
Efficient Production of Self-Assembled Bioconjugate Nanovaccines against Klebsiella pneumoniae O2 Serotype in Engineered Escherichia coli


 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Strains and Plasmids
2.2. Bacterial Culture
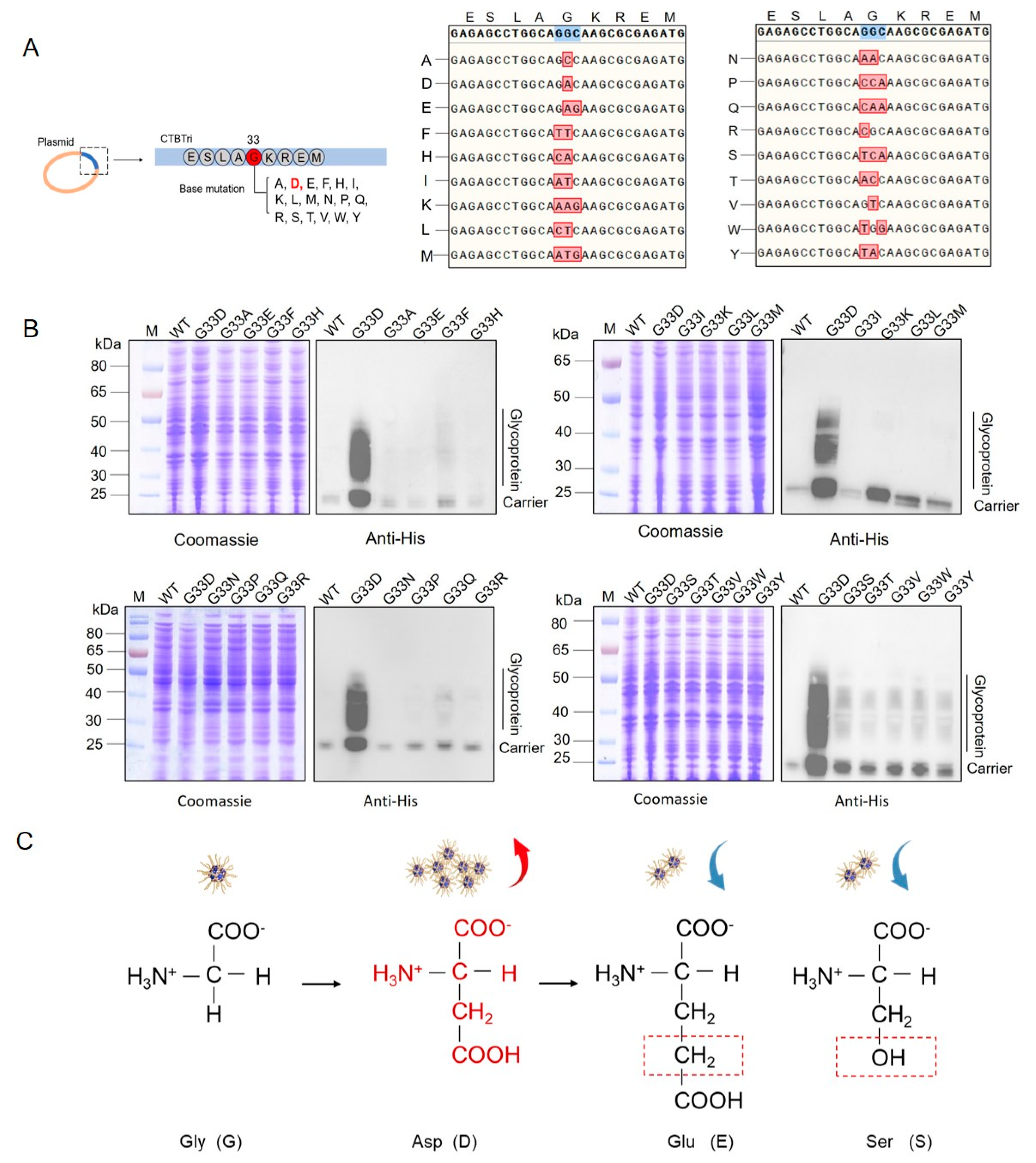
2.3. Base Mutation
2.4. Immunoblotting
2.5. Glycoprotein Purification
2.6. Experimental Animals
2.7. Immunization Experiments
2.8. ELISA
2.9. Statistical Analysis
3. Results
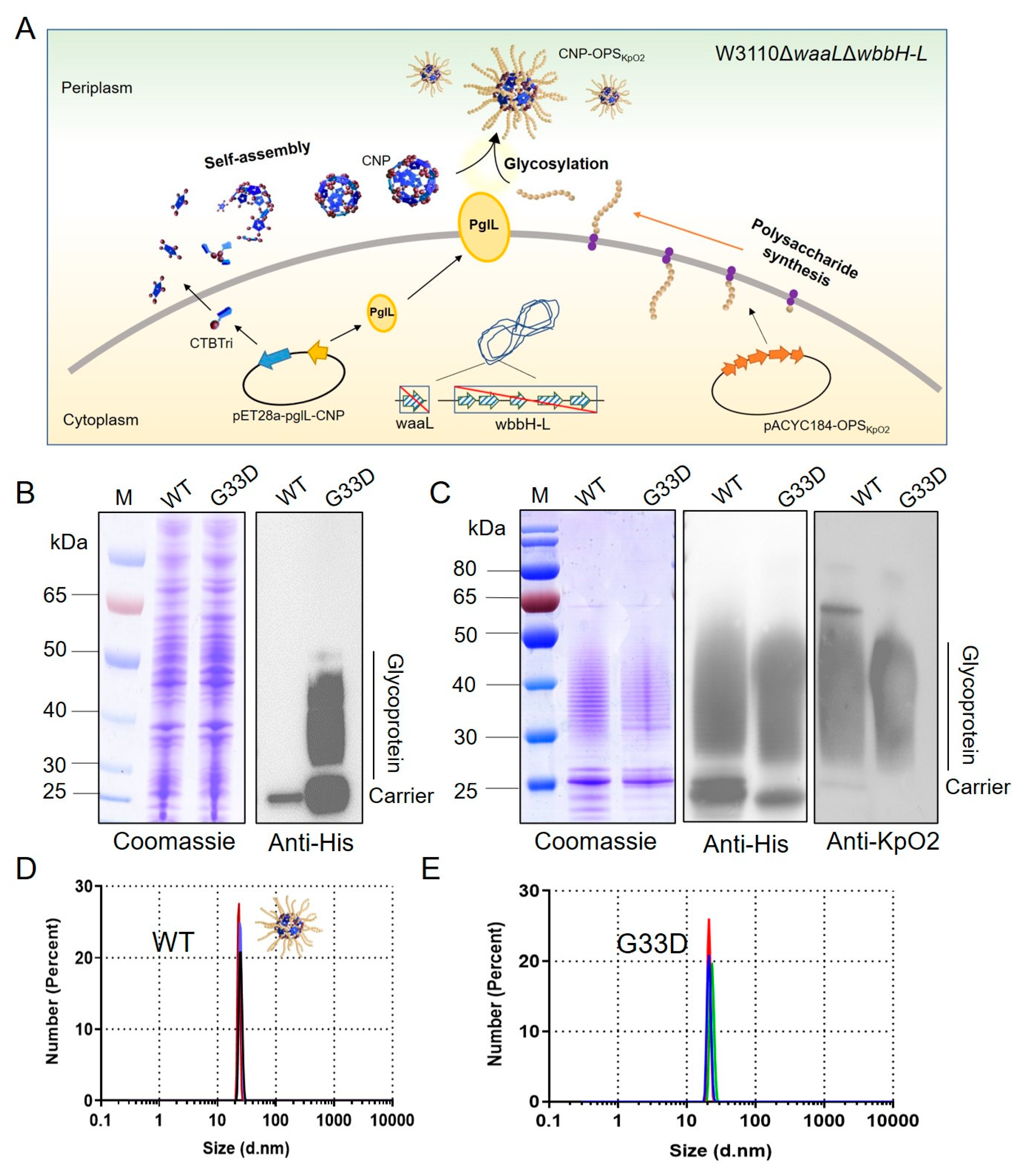
3.1. G33D Mutation of CTBTri Dramatically Increased the Expression of Glycoprotein
3.2. Analysis of Glycosylation of the Various Mutations at the 33rd Position in the CNP
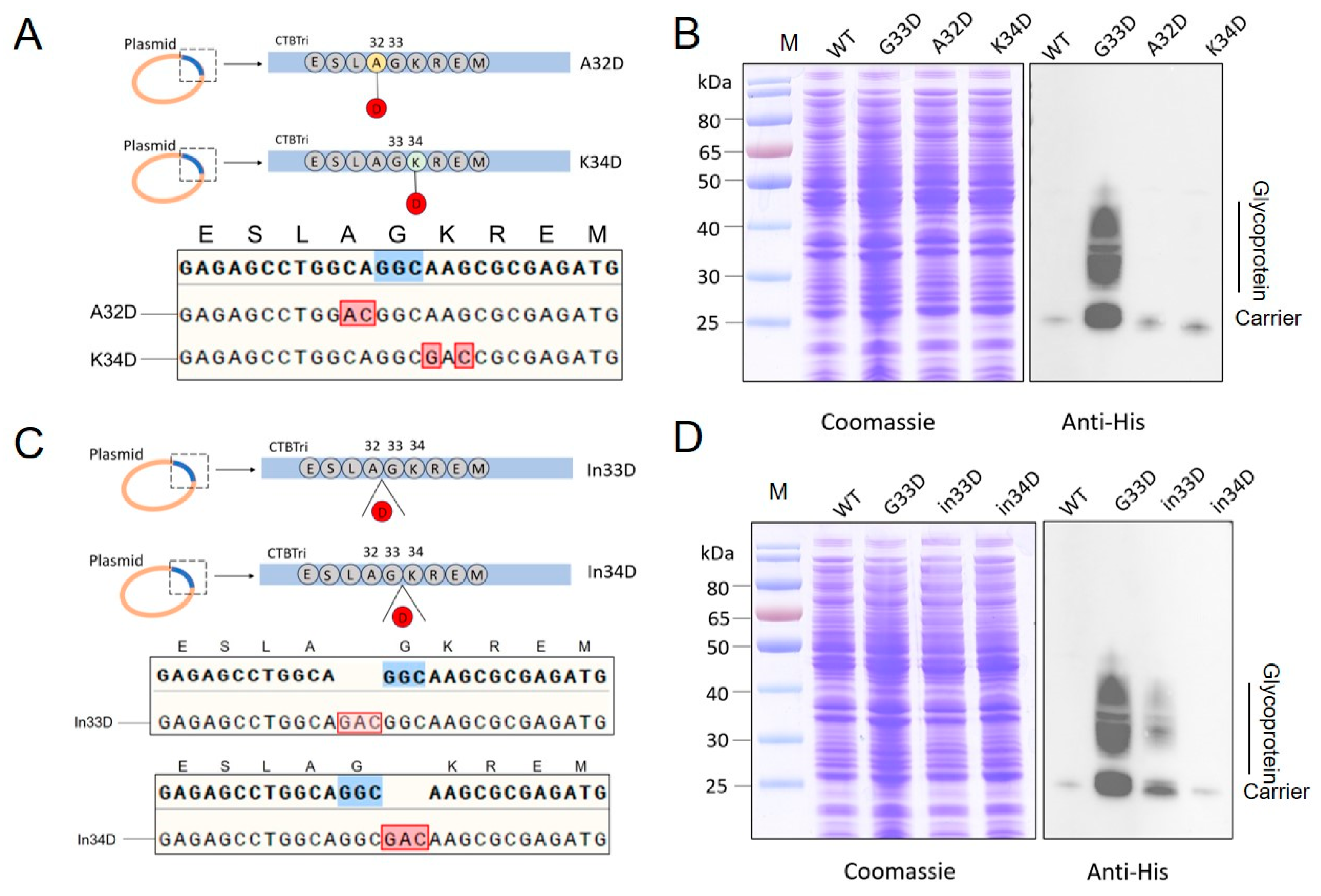
3.3. Evaluation of the Glycosylation by Mutating Animo Acids to D around the 33rd Position
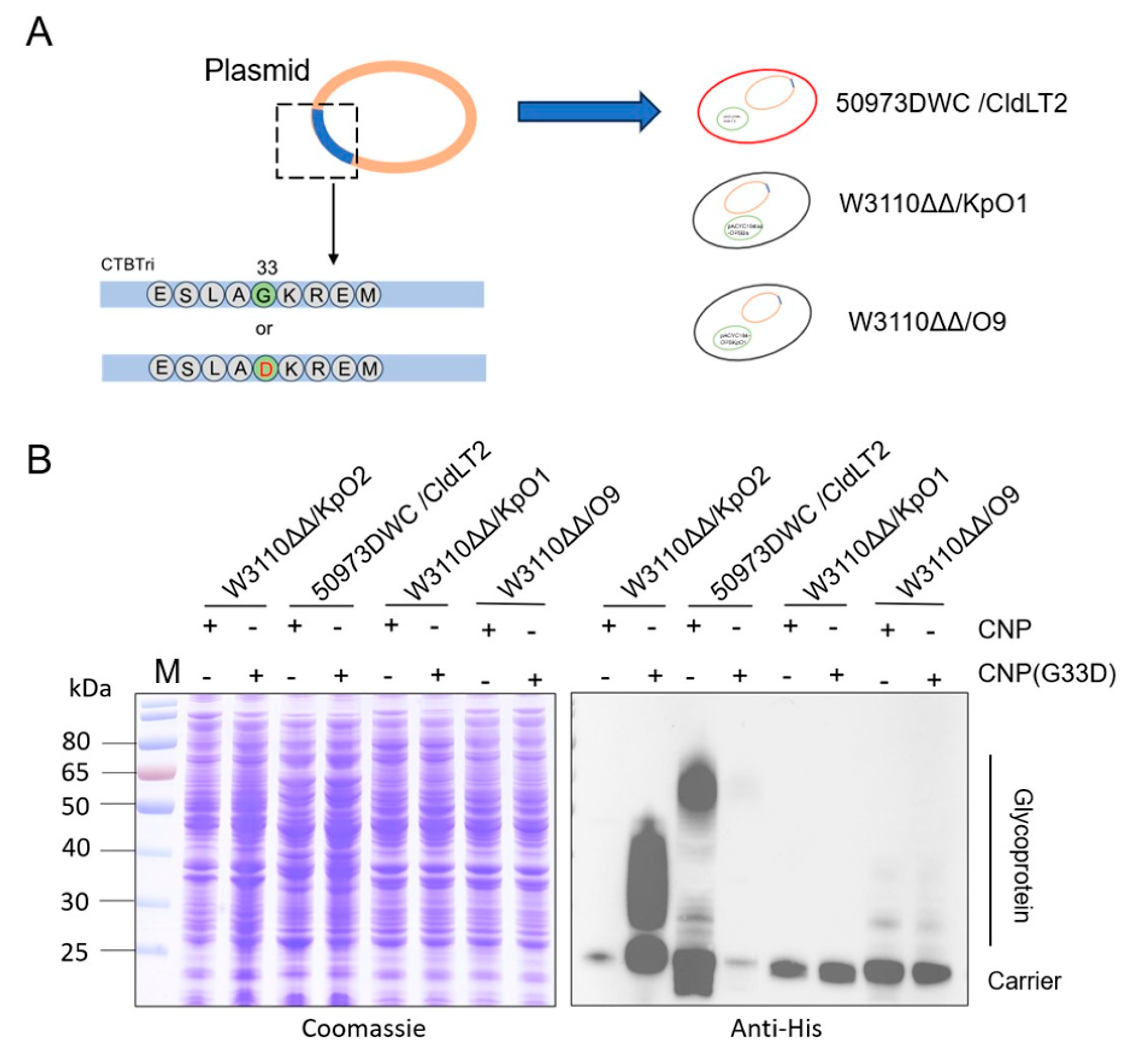
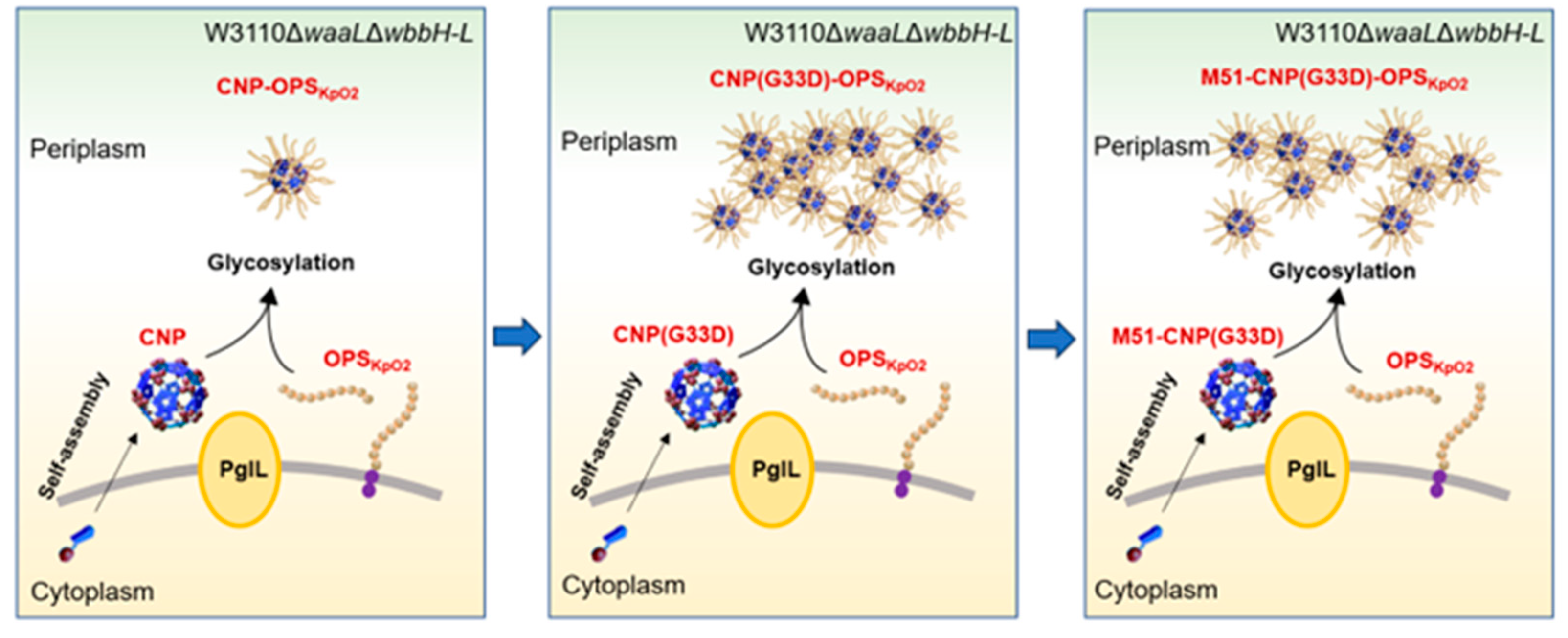
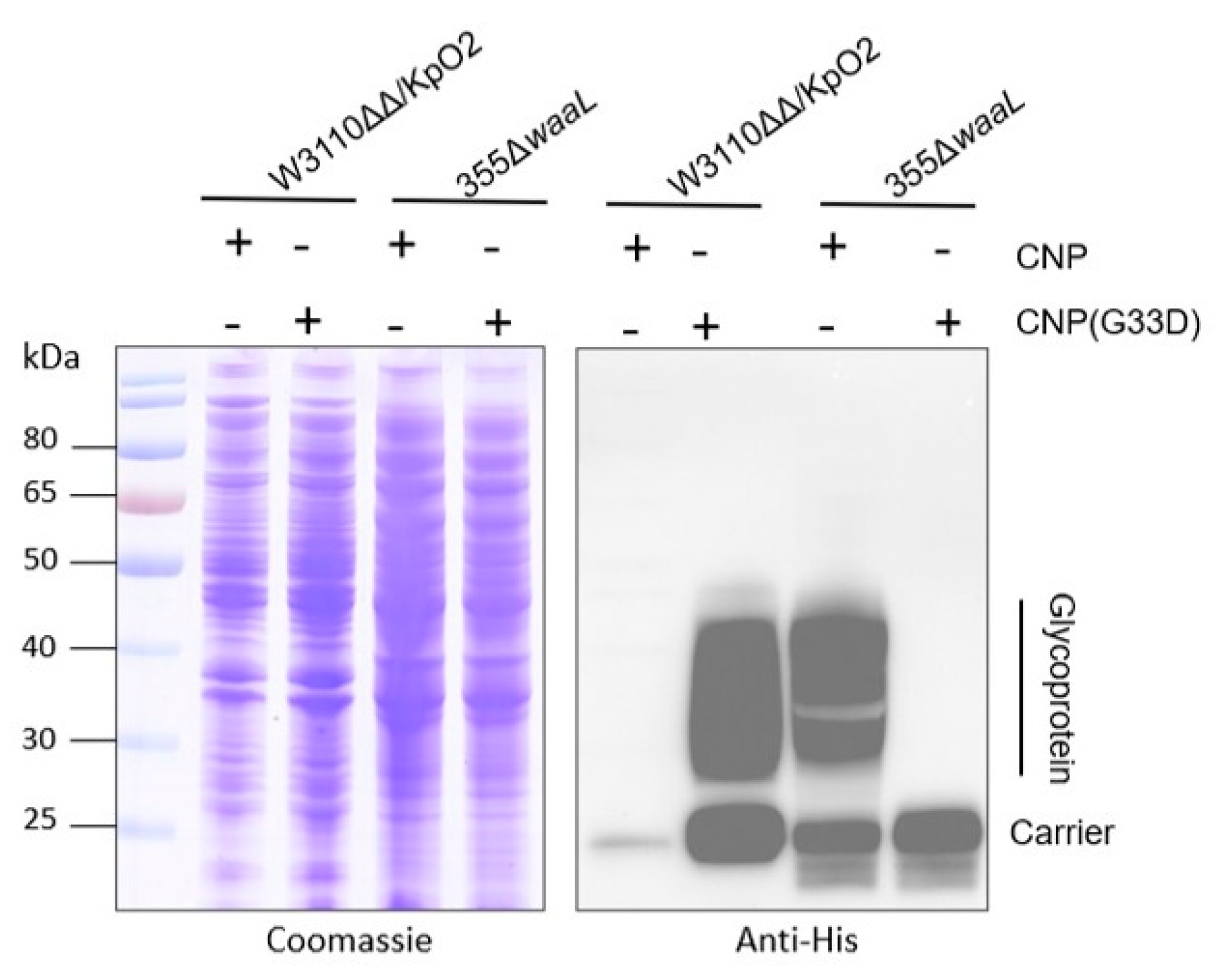
3.4. Glycosylation Analysis of CNP(G33D) for Various OPSs
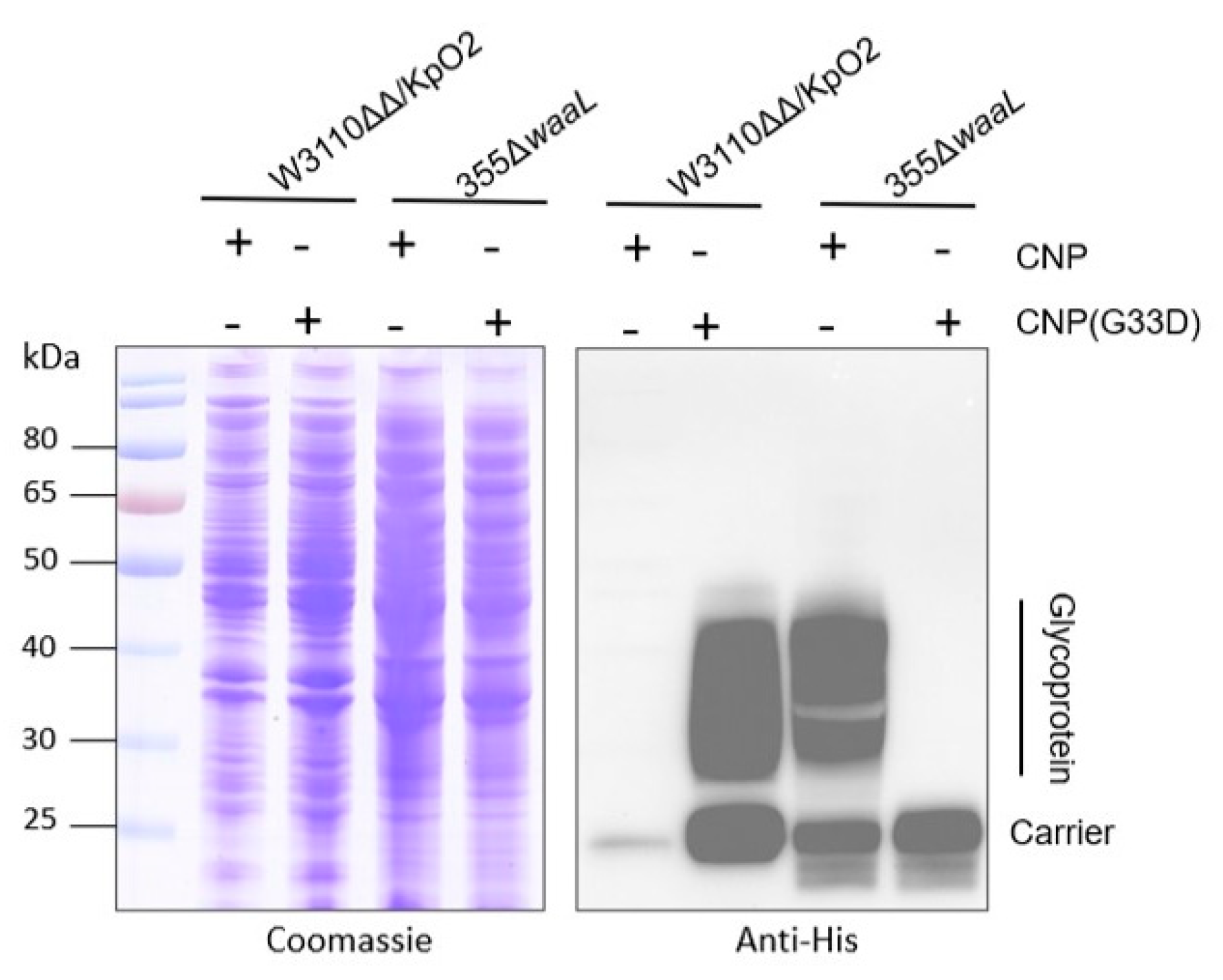
3.5. Analysis of Glycosylation of CNP(G33D) in Kp355
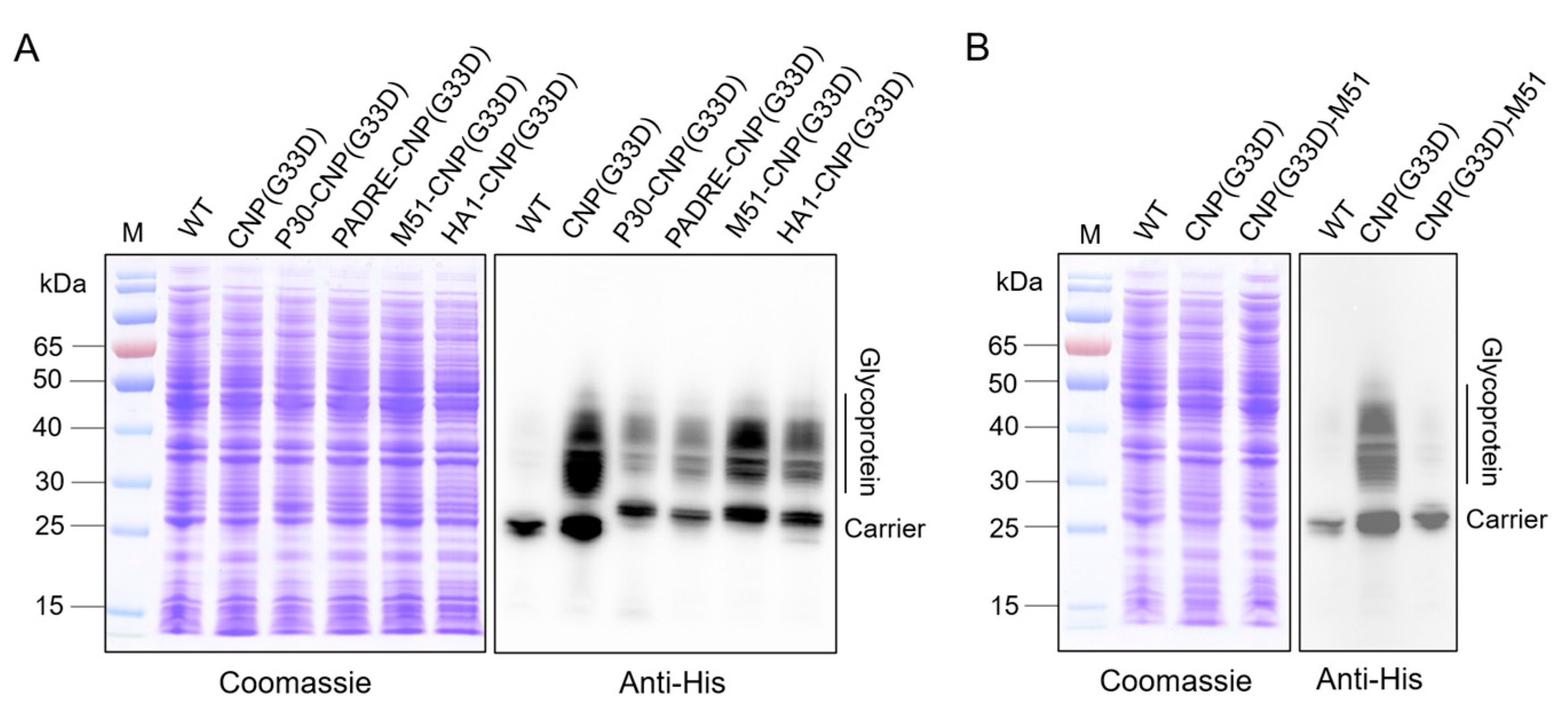
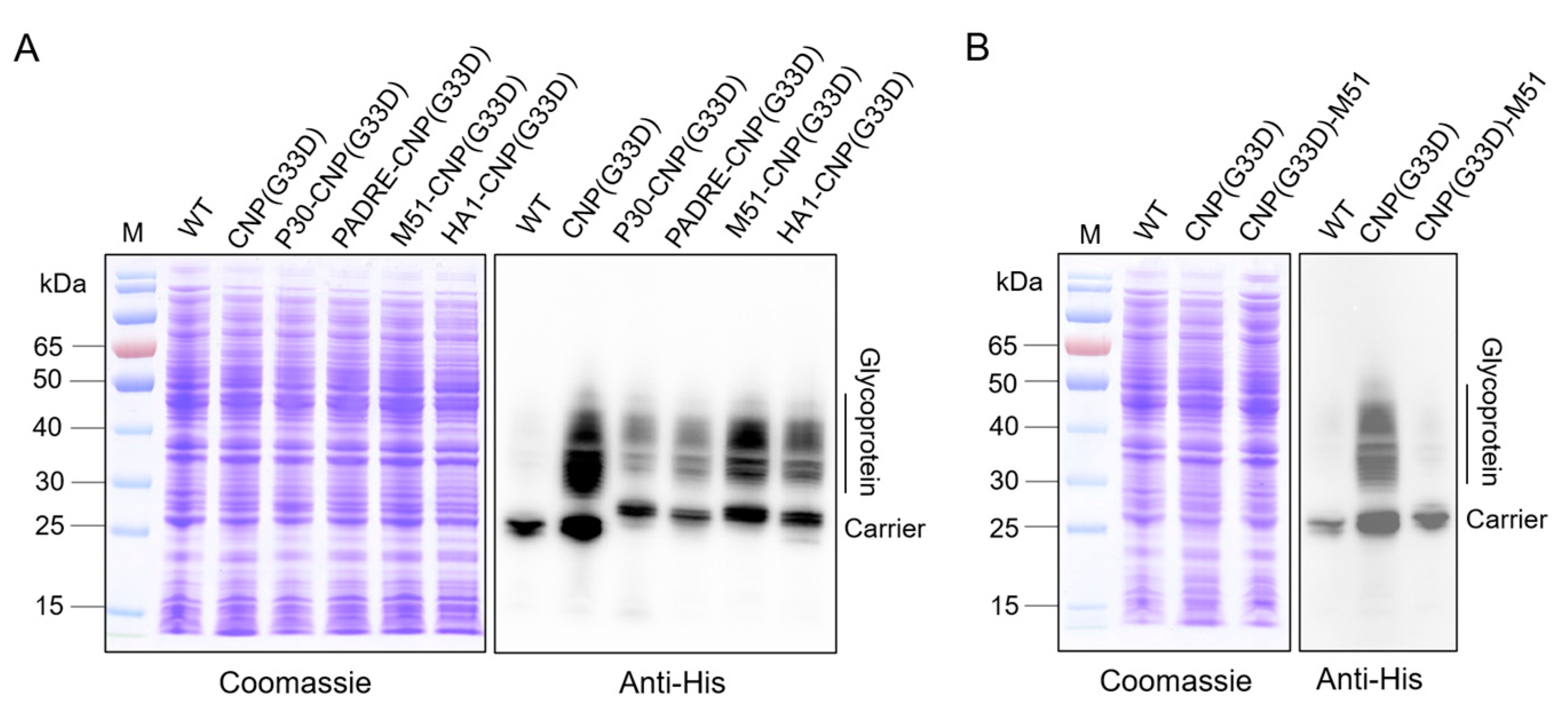
3.6. Analysis of Glycosylation of CNP(G33D) Fused with Different T-Cell Epitopes
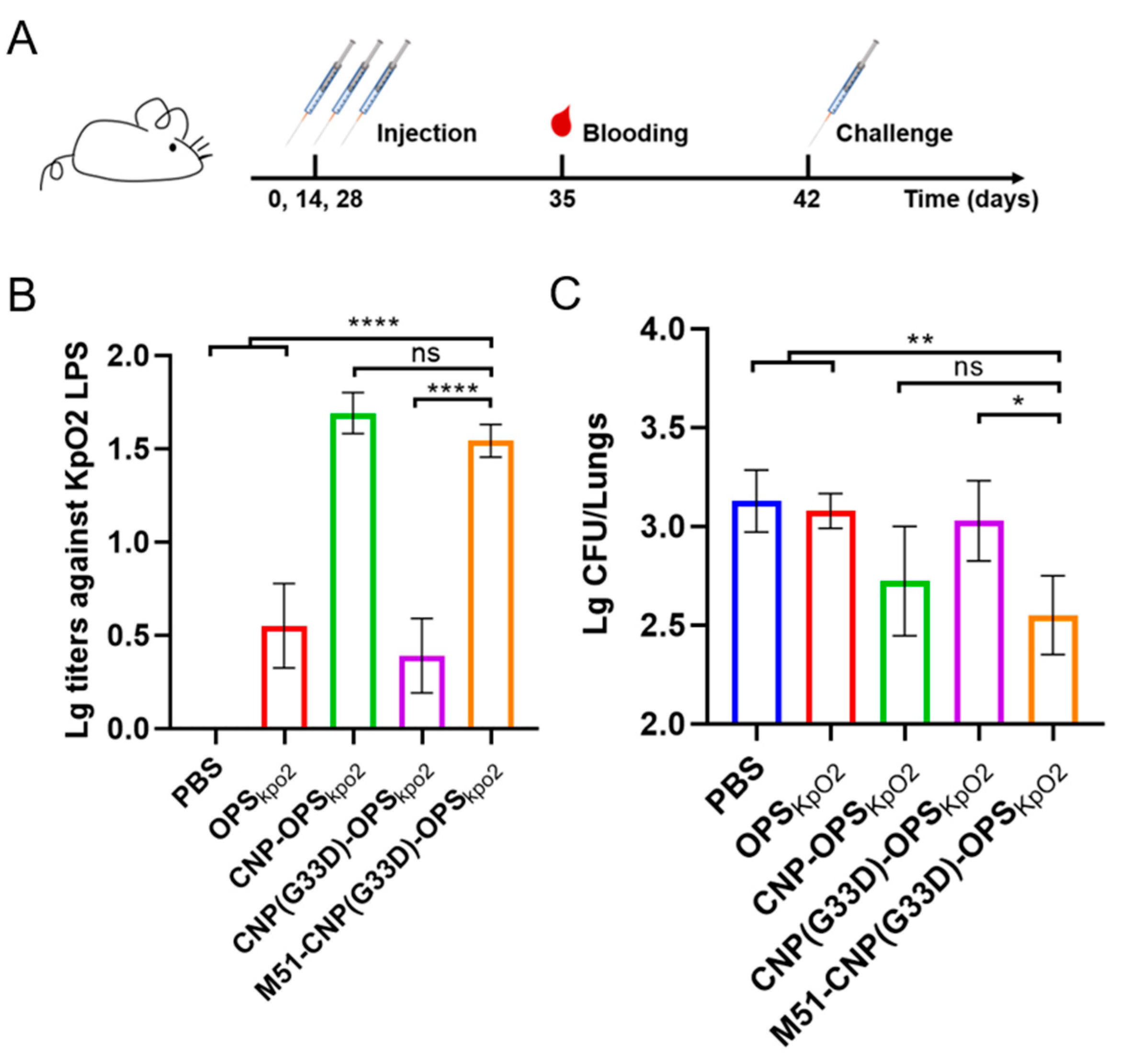
3.7. Evaluation of the Efficacy of Various Nanovaccines
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Pennini, M.E.; De Marco, A.; Pelletier, M.; Bonnell, J.; Cvitkovic, R.; Beltramello, M.; Cameroni, E.; Bianchi, S.; Zatta, F.; Zhao, W.; et al. Immune stealth-driven O2 serotype prevalence and potential for therapeutic antibodies against multidrug resistant Klebsiella pneumoniae. Nat. Commun. 2017, 8, 1991. [Google Scholar] [CrossRef]
- Castanheira, M.; Deshpande, L.M.; Mendes, R.E.; Canton, R.; Sader, H.S.; Jones, R.N. Variations in the Occurrence of Resistance Phenotypes and Carbapenemase Genes Among Enterobacteriaceae Isolates in 20 Years of the SENTRY Antimicrobial Surveillance Program. Open Forum Infect. Dis. 2019, 6, S23–S33. [Google Scholar] [CrossRef]
- Pitout, J.D.; Nordmann, P.; Poirel, L. Carbapenemase-Producing Klebsiella pneumoniae, a Key Pathogen Set for Global Nosocomial Dominance. Antimicrob. Agents Chemother. 2015, 59, 5873–5884. [Google Scholar] [CrossRef]
- Munoz-Price, L.S.; Poirel, L.; Bonomo, R.A.; Schwaber, M.J.; Daikos, G.L.; Cormican, M.; Cornaglia, G.; Garau, J.; Gniadkowski, M.; Hayden, M.K.; et al. Clinical epidemiology of the global expansion of Klebsiella pneumoniae carbapenemases. Lancet Infect. Dis. 2013, 13, 785–796. [Google Scholar] [CrossRef]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Raetz, C.R.; Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef] [PubMed]
- Soliman, C.; Pier, G.B.; Ramsland, P.A. Antibody recognition of bacterial surfaces and extracellular polysaccharides. Curr. Opin. Struct. Biol. 2020, 62, 48–55. [Google Scholar] [CrossRef]
- Jones, C. Vaccines based on the cell surface carbohydrates of pathogenic bacteria. An. Da Acad. Bras. De. Cienc. 2005, 77, 293–324. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Bai, J.; Yuan, J.; Zhang, H.; Lu, G.; Wang, Y.; Jiang, L.; Liu, B.; Wang, L.; Huang, D.; et al. High efficiency biosynthesis of O-polysaccharide-based vaccines against extraintestinal pathogenic Escherichia coli. Carbohydr. Polym. 2021, 255, 117475. [Google Scholar] [CrossRef]
- Rappuoli, R. Glycoconjugate vaccines: Principles and mechanisms. Sci. Transl. Med. 2018, 10, eaat4615. [Google Scholar] [CrossRef]
- Szymanski, C.M.; Yao, R.; Ewing, C.P.; Trust, T.J.; Guerry, P. Evidence for a system of general protein glycosylation in Campylobacter jejuni. Mol. Microbiol. 1999, 32, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- Terra, V.S.; Mills, D.C.; Yates, L.E.; Abouelhadid, S.; Cuccui, J.; Wren, B.W. Recent developments in bacterial protein glycan coupling technology and glycoconjugate vaccine design. J. Med. Microbiol. 2012, 61, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Kay, E.; Cuccui, J.; Wren, B.W. Recent advances in the production of recombinant glycoconjugate vaccines. NPJ Vaccines 2019, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Hug, I.; Feldman, M.F. Analogies and homologies in lipopolysaccharide and glycoprotein biosynthesis in bacteria. Glycobiology 2011, 21, 138–151. [Google Scholar] [CrossRef]
- Pan, C.; Sun, P.; Liu, B.; Liang, H.; Peng, Z.; Dong, Y.; Wang, D.; Liu, X.; Wang, B.; Zeng, M.; et al. Biosynthesis of Conjugate Vaccines Using an O-Linked Glycosylation System. mBio 2016, 7, e00443-00416. [Google Scholar] [CrossRef]
- Harding, C.M.; Feldman, M.F. Glycoengineering bioconjugate vaccines, therapeutics, and diagnostics in E. coli. Glycobiology 2019, 29, 519–529. [Google Scholar] [CrossRef]
- Feldman, M.F.; Wacker, M.; Hernandez, M.; Hitchen, P.G.; Marolda, C.L.; Kowarik, M.; Morris, H.R.; Dell, A.; Valvano, M.A.; Aebi, M. Engineering N-linked protein glycosylation with diverse O antigen lipopolysaccharide structures in Escherichia coli. Proc. Natl. Acad. Sci. USA 2005, 102, 3016–3021. [Google Scholar] [CrossRef] [PubMed]
- Cuccui, J.; Wren, B. Hijacking bacterial glycosylation for the production of glycoconjugates, from vaccines to humanised glycoproteins. J. Pharm. Pharmacol. 2015, 67, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Dow, J.M.; Mauri, M.; Scott, T.A.; Wren, B.W. Improving protein glycan coupling technology (PGCT) for glycoconjugate vaccine production. Expert. Rev. Vaccines 2020, 19, 507–527. [Google Scholar] [CrossRef]
- Faridmoayer, A.; Fentabil, M.A.; Mills, D.C.; Klassen, J.S.; Feldman, M.F. Functional characterization of bacterial oligosaccharyltransferases involved in O-linked protein glycosylation. J. Bacteriol. 2007, 189, 8088–8098. [Google Scholar] [CrossRef]
- Faridmoayer, A.; Fentabil, M.A.; Haurat, M.F.; Yi, W.; Woodward, R.; Wang, P.G.; Feldman, M.F. Extreme substrate promiscuity of the Neisseria oligosaccharyl transferase involved in protein O-glycosylation. J. Biol. Chem. 2008, 283, 34596–34604. [Google Scholar] [CrossRef] [PubMed]
- Harding, C.M.; Nasr, M.A.; Scott, N.E.; Goyette-Desjardins, G.; Nothaft, H.; Mayer, A.E.; Chavez, S.M.; Huynh, J.P.; Kinsella, R.L.; Szymanski, C.M.; et al. A platform for glycoengineering a polyvalent pneumococcal bioconjugate vaccine using E. coli as a host. Nat. Commun. 2019, 10, 891. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Wu, J.; Wang, K.; Li, X.; Sun, P.; Zhang, L.; Huang, J.; Liu, Y.; Hua, X.; Yu, Y.; et al. Production of a Promising Biosynthetic Self-Assembled Nanoconjugate Vaccine against Klebsiella Pneumoniae Serotype O2 in a General Escherichia Coli Host. Adv. Sci. 2021, 8, e2100549. [Google Scholar] [CrossRef] [PubMed]
- Ihssen, J.; Haas, J.; Kowarik, M.; Wiesli, L.; Wacker, M.; Schwede, T.; Thöny-Meyer, L. Increased efficiency of Campylobacter jejuni N-oligosaccharyltransferase PglB by structure-guided engineering. Open Biol. 2015, 5, 140227. [Google Scholar] [CrossRef] [PubMed]
- Pichichero, M.E. Protein carriers of conjugate vaccines: Characteristics, development, and clinical trials. Hum. Vaccines Immunother. 2013, 9, 2505–2523. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.M.; Simon, J.K.; Baker, J.R., Jr. Applications of nanotechnology for immunology. Nat. Rev. Immunol. 2013, 13, 592–605. [Google Scholar] [CrossRef]
- Pan, C.; Yue, H.; Zhu, L.; Ma, G.H.; Wang, H.L. Prophylactic vaccine delivery systems against epidemic infectious diseases. Adv. Drug Deliv. Rev. 2021, 176, 113867. [Google Scholar] [CrossRef]
- Reddy, S.T.; van der Vlies, A.J.; Simeoni, E.; Angeli, V.; Randolph, G.J.; O’Neil, C.P.; Lee, L.K.; Swartz, M.A.; Hubbell, J.A. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotechnol. 2007, 25, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Manolova, V.; Flace, A.; Bauer, M.; Schwarz, K.; Saudan, P.; Bachmann, M.F. Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol. 2008, 38, 1404–1413. [Google Scholar] [CrossRef]
- Cho, N.H.; Cheong, T.C.; Min, J.H.; Wu, J.H.; Lee, S.J.; Kim, D.; Yang, J.S.; Kim, S.; Kim, Y.K.; Seong, S.Y. A multifunctional core-shell nanoparticle for dendritic cell-based cancer immunotherapy. Nat. Nanotechnol. 2011, 6, 675–682. [Google Scholar] [CrossRef]
- Zhou, J.; Kroll, A.V.; Holay, M.; Fang, R.H.; Zhang, L. Biomimetic Nanotechnology toward Personalized Vaccines. Adv. Mater. 2020, 32, e1901255. [Google Scholar] [CrossRef] [PubMed]
- Fuenmayor, J.; Gòdia, F.; Cervera, L. Production of virus-like particles for vaccines. New Biotechnol. 2017, 39, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, X.; Bian, Y.; Wang, S.; Chai, Q.; Guo, Z.; Wang, Z.; Zhu, P.; Peng, H.; Yan, X.; et al. Dual-targeting nanoparticle vaccine elicits a therapeutic antibody response against chronic hepatitis B. Nat. Nanotechnol. 2020, 15, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Pan, C.; Wang, K.; Guo, Y.; Sun, Y.; Li, X.; Sun, P.; Wu, J.; Wang, H.; Zhu, L. Preparation of a Klebsiella pneumoniae conjugate nanovaccine using glycol-engineered Escherichia coli. Microb. Cell Factories 2023, 22, 95. [Google Scholar] [CrossRef] [PubMed]
- Raman, S.; Machaidze, G.; Lustig, A.; Aebi, U.; Burkhard, P. Structure-based design of peptides that self-assemble into regular polyhedral nanoparticles. Nanomed. Nanotechnol. Biol. Med. 2006, 2, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.S.; Boyken, S.E.; Baker, D. The coming of age of de novo protein design. Nature 2016, 537, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Wu, J.; Qing, S.; Zhang, X.; Zhang, L.; Yue, H.; Zeng, M.; Wang, B.; Yuan, Z.; Qiu, Y.; et al. Biosynthesis of Self-Assembled Proteinaceous Nanoparticles for Vaccination. Adv. Mater. 2020, 32, e2002940. [Google Scholar] [CrossRef] [PubMed]
- Ye, T.; Jiao, Z.; Li, X.; He, Z.; Li, Y.; Yang, F.; Zhao, X.; Wang, Y.; Huang, W.; Qin, M.; et al. Inhaled SARS-CoV-2 vaccine for single-dose dry powder aerosol immunization. Nature 2023, 624, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Reglinski, M.; Ercoli, G.; Plumptre, C.; Kay, E.; Petersen, F.C.; Paton, J.C.; Wren, B.W.; Brown, J.S. A recombinant conjugated pneumococcal vaccine that protects against murine infections with a similar efficacy to Prevnar-13. NPJ Vaccines 2018, 3, 53. [Google Scholar] [CrossRef]
- Sánchez, J.; Holmgren, J. Cholera toxin structure, gene regulation and pathophysiological and immunological aspects. Cell. Mol. Life Sci. CMLS 2008, 65, 1347–1360. [Google Scholar] [CrossRef]
- Jobling, M.G.; Holmes, R.K. Mutational analysis of ganglioside GM1-binding ability, pentamer formation, and epitopes of cholera toxin B (CTB) subunits and CTB/heat-labile enterotoxin B subunit chimeras. Infect. Immun. 2002, 70, 1260–1271. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Quinn, P.J. Lipopolysaccharide: Biosynthetic pathway and structure modification. Prog. Lipid Res. 2010, 49, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zu, S.; Zhang, D.; Zhao, Z.; Ji, Y.; Xi, H.; Shan, X.; Qian, A.; Han, W.; Gu, J. Oral vaccination with recombinant Lactobacillus casei expressing Aha1 fused with CTB as an adjuvant against Aeromonas veronii in common carp (Cyprinus carpio). Microb. Cell Fact. 2022, 21, 114. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, J.; Lycke, N.; Czerkinsky, C. Cholera toxin and cholera B subunit as oral-mucosal adjuvant and antigen vector systems. Vaccine 1993, 11, 1179–1184. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, T. Cholera Toxin Subunit B as Adjuvant—An Accelerator in Protective Immunity and a Break in Autoimmunity. Vaccines 2015, 3, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Firdaus, F.Z.; Skwarczynski, M.; Toth, I. Developments in Vaccine Adjuvants. Methods Mol. Biol. 2022, 2412, 145–178. [Google Scholar] [CrossRef] [PubMed]
- Qiao, N.; Du, G.; Zhong, X.; Sun, X. Recombinant lactic acid bacteria as promising vectors for mucosal vaccination. Exploration 2021, 1, 20210026. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, X.; Guo, Y.; Wu, Y.; Yin, L.; Tu, L.; Hong, S.; Cai, H.; Ding, F. Synthetic self-adjuvanted multivalent Mucin 1 (MUC1) glycopeptide vaccines with improved in vivo antitumor efficacy. MedComm 2024, 5, e484. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Sun, Z.Y.; Huang, Z.H.; Shi, L.; Zhao, Y.F.; Kunz, H.; Li, Y.M. Fully synthetic self-adjuvanting thioether-conjugated glycopeptide-lipopeptide antitumor vaccines for the induction of complement-dependent cytotoxicity against tumor cells. Chemistry 2013, 19, 1962–1970. [Google Scholar] [CrossRef]
- Gaidzik, N.; Westerlind, U.; Kunz, H. The development of synthetic antitumour vaccines from mucin glycopeptide antigens. Chem. Soc. Rev. 2013, 42, 4421–4442. [Google Scholar] [CrossRef]
- Nuhn, L.; Hartmann, S.; Palitzsch, B.; Gerlitzki, B.; Schmitt, E.; Zentel, R.; Kunz, H. Water-soluble polymers coupled with glycopeptide antigens and T-cell epitopes as potential antitumor vaccines. Angew. Chem. Int. Ed. Engl. 2013, 52, 10652–10656. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Pan, C.; Cheng, P.; Wang, J.; Zhao, G.; Wu, X. Peptide-Based Vaccines for Tuberculosis. Front. Immunol. 2022, 13, 830497. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid | Characteristic | Source |
|---|---|---|
| pACYC184-OPSKpO1 | Encoded O1 serotype OPS of K. pneumoniae, Cmr | Laboratory stock |
| pACYC184-OPSKpO2 | Encoded O2 serotype OPS of K. pneumoniae, Cmr | Laboratory stock |
| pET28a-pglL-CNP | Encoded PglL and CNP, Kanr | Laboratory stock |
| pACYC184tac-OPSBa | Encoded OPS of YeO9, Cmr | Laboratory stock |
| pET28a-pglL-CNP(G33D) | Encoded PglL and CNP(G33D), Kanr | This work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Sun, P.; Li, T.; Li, J.; Ye, J.; Li, X.; Wu, J.; Lu, Y.; Zhu, L.; Wang, H.; et al. Efficient Production of Self-Assembled Bioconjugate Nanovaccines against Klebsiella pneumoniae O2 Serotype in Engineered Escherichia coli. Nanomaterials 2024, 14, 728. https://doi.org/10.3390/nano14080728
Zhang Y, Sun P, Li T, Li J, Ye J, Li X, Wu J, Lu Y, Zhu L, Wang H, et al. Efficient Production of Self-Assembled Bioconjugate Nanovaccines against Klebsiella pneumoniae O2 Serotype in Engineered Escherichia coli. Nanomaterials. 2024; 14(8):728. https://doi.org/10.3390/nano14080728
Chicago/Turabian StyleZhang, Yan, Peng Sun, Ting Li, Juntao Li, Jingqin Ye, Xiang Li, Jun Wu, Ying Lu, Li Zhu, Hengliang Wang, and et al. 2024. "Efficient Production of Self-Assembled Bioconjugate Nanovaccines against Klebsiella pneumoniae O2 Serotype in Engineered Escherichia coli" Nanomaterials 14, no. 8: 728. https://doi.org/10.3390/nano14080728
APA StyleZhang, Y., Sun, P., Li, T., Li, J., Ye, J., Li, X., Wu, J., Lu, Y., Zhu, L., Wang, H., & Pan, C. (2024). Efficient Production of Self-Assembled Bioconjugate Nanovaccines against Klebsiella pneumoniae O2 Serotype in Engineered Escherichia coli. Nanomaterials, 14(8), 728. https://doi.org/10.3390/nano14080728