

Improved Method for Electron Powder Diffraction-Based Rietveld Analysis of Nanomaterials



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
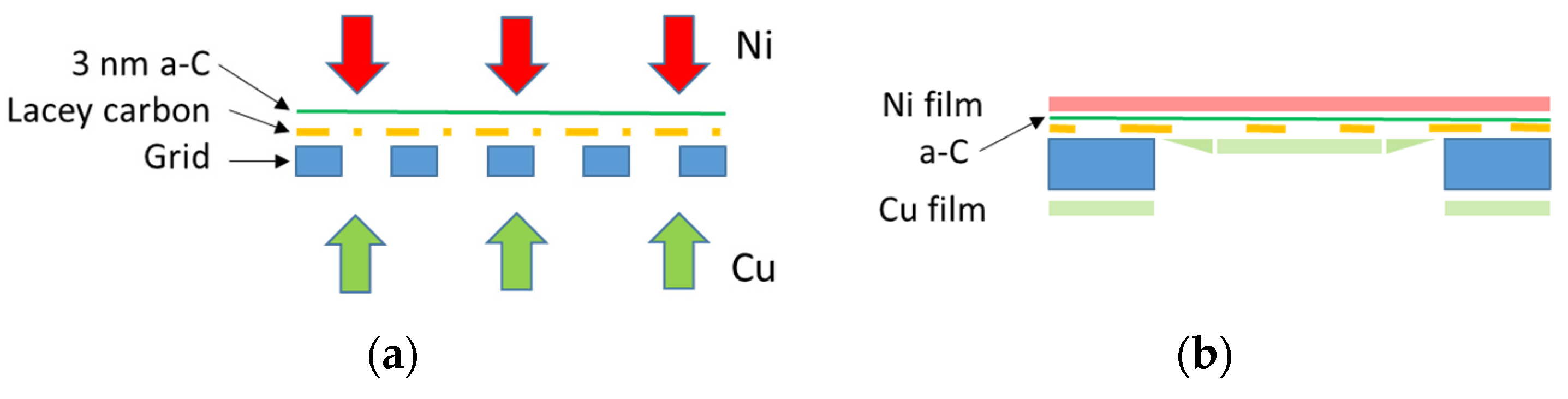
2.2. Electron Diffraction
2.3. Diffraction Data Pre-Processing and Rietveld Analysis
3. Results
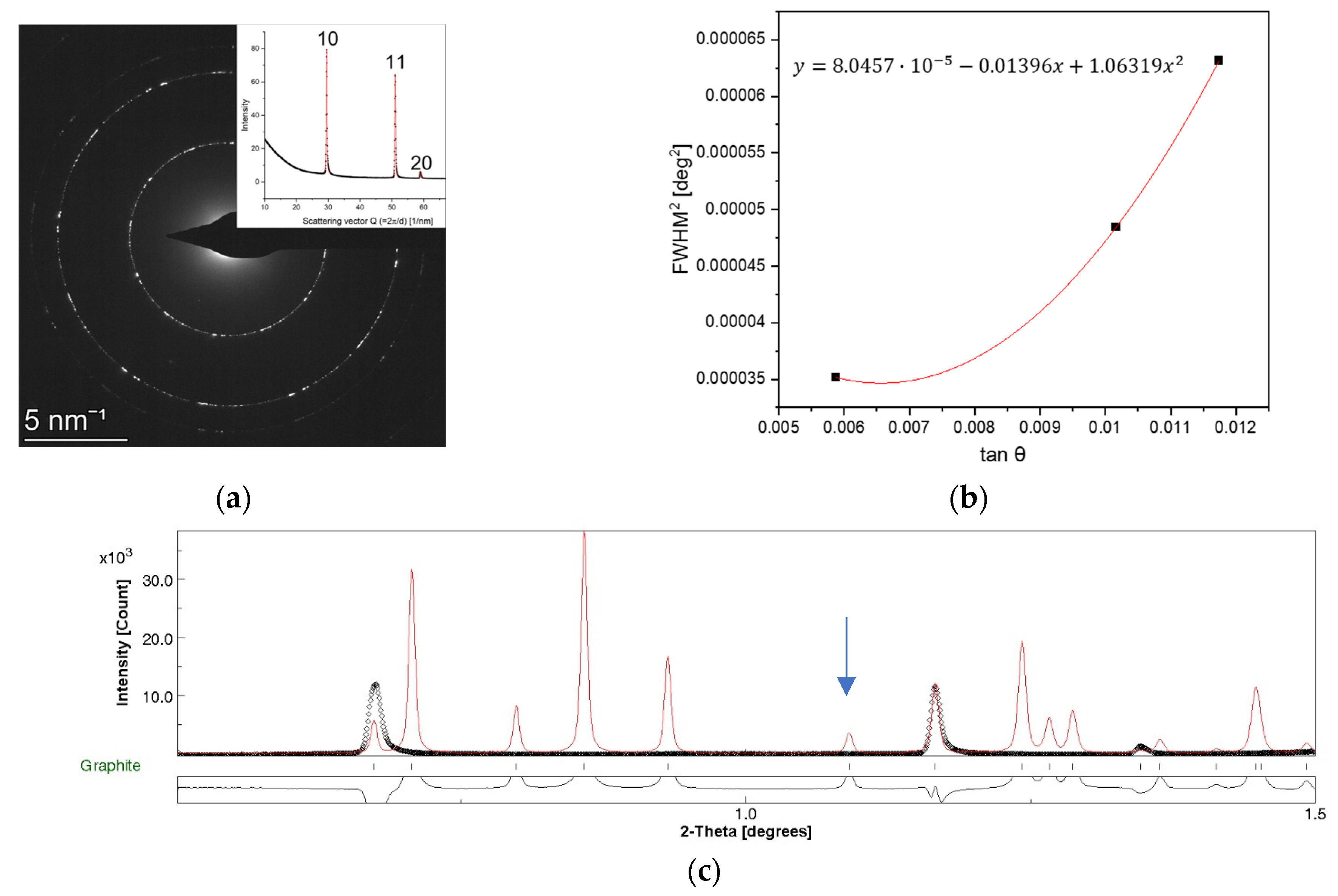
3.1. Experimental Determination of Instrumental Broadening of TEM
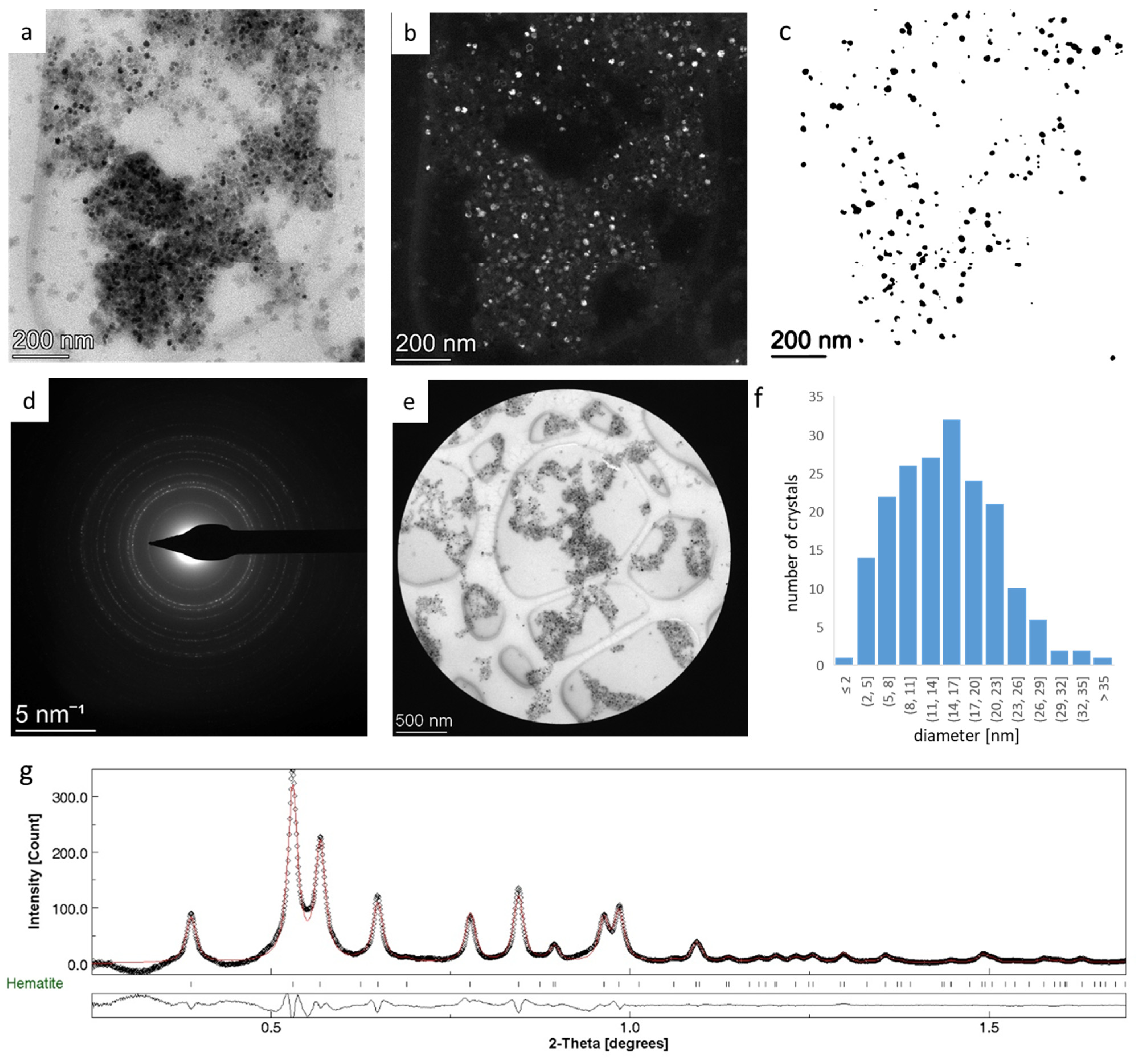
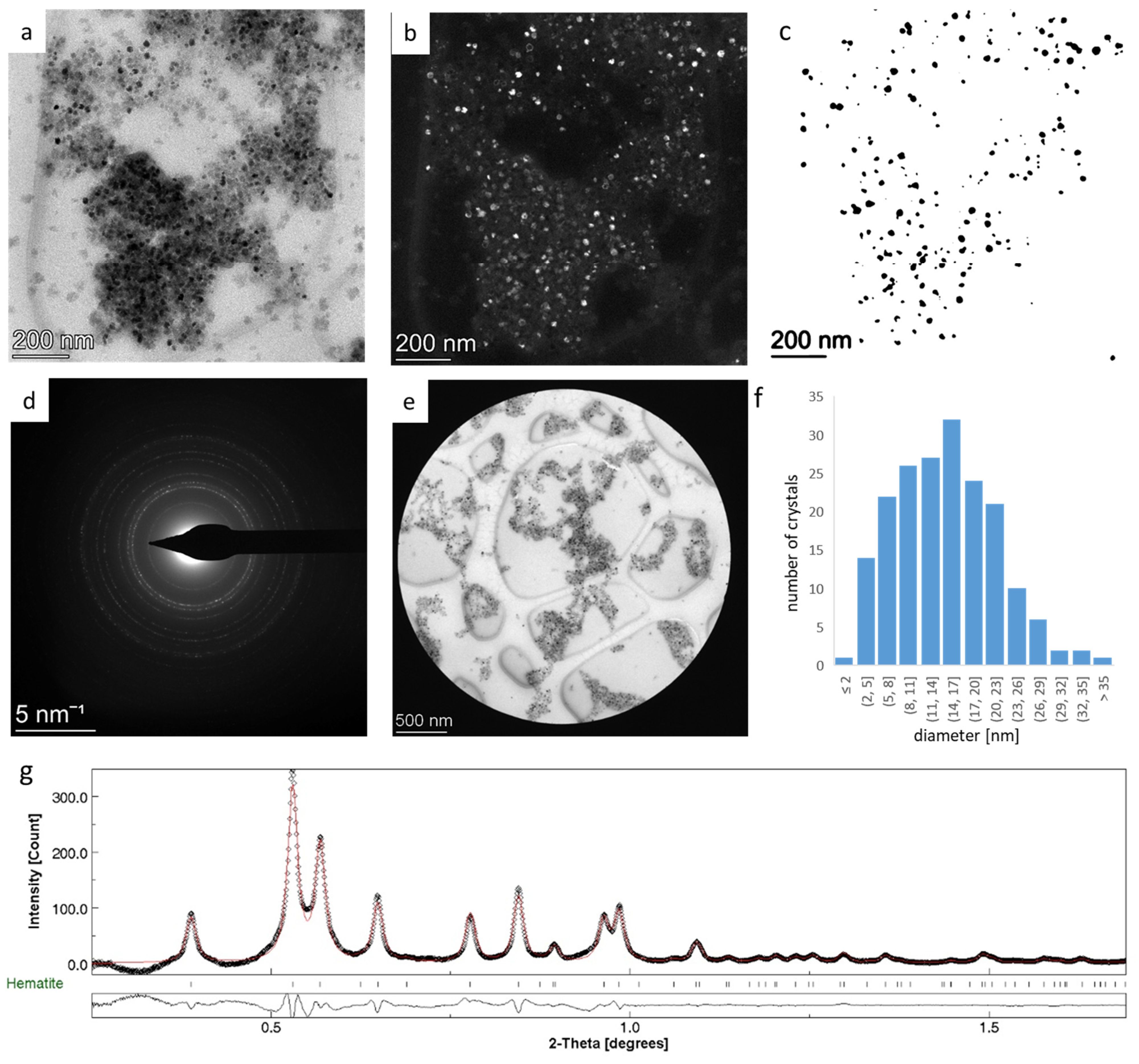
3.2. Rietveld Analysis of Nanocrystalline Hematite—Refining G0
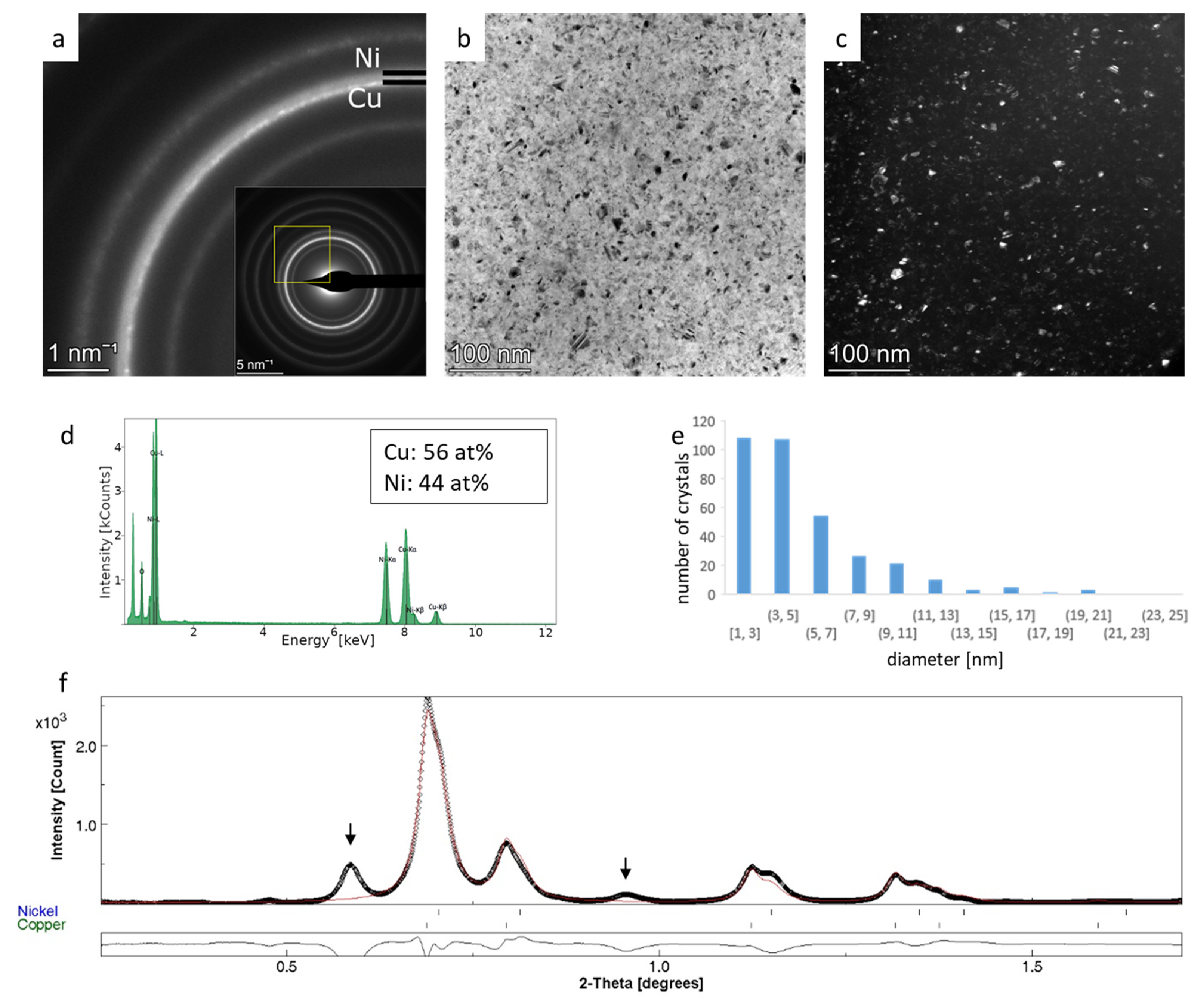
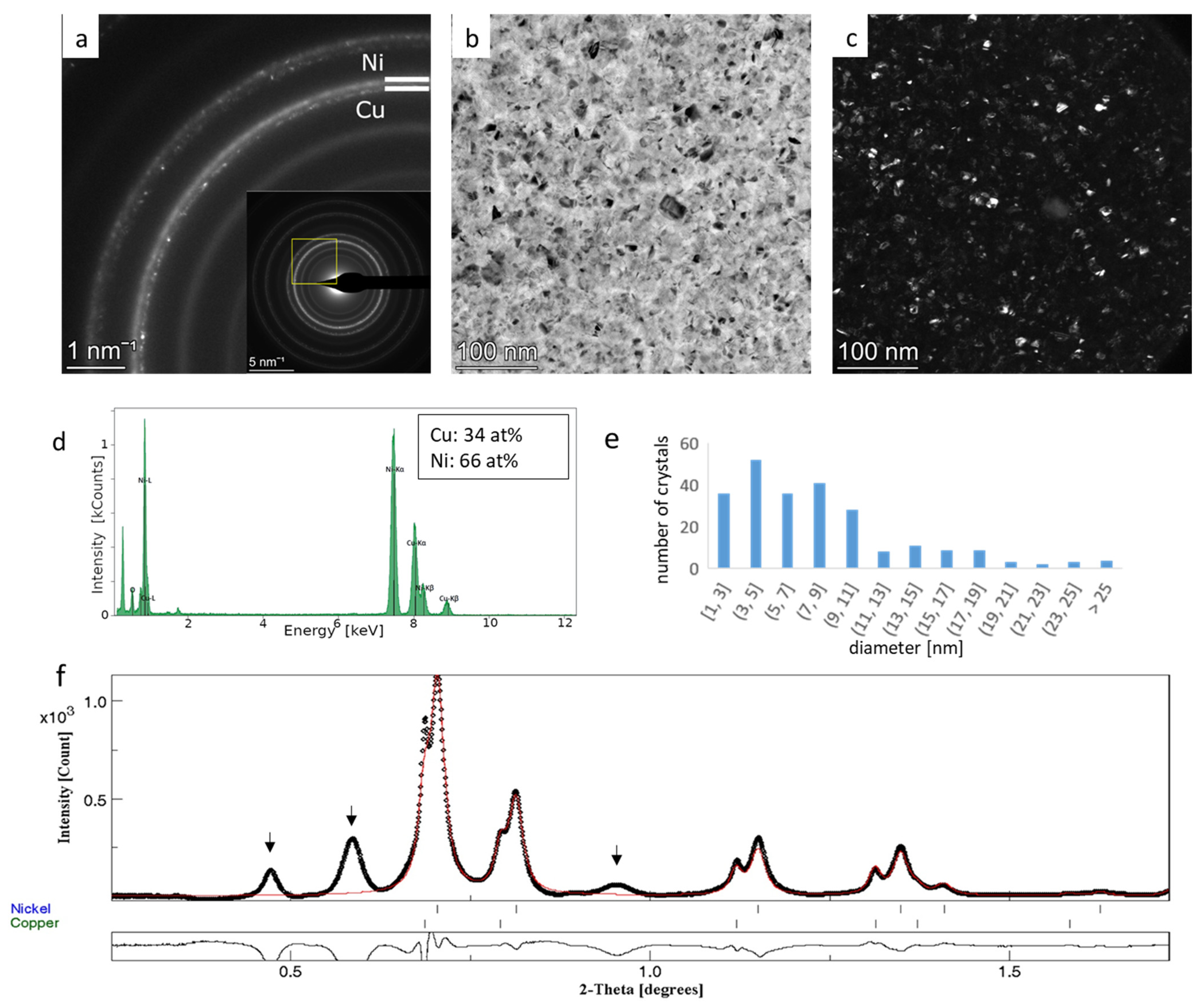
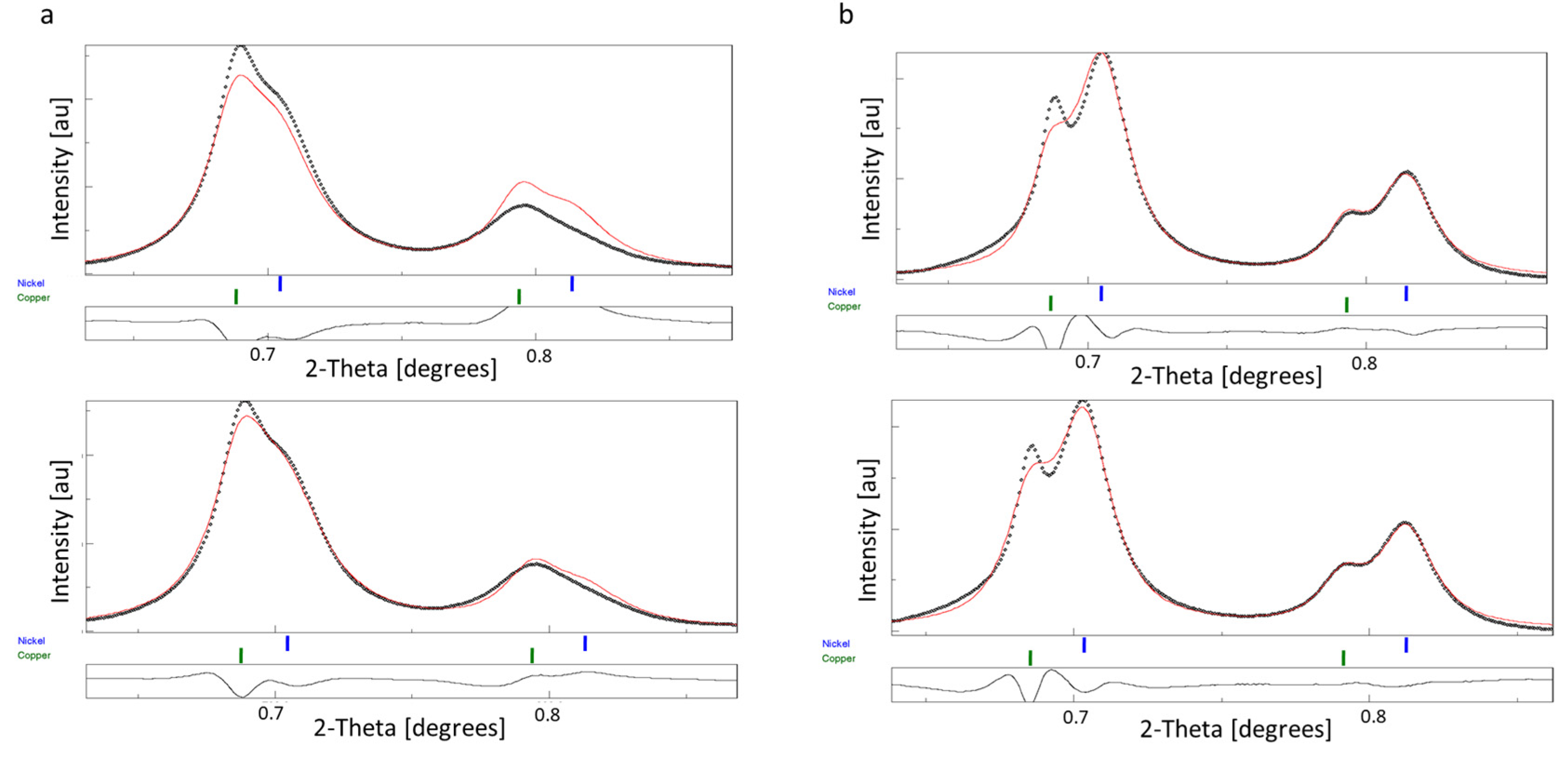
3.3. Rietveld Analysis of Cu-Ni Thin Films—Crystallite Size and Phase Ratio from Overlapping Rings
4. Discussion
4.1. Validation of the Peak Broadening Determination of TEM
4.2. Benefits of the Single Step in-TEM Determination of Instrumental Broadening
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kipp, J.E. The role of solid nanoparticle technology in the parenteral delivery of poorly water–soluble drugs. Int. J. Pharm. 2004, 284, 109–122. [Google Scholar] [CrossRef]
- Feng, X.; Chen, A.; Zhang, Y.; Wang, J.; Shao, L.; Wei, L. Application of dental nanomaterials: Potential toxicity to the central nervous system. Int. J. Nanomed. 2015, 10, 3447–3565. [Google Scholar]
- Czigány, Z.; Kis, V.K. Acquisition and evaluation procedure to improve the accuracy of SAED. Microsc. Res. Tech. 2023, 86, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Rietveld, H.M. Line Profiles of Neutron Powder–Diffraction Peaks for Structure Refinement. Acta Crystallogr. 1967, 22, 151–152. [Google Scholar] [CrossRef]
- Zuo, J.-M.; Lábár, J.L.; Zhang, J.; Gorelik, T.E.; Kolb, U. Electron powder diffraction. In International Tables for Crystallography Vol. H; John Wiley & Sons: Hoboken, NJ, USA, 2019; pp. 102–117. ISBN 978-1-118-41628-0. [Google Scholar]
- Chen, H.; Zuo, J.-M. Structure and phase separation of Ag–Cu alloy thin films. Acta Mater. 2007, 55, 1617–1628. [Google Scholar] [CrossRef]
- Weirich, T.E.; Winterer, M.; Seifried, S.; Hahn, H.; Fuess, H. Rietveld analysis of electron powder diffraction data from nanocrystalline anatase, TiO2. Ultramicroscopy 2000, 81, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Vainshtein, B.K. Structure Analysis by Electron Diffraction; Pergamon Press: Oxford, UK, 1964. [Google Scholar]
- Weirich, T.E.; Winterer, M.; Seifried, S.; Mayer, J. Structure of nanocrystalline anatase solved and refined from electron powder data. Acta Crystallogr. 2002, A58, 308–315. [Google Scholar] [CrossRef]
- Kim, J.-G.; Seo, J.-W.; Cheon, J.-W.; Kim, Y.-J. Rietveld Analysis of Nano–crystalline MnFe2O4 with Electron Powder Diffraction. Bull. Korean Chem. Soc. 2009, 30, 183–187. [Google Scholar]
- Luo, Z.; Vasquez, Y.; Bondi, J.F.; Schaak, R.E. Pawley and Rietveld refinements using electron diffraction from L12-type intermetallic Au3Fe1−x nanocrystals during their in-situ order–disorder transition. Ultramicroscopy 2011, 111, 1295–1304. [Google Scholar] [CrossRef]
- Tonejc, A.M.; Djerdj, I.; Tonejc, A. An analysis of evolution of grain size–lattice parameters dependence in nanocrystalline TiO2 anatase. Mater. Sci. Eng. C 2002, 19, 85–89. [Google Scholar] [CrossRef]
- Song, K.; Kim, Y.-J.; Kim, Y.-I.; Kim, J.-G. Application of theta–scan precession electron diffraction to structure analysis of hydroxyapatite nanopowder. J. Electron. Microsc. 2012, 61, 9–15. [Google Scholar] [CrossRef]
- Gemmi, M.; Voltolini, M.; Ferretti, A.M.; Ponti, A. Quantitative texture analysis from powder–like electron diffraction data. J. Appl. Cryst. 2011, 44, 454–461. [Google Scholar] [CrossRef]
- Boullay, P.; Lutterotti, L.; Chateigner, D.; Sicard, L. Fast microstructure and phase analyses of nanopowders using combined analysis of transmission electron microscopy scattering patterns. Acta Crystallogr. A 2014, 70, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Serafini, A.; Lutterotti, L.; Gross, S.; Gialanella, S. Characterization of nanograined powder samples using the Rietveld method applied to electron diffraction ring patterns. Powder Diffr. 2017, 32, S63–S68. [Google Scholar] [CrossRef]
- Sinha, A.; Bortolotti, M.; Ischia, G.; Lutterotti, L.; Gialanella, S. Electron diffraction characterization of nanocrystalline materials using a Rietveld–based approach. Part I. Methodology. J. Appl. Cryst. 2022, 55, 953–965. [Google Scholar] [CrossRef]
- Gracheva, M.; Klencsár, Z.; Kis, V.K.; Béres, K.A.; May, Z.; Halasy, V.; Singh, A.; Fodor, F.; Solti, Á.; Kiss, L.F.; et al. Iron nanoparticles for plant nutrition: Synthesis, transformation, and utilization by the roots of Cucumis sativus. J. Mater. Res. 2023, 38, 1035–1047. [Google Scholar] [CrossRef]
- Lábár, J.L.; Adamik, M.; Barna, B.P.; Czigány, Z.; Fogarassy, Z.; Horváth, Z.E.; Geszti, O.; Misják, F.; Morgiel, J.; Radnóczi, G.; et al. Electron diffraction based analysis of phase fractions and texture in nanocrystalline thin films. Part III: Application examples. Microsc. Microanal. 2012, 18, 406–420. [Google Scholar] [CrossRef]
- Lábár, J.L.; Das, P.P. Pattern center and distorsion determined from faint, diffuse electron diffraction rings from amorphous materials. Microsc. Microanal. 2017, 18, 406–420. [Google Scholar] [CrossRef]
- Lutterotti, L.; Matthies, S.; Wenk, H.-R.; Schultz, A.S.; Richardson, J.W. Combined texture and structure analysis of deformed limestone from time–of–flight neutron diffraction spectra. J. Appl. Phys. 1997, 81, 594–600. [Google Scholar] [CrossRef]
- Lutterotti, L. Total pattern fitting for the combined size–strain–stress–texture determination in thin film diffraction. Nucl. Instrum. Methods Phys. Res. B 2010, 268, 334–340. [Google Scholar] [CrossRef]
- Peng, L.-M.; Ren, G.; Dudarev, S.L.; Whelan, M.J. Robust parameterization of elastic and absorptive electron atomic scattering factors. Acta Crystallogr. A 1996, 52, 257–276. [Google Scholar] [CrossRef]
- Ischia, G.; Wenk, H.-R.; Lutterotti, L.; Berberich, F. Quantitative Rietveld texture analysis of zirconium from single synchrotron diffraction Images. J. Appl. Cryst. 2005, 38, 377–380. [Google Scholar] [CrossRef]
- Shinotsuka, H.; Tanuma, S.; Powell, C.J.; Penn, D.R. Calculations of electron inelastic mean free paths. X. Data for 41 elemental solids over the 50eV to 200keV range with the relativistic full Penn algorithm. Surf. Interface Anal. 2015, 47, 871–888. [Google Scholar] [CrossRef]
- Meyer, Y.C.; Geim, A.K.; Katsnelson, M.I.; Novoselov, K.S.; Booth, T.J.; Roth, S. The structure of suspended graphene sheets. Nature 2007, 446, 60–63. [Google Scholar] [CrossRef]
- Caglioti, G.; Paoletti, A.; Ricci, F.P. Choice of collimators for a crystal spectrometer for neutron diffraction. Nucl. Instrum. 1958, 3, 223–228. [Google Scholar] [CrossRef]
- Brindley, G.W. Order-disorder in clay mineral structures. In Crystal Structures of Clay Minerals and Their X-ray Identification; Brindley, G.W., Brown, G., Eds.; The Mineralogical Society of Great Britain and Ireland: London, UK, 1980; pp. 125–195. [Google Scholar]
- Warren, B.E. X-ray Diffraction in Random Layer Lattices. Phys. Rev. 1941, 59, 693–698. [Google Scholar] [CrossRef]
- Tapasztó, L.; Dumitrica, T.; Kim, S.J.; Nemes-Incze, P.; Hwang, C.; Biró, L.P. Breakdown of continuum mechanics for nanometre-wavelength rippling of graphene. Nat. Phys. 2012, 8, 739–742. [Google Scholar] [CrossRef]
- Zhang, D.-B.; Akatyeva, E.; Dumitrica, T. Bending ultrathin graphene at the margins of continuum mechanics. Phys. Rev. Lett. 2011, 106, 255503. [Google Scholar] [CrossRef]
- Maslen, E.N.; Strelstov, V.A.; Streltsova, N.R. Synchrotron X–ray study of the electron density in α–Fe2O3. Acta Crystallogr. B 1994, 50, 435–441. [Google Scholar] [CrossRef]
- Zolotoyabko, E. Determination of the degree of preferred orientation within the March–Dollase approach. J. Appl. Cryst. 2009, 42, 513–518. [Google Scholar] [CrossRef]
- Wenk, H.-R.; Lutterotti, L.; Kaercher, P.; Kanitpanyacharoen, W.; Miyagi, L.; Vasin, R. Rietveld texture analysis from synchrotron diffraction images. II. Complex multiphase materials and diamond anvil cell experiments. Powder Diffr. 2014, 29, 220–232. [Google Scholar] [CrossRef]
- Lutterotti, L.; Vasin, R.N.; Wenk, H.-R. Rietveld texture analysis from synchrotron diffraction images: I. Basic analysis. Powder Diffr. 2014, 29, 76–84. [Google Scholar] [CrossRef]
- Kulovits, A.K.; Facco, G.; Wiezorek, J.M.K. Grain size determination in nano–scale polycrystalline aggregates by precession illumination–hollow cone dark field imaging in the transmission electron microscope. Mater. Charact. 2012, 63, 17–26. [Google Scholar] [CrossRef]
- Gammer, C.; Mangler, C.; Karnthaler, H.P.; Rentenberger, C. Growth of nanosized chemically ordered domains in intermetallic FeAl made nanocrystalline by severe plastic deformation. Scr. Mater. 2011, 65, 57–60. [Google Scholar] [CrossRef]
- Gorelik, T.E.; Neder, R.; Terban, M.W.; Lee, Z.; Mu, X.; Jung, C.; Jacob, T.; Kaiser, U. Towards quantitative treatment of electron pair distribution function. Acta Crystallogr. B 2019, 75, 532–549. [Google Scholar]
- Carvalho, D.; Morales, F.M. High–resolution electron diffraction: Accounting for radially and angularly invariant distortions. Microsc. Microanal. 2012, 18, 638–644. [Google Scholar] [CrossRef]
- Saitoh, K.; Nakahara, H.; Tanaka, N. Improvement of the precision of lattice parameter determination by nano–beam electron diffraction. Microscopy 2013, 62, 533–539. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peak 10 | Peak 11 | Peak 20 | |
|---|---|---|---|
| FWHM [nm−1] | 0.26 | 0.306 | 0.348 |
| Gaussianity, G0 | 0.62 | 0.56 | 0.72 |
| U | 1.06319 |
| V | −0.01396 |
| W | 8.0457·10−5 |
| G0 | 0.64377 |
| G1 | 0.02128 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kis, V.K.; Kovács, Z.; Czigány, Z. Improved Method for Electron Powder Diffraction-Based Rietveld Analysis of Nanomaterials. Nanomaterials 2024, 14, 444. https://doi.org/10.3390/nano14050444
Kis VK, Kovács Z, Czigány Z. Improved Method for Electron Powder Diffraction-Based Rietveld Analysis of Nanomaterials. Nanomaterials. 2024; 14(5):444. https://doi.org/10.3390/nano14050444
Chicago/Turabian StyleKis, Viktória K., Zsolt Kovács, and Zsolt Czigány. 2024. "Improved Method for Electron Powder Diffraction-Based Rietveld Analysis of Nanomaterials" Nanomaterials 14, no. 5: 444. https://doi.org/10.3390/nano14050444
APA StyleKis, V. K., Kovács, Z., & Czigány, Z. (2024). Improved Method for Electron Powder Diffraction-Based Rietveld Analysis of Nanomaterials. Nanomaterials, 14(5), 444. https://doi.org/10.3390/nano14050444