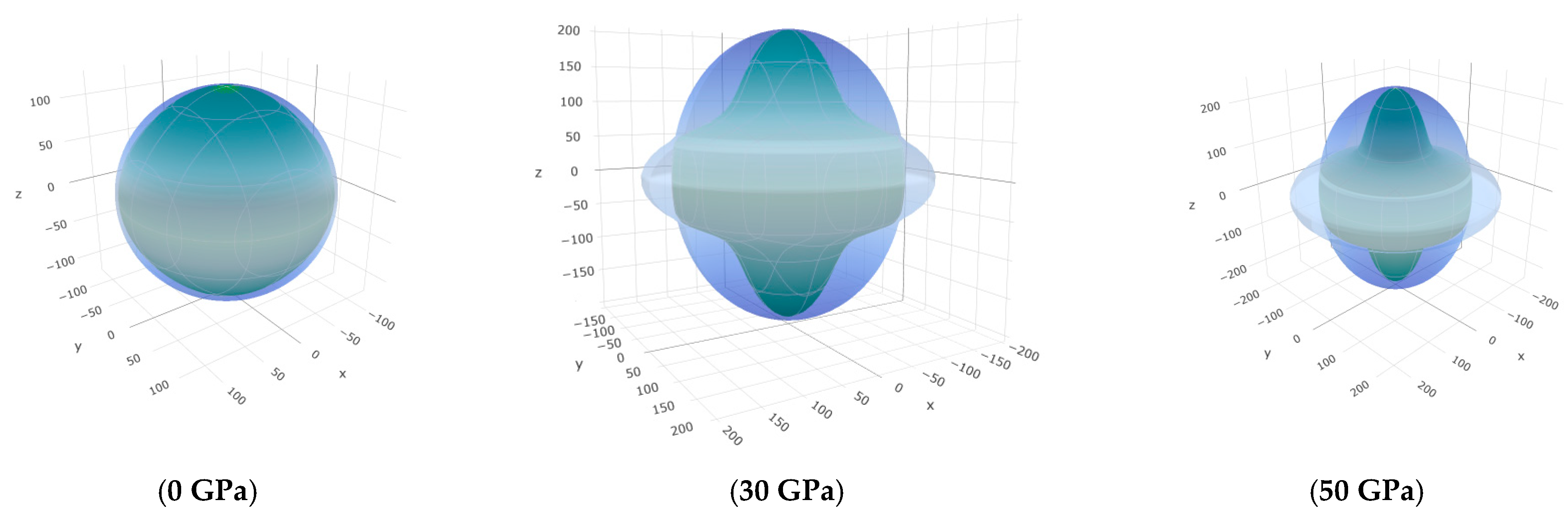
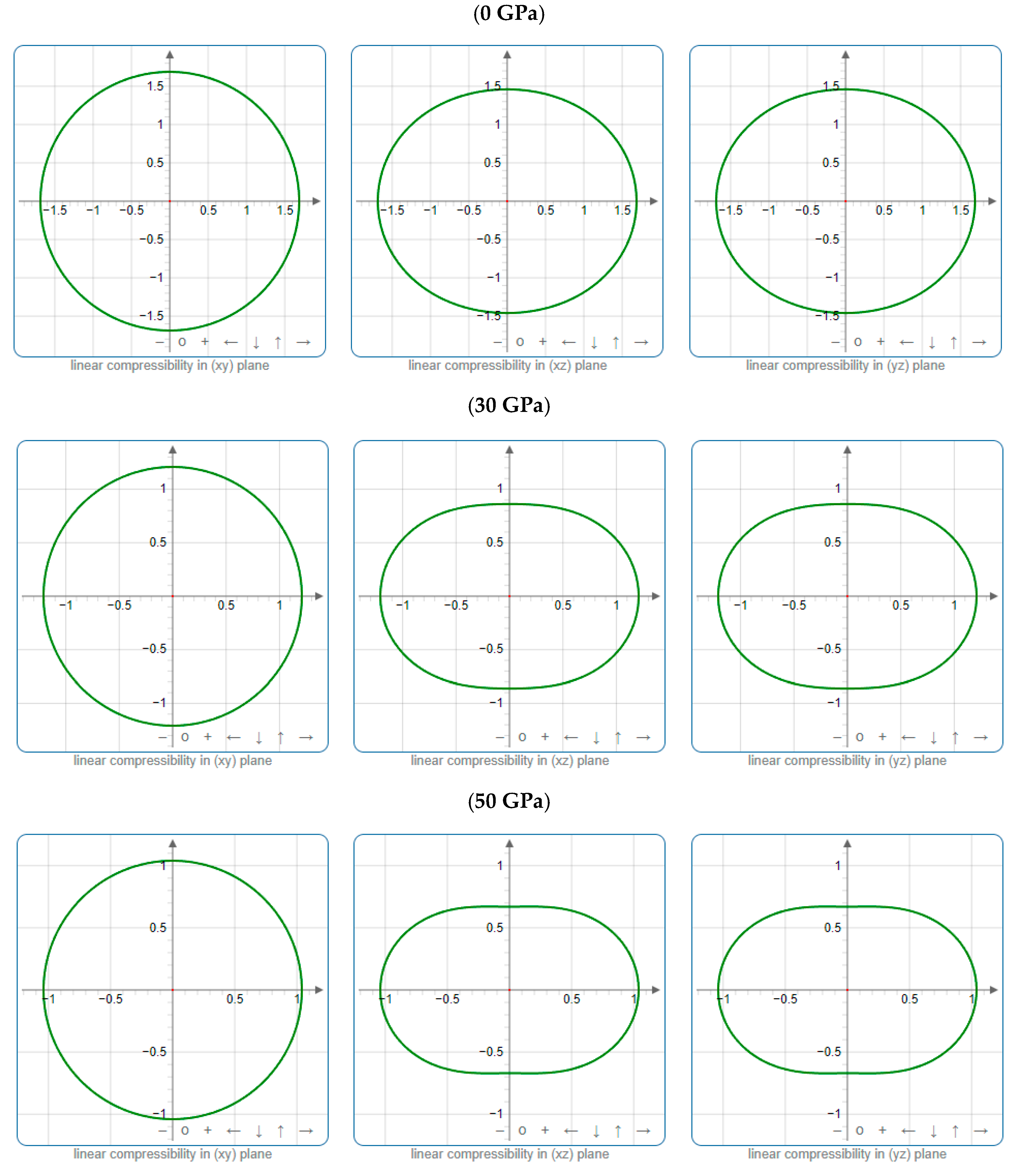
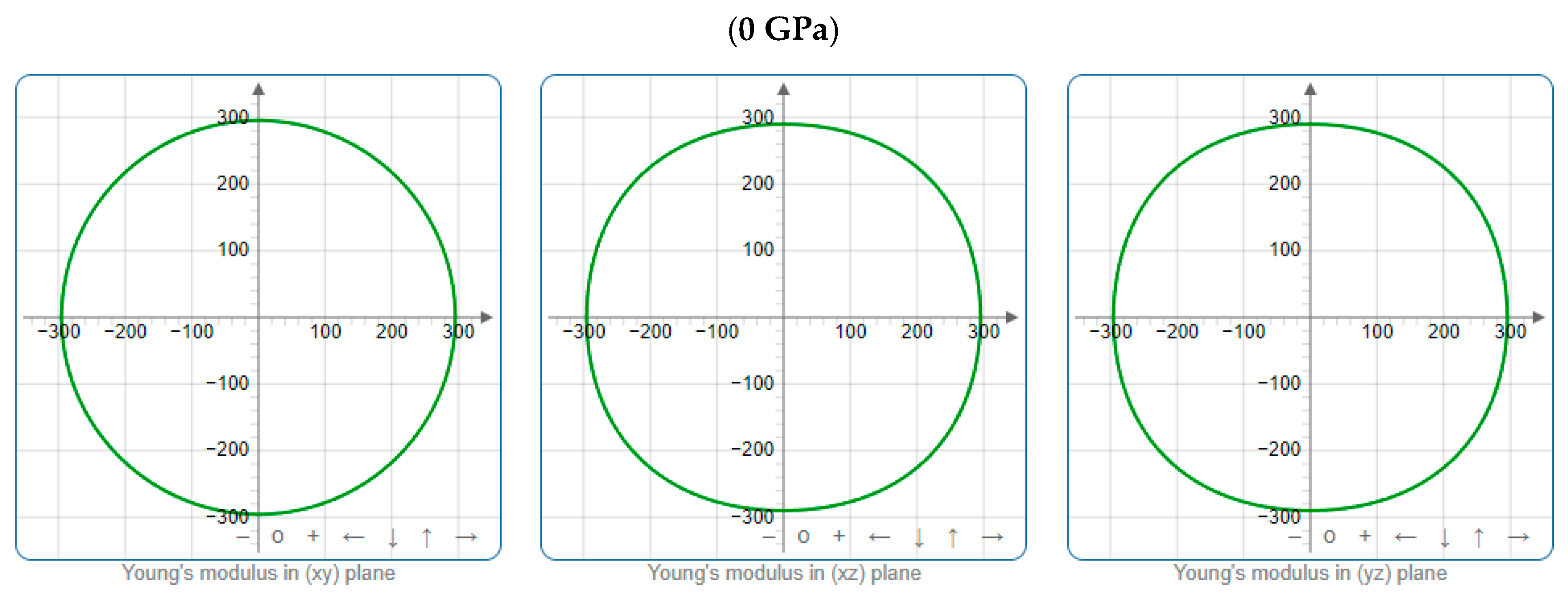
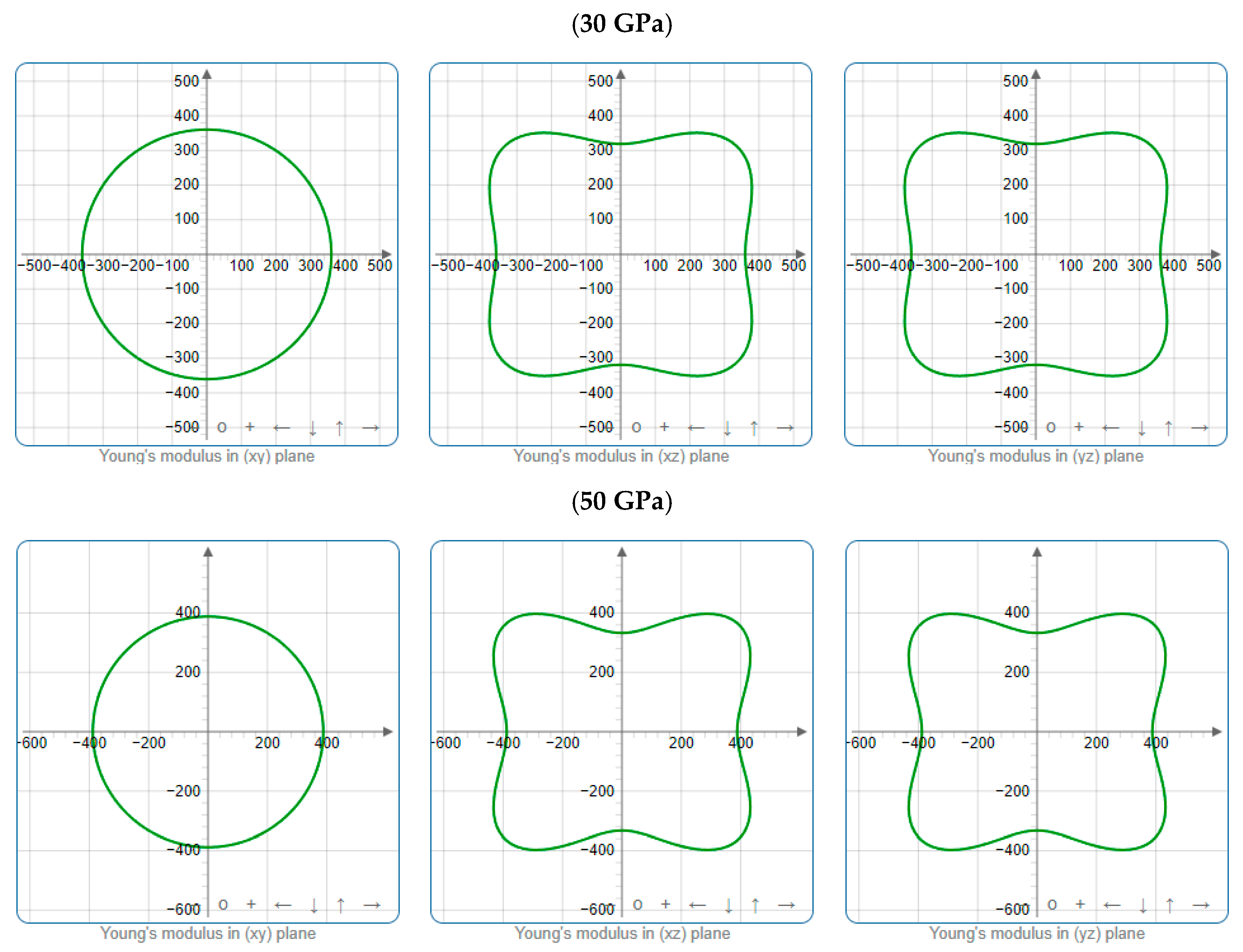
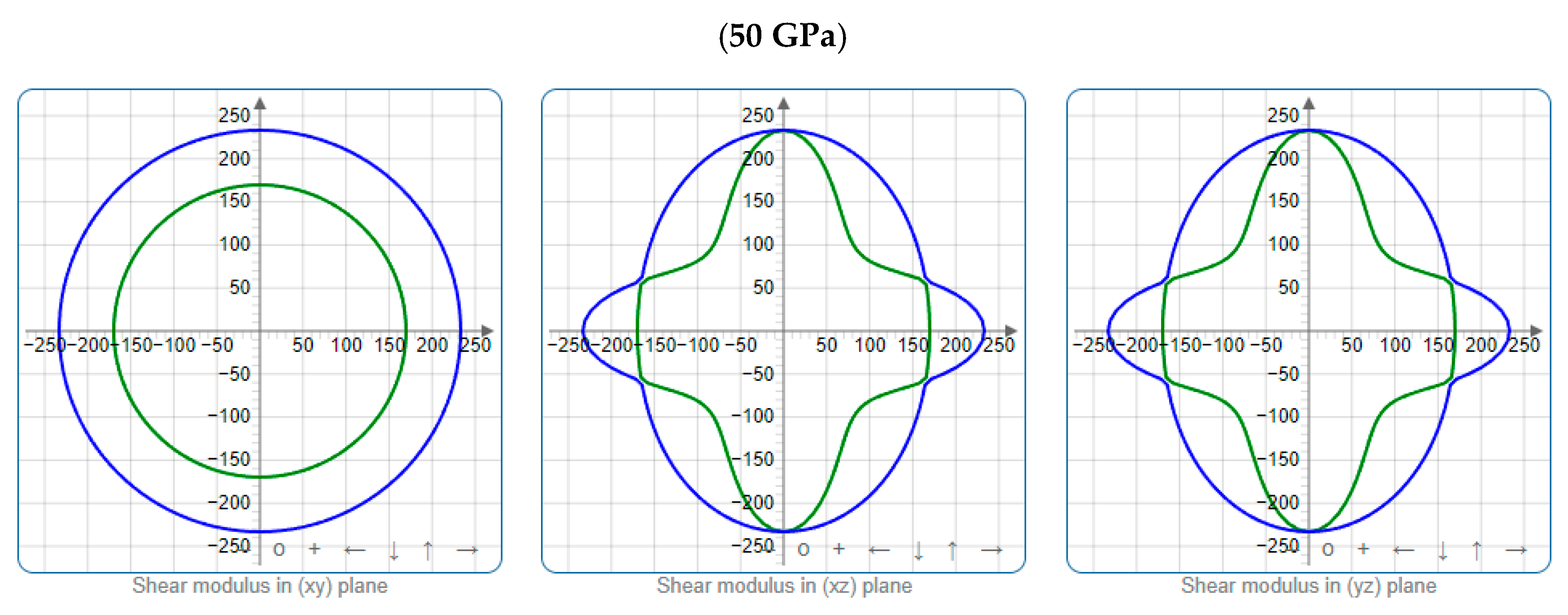
3.3. Mechanical Properties
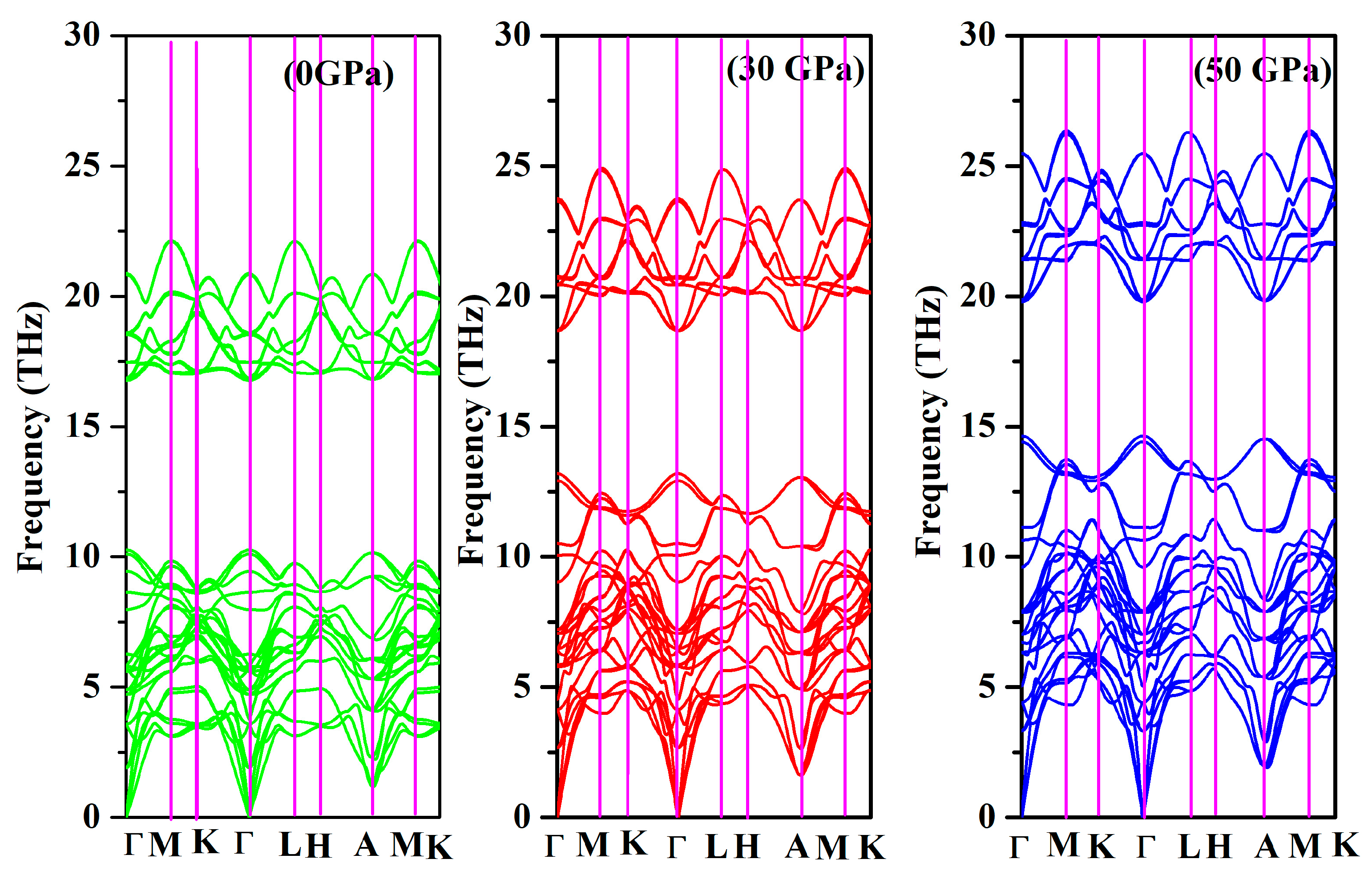
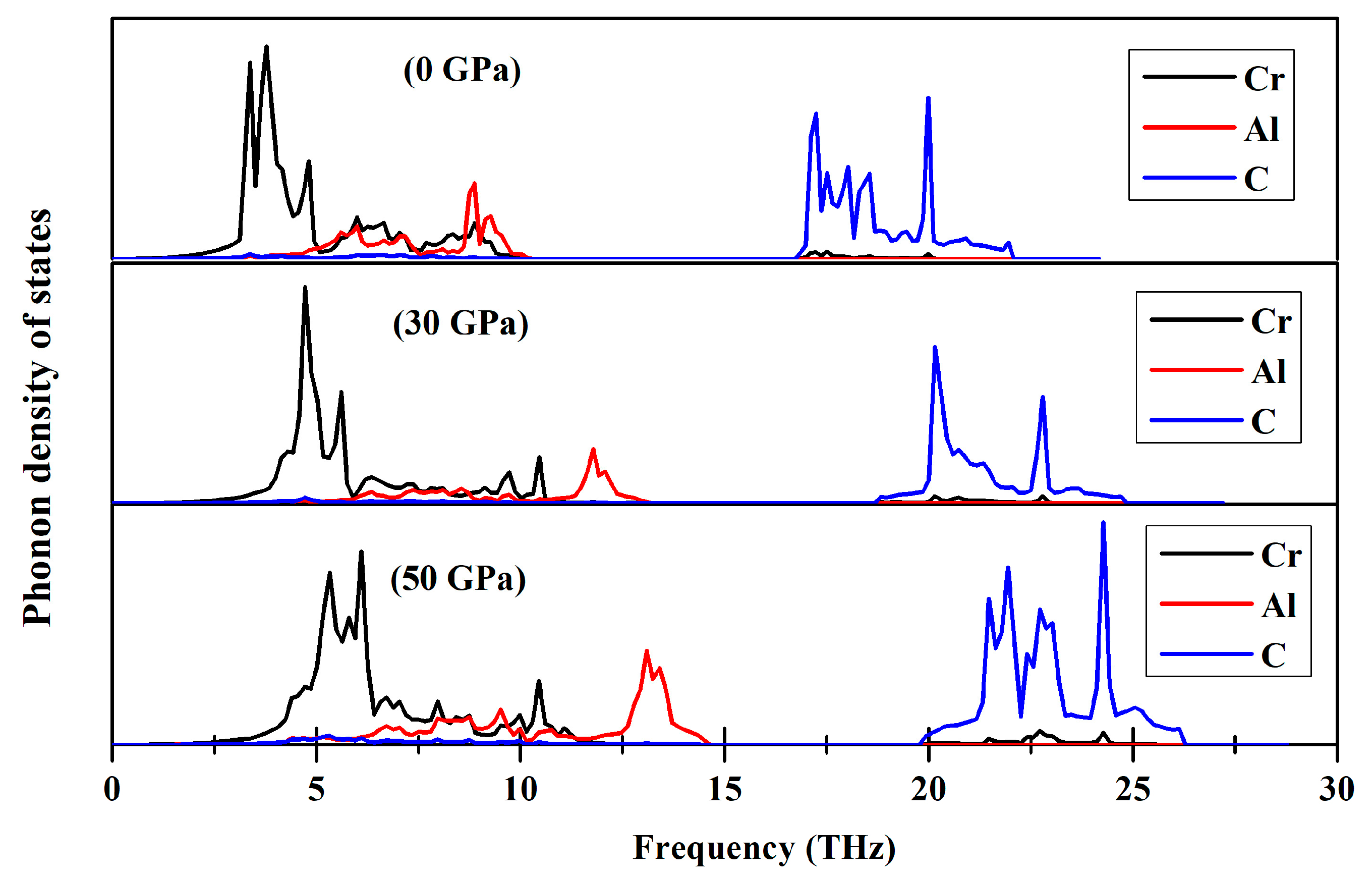
3.4. Dynamical Properties
3.5. Thermal Properties
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Barsoum, M.W.; Brodkin, D.; El-Raghy, T. Layered machinable ceramics for high temperature applications. Scr. Mater. 1997, 36, 535. [Google Scholar] [CrossRef]
- Barsoum, M.W. Thermodynamically stable nanolaminates. Prog. Solid State Chem. Prog. Solid State Chem. 2000, 28, 201. [Google Scholar] [CrossRef]
- Tzenovm, N.V.; Barsoum, M.W. Synthesis and Characterization of Ti3AlC2. J. Am. Ceram. Soc. 2000, 83, 825. [Google Scholar] [CrossRef]
- Sun, Z.; Zhou, Y.; Li, M. High temperature oxidation behavior of Ti3SiC2-based material in air. Acta Mater. 2001, 49, 4347–4353. [Google Scholar] [CrossRef]
- Palmquist, J.P.; Li, S.; Persson, P.O.A.; Emmerlich, J.; Wilhelmsson, O.; Hogberg, H.; Katsnelson, M.I.; Johansson, B.; Ahuja, O.; Eriksson, L.; et al. Mn+1AXn phases in the Ti-Si-C system studied by thin-film synthesis and ab initio calculations. Phys. Rev. B 2004, 70, 165401. [Google Scholar] [CrossRef]
- Xie, X.; Li, X.Q.; Jia, Q.; Bai, C.G.; Malzbende, J.; Cui, Y.Y.; Yang, R. Mechanical properties and toughening mechanisms of highly textured Ti3AlC2 composite material. J. Am. Ceram. Soc. 2022, 42, 5493–5504. [Google Scholar] [CrossRef]
- Wang, J.Y.; Zhou, Y.C. First-principles study of equilibrium properties and electronic structure of Ti3Si0.75Al0.25C2 solid solution. J. Phys. Condens. Matter 2003, 15, 5959. [Google Scholar] [CrossRef]
- Barsoum, M.W.; El-Raghy, T. Synthesis and Characterization of a Remarkable Ceramic: Ti3SiC2. J. Am. Ceram. Soc. 1996, 79, 1953–1956. [Google Scholar] [CrossRef]
- Sun, Z.M.; Music, D.; Ahuja, R.; Schneider, J.M. Theoretical investigation of the bonding and elastic properties of nanolayered ternary nitrides. Phys. Rev. B 2005, 71, 193402. [Google Scholar] [CrossRef]
- Hug, G. Electronic structures of and composition gaps among the ternary carbides Ti2MC. Phys. Rev. B 2006, 74, 184113. [Google Scholar] [CrossRef]
- Daoudi, B.; Yakoubi, A.; Beldi, L.; Bouhafs, B. Full-potential electronic structure of Hf2AlC and Hf2AlN. Acta Mater. 2007, 55, 4161–4165. [Google Scholar] [CrossRef]
- Bai, Y.; He, X.; Li, M.; Sun, Y.; Zhu, C.; Li, Y. Chemical bonding and elastic properties of Ti3AC2 phases (A = Si, Ge, and Sn): A first-principle study. Solid State Sci. 2009, 12, 144–147. [Google Scholar] [CrossRef]
- Ali, M.S.; Islam, A.K.M.A.; Hossain, M.M.; Parvin, F. Phase stability, elastic, electronic, thermal and optical properties of Ti3Al1-xSixC2 (0≤ x ≤1): First principle study. Phys. B 2012, 407, 4221–4228. [Google Scholar] [CrossRef]
- Etzkorn, J.; Ade, M.; Hillebrecht, H. Ta3AlC2 and Ta4AlC3-Single-Crystal Investigations of Two New Ternary Carbides of Tantalum Synthesized by the Molten Metal Technique. Inorg. Chem. 2007, 46, 1410–1418. [Google Scholar] [CrossRef]
- Dubois, S.; Cabioch, T.; Chartier, P.; Gauthier, V.; Jaouen, M. A New Ternary Nanolaminate Carbide: Ti3SnC2. J. Am. Ceram. Soc. 2007, 90, 2642–2644. [Google Scholar] [CrossRef]
- Wang, X.H.; Zhou, Y.C. Oxidation behavior of Ti3AlC2 powders in flowing air. J. Mater. Chem. 2002, 12, 2781–2785. [Google Scholar] [CrossRef]
- Lee, D.B. High-temperature oxidation of Ti3AlC2 between 1173 and 1473 K in air. Mater. Sci. Eng. A Struct. Mater. Prop. Microstruct. Process. 2006, 434, 147–154. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K. Ernzerhof, Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Togo, A.; Oba, F.; Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type and -type at high pressures. Phys. Rev. B 2008, 78, 134106. [Google Scholar] [CrossRef]
- Fan, Q.; Hou, H.J.; Yang, J.H. Effect of pressures on the structural, electronic, optical, elastic, dynamical properties and thermal properties of Mo2C: A study explored by theoretical simulation. Int. J. Refract. Met. H. 2024, 119, 106522. [Google Scholar] [CrossRef]
- He, X.D.; Bai, Y.L.; Zhu, C.C.; Sun, Y.; Li, M.; Barsoum, M.W. General trends in the structural, electronic and elastic properties of the M3AlC2 phases (M = transition metal): A first-principle study. Comp. Mate. Sci. 2010, 49, 691–698. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef]
- Reuss, A. Berechnung der Fließgrenze von Mischkristallen auf Grund der Plastizitätsbedingung für Einkristalle. ZAMM J. Appl. Math. Mech. Zeitschrif Für Angew. Math. Mech. 1929, 9, 49. [Google Scholar] [CrossRef]
- Brazhkin, V.V. High-pressure synthesized materials: Treasures and hints. High Press. Res. 2007, 27, 333. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. Sect. A 1952, 65, 349. [Google Scholar] [CrossRef]
- Pugh, S.F. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos. Mag. 1954, 45, 823. [Google Scholar] [CrossRef]
- Vasilyev, D.; Ikhsanov, R.S.; Zheleznyi, M.; Kartsev, A. Calculations of elastic and thermal properties of the strengthening C14 Fe6Nb4Al2 Laves phase using the density functional theory. J. Mater. Sci. 2025, 60, 5427. [Google Scholar] [CrossRef]
- Chung, D.H.; Buessem, W.R.; Vahldiek, F.W.; Mersol, S.A. Anisotropy in Single Crystal Refractory Compound; Plenum: New York, NY, USA, 1968. [Google Scholar]
- Ranganathan, S.I.; Ostoja-Starzewski, M. Universal elastic anisotropy index. Phys. Rev. Lett. 2008, 1010, 55504. [Google Scholar] [CrossRef]
- Fan, Q.; Zhang, S.R.; Hou, H.J.; Yang, J.H. First-Principles to Explore the Pressure on the Structural, Mechanical, Electronic Structure and Thermodynamic Properties of Nb2C. Int. J. Quantum Chem. 2025, 125, e70040. [Google Scholar] [CrossRef]
- Fan, Q.; Zhang, S.R.; Hou, H.J.; Yang, J.H. Predictions of structural stability, elastic anisotropy and thermodynamic properties of TM5Si3C (TM = Cr, Mo, and W). Vacuum 2023, 208, 111648. [Google Scholar] [CrossRef]
- Gaillac, R.; Pullumbi, P.; Coudert, F.-X. ELATE: An open-source online application for analysis and visualization of elastic tensors. J. Phys. Condens. Matter 2016, 28, 275201. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, M.W.; Ma, X.X.; Tang, G.Z.; Paude, R. Ab initio predictions of structure and physical properties of the Zr2GaC and Hf2GaC MAX phases under pressure. Sci. Rep. 2021, 11, 3260. [Google Scholar] [CrossRef]
- Kabir, M.H.; Hossain, M.M.; Ali, M.A.; Uddin, M.M.; Ali, M.L.; Hasan, M.Z.; Islam, A.K.M.A.; Naqib, S.H. First principles study of mechanical, thermal, electronic, optical and superconducting properties of C40-type germanide-based MGe2 (M = V, Nb and Ta). Results Phys. 2023, 51, 106701. [Google Scholar] [CrossRef]
- Ali, M.A.; Hossain, M.M.; Uddin, M.M.; Islam, A.K.M.A.; Naqib, S.H. Understanding the improvement of thermo-mechanical and optical properties of 212 MAX phase borides Zr2AB2 (A = In, Tl). J. Mater. Res. Technol. 2021, 15, 2227. [Google Scholar] [CrossRef]
- Rana, M.R.; Islam, S.; Hoque, K.; Biswas, G.G.; Hossain, M.E.; Naqib, S.H.; Ali, M.A. DFT prediction of the stability and physical properties of M2GaB (M = Sc, V, Nb, Ta). J. Mater. Res. Technol. 2023, 24, 7795. [Google Scholar] [CrossRef]
- Ali, M.A. Muhammad Waqas Qureshi, DFT insights into the new Hf-based chalcogenide MAX phase Hf2SeC. Vacuum 2022, 201, 111072. [Google Scholar] [CrossRef]
- Ali, M.A.; Hossain, M.M.; Uddin, M.M.; Hossain, M.A.; Islam, A.K.M.A.; Naqib, S.H. Physical properties of new MAX phase borides M2SB (M = Zr, Hf and Nb) in comparison with conventional MAX phase carbides M2SC (M = Zr, Hf and Nb): Comprehensive insights. J. Mater. Res. Technol. 2021, 11, 1000. [Google Scholar] [CrossRef]
- Arab, F.; Sahraoui, F.A.; Haddadi, K.; Bouhemadou, A.; Louail, L. Phase stability, mechanical and thermodynamic properties of orthorhombic and trigonal MgSiN2: An ab initio study. Phase Transit. 2016, 89, 480. [Google Scholar] [CrossRef]
- Lee, C.; Gonze, X. Ab initio calculation of the thermodynamic properties and atomic temperature factors of SiO2 α-quartz and stishovite. Phys. Rev. B 1995, 51, 8610. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pressure (GPa) | a | c | ρ | V | |
|---|---|---|---|---|---|
| 0 | Present | 2.8699 | 17.3922 | 5.5415 | 124.052 |
| 0 | Ref. [23] | 2.8508 | 17.272 | 5.66 | 121.56 |
| 10 | Present | 2.8260 | 17.1720 | 5.7881 | 118.767 |
| 20 | Present | 2.7896 | 16.9888 | 5.9269 | 115.986 |
| 30 | Present | 2.7572 | 16.8486 | 6.0464 | 113.693 |
| 40 | Present | 2.7287 | 16.7252 | 6.1557 | 111.694 |
| 50 | Present | 2.7040 | 16.6027 | 6.2567 | 109.872 |
| Pressure (GPa) | C11 | C12 | C13 | C33 | C44 | C66 | BV | BR | B | GV | GR | G | E | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Present | 0 | 359.5 | 114.8 | 136.8 | 368.9 | 119.0 | 122.4 | 207.2 | 206.9 | 207.0 | 118.7 | 118.5 | 118.6 | 298.8 |
| Theo. [23] | 0 | 381 | 100 | 136 | 381 | 118 | 141 | 210 | 127 | 317 | ||||
| Present | 10 | 408.9 | 134.9 | 169.4 | 415.9 | 148.9 | 137.0 | 242.3 | 241.8 | 242.1 | 137.6 | 136.4 | 137.0 | 345.8 |
| Present | 20 | 446.3 | 152 | 205.3 | 460.2 | 173.6 | 147.2 | 275.3 | 273.9 | 274.6 | 151.6 | 148.1 | 149.8 | 380.3 |
| Present | 30 | 484.7 | 172.7 | 238.4 | 491.7 | 195.3 | 156.0 | 306.7 | 305.0 | 305.8 | 163.4 | 157.0 | 160.2 | 409.2 |
| Present | 40 | 524.5 | 195.5 | 270.7 | 528.5 | 214.8 | 164.5 | 339.0 | 337.0 | 338.0 | 174.9 | 165.8 | 170.3 | 437.5 |
| Present | 50 | 554.4 | 214.9 | 299.0 | 565.0 | 233.2 | 169.7 | 366.6 | 363.8 | 365.2 | 184.6 | 172.7 | 178.6 | 460.8 |
| Pressure (GPa) | G/B | v | C13–C44 | C12–C66 | |
|---|---|---|---|---|---|
| 0 | Present | 0.573 | 0.2595 | 17.8 | −7.6 |
| 0 | Theo. [23] | 0.61 | 0.25 | ||
| 10 | Present | 0.566 | 0.2619 | 20.5 | −2.1 |
| 20 | Present | 0.546 | 0.2692 | 31.7 | 4.8 |
| 30 | Present | 0.524 | 0.2770 | 43.1 | 16.7 |
| 40 | Present | 0.504 | 0.2843 | 55.9 | 31 |
| 50 | Present | 0.493 | 0.2897 | 65.8 | 45.2 |
| Pressure (GPa) | AU | AB (%) | AG (%) | A1 | A2 | A3 |
|---|---|---|---|---|---|---|
| 0 | 0.0064 | 0.0006 | 0.0001 | 1.0466 | 1.0466 | 1 |
| 10 | 0.0508 | 0.0044 | 0.0034 | 1.2255 | 1.2255 | 1 |
| 20 | 0.1206 | 0.0111 | 0.0040 | 1.4003 | 1.4003 | 1 |
| 30 | 0.2135 | 0.0203 | 0.0031 | 1.5637 | 1.5637 | 1 |
| 40 | 0.2817 | 0.0271 | 0.0017 | 1.6794 | 1.6794 | 1 |
| 50 | 0.3588 | 0.0338 | 0.0046 | 1.7890 | 1.7890 | 1 |
| Pressure (GPa) | βmax/βmin | Gmax/Gmin | Emax/Emin |
|---|---|---|---|
| 0 | 1.1565 | 1.08 | 1.042 |
| 10 | 1.2024 | 1.234 | 1.175 |
| 20 | 1.3580 | 1.421 | 1.302 |
| 30 | 1.4025 | 1.599 | 1.44 |
| 40 | 1.4434 | 1.727 | 1.536 |
| 50 | 1.5528 | 1.847 | 1.607 |
| Pressure (GPa) | HChen | HMiao | vl | vt | vm | θ | γa | Tm | k |
|---|---|---|---|---|---|---|---|---|---|
| 0 | 14.03 | 19.02 | 8.1173 | 4.6262 | 5.1417 | 702.4 | 1.5465 | 1985.9 | 1.8586 |
| Ti3AlC2 0 [13] | 9.2 | ||||||||
| Ti3SiC2 0 [13] | 11.1 | ||||||||
| 10 | 15.27 | 21.75 | 8.5666 | 4.8651 | 5.4088 | 749.7 | 1.5591 | 2204.6 | 2.0140 |
| 20 | 15.44 | 23.05 | 8.9460 | 5.0274 | 5.5941 | 781.6 | 1.5966 | 2383.2 | 2.1205 |
| 30 | 15.29 | 23.82 | 9.2684 | 5.1473 | 5.7330 | 806.3 | 1.6386 | 2545.7 | 2.2069 |
| 40 | 15.11 | 24.49 | 9.5810 | 5.2598 | 5.8635 | 829.6 | 1.6794 | 2720.3 | 2.2888 |
| 50 | 14.99 | 25.05 | 9.8199 | 5.3428 | 5.9601 | 847.9 | 1.7109 | 2864.7 | 2.3553 |
| Temperature/K | 0 GPa | 30 GPa | 50 GPa | |||
|---|---|---|---|---|---|---|
| F | E | F | E | F | E | |
| 300 | 43.31 | 114.81 | 62.43 | 122.38 | 72.42 | 126.92 |
| 400 | 15.85 | 139.34 | 39.03 | 145.54 | 50.96 | 149.30 |
| 500 | −18.04 | 165.71 | 9.51 | 170.89 | 23.55 | 174.06 |
| 600 | −57.36 | 193.14 | −25.26 | 197.57 | −9.00 | 200.30 |
| 700 | −101.32 | 221.23 | −64.57 | 225.09 | −46.02 | 227.47 |
| 800 | −149.34 | 249.75 | −107.86 | 253.17 | −86.99 | 255.28 |
| 900 | −200.95 | 278.57 | −154.69 | 281.63 | −131.46 | 283.53 |
| 1000 | −255.79 | 307.61 | −204.71 | 310.38 | −179.10 | 312.09 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Fan, S.; Hou, H.; Fan, Q. DFT Prediction of Structural and Physical Properties of Cr3AlC2 Under Pressure. Nanomaterials 2025, 15, 1082. https://doi.org/10.3390/nano15141082
Yang J, Fan S, Hou H, Fan Q. DFT Prediction of Structural and Physical Properties of Cr3AlC2 Under Pressure. Nanomaterials. 2025; 15(14):1082. https://doi.org/10.3390/nano15141082
Chicago/Turabian StyleYang, Jianhui, Shenghai Fan, Haijun Hou, and Qiang Fan. 2025. "DFT Prediction of Structural and Physical Properties of Cr3AlC2 Under Pressure" Nanomaterials 15, no. 14: 1082. https://doi.org/10.3390/nano15141082
APA StyleYang, J., Fan, S., Hou, H., & Fan, Q. (2025). DFT Prediction of Structural and Physical Properties of Cr3AlC2 Under Pressure. Nanomaterials, 15(14), 1082. https://doi.org/10.3390/nano15141082