
Molecular Dynamic Simulation of Primary Damage with Electronic Stopping in Indium Phosphide

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Molecular Dynamics Modeling
2.2. Interatomic Potential
3. Results
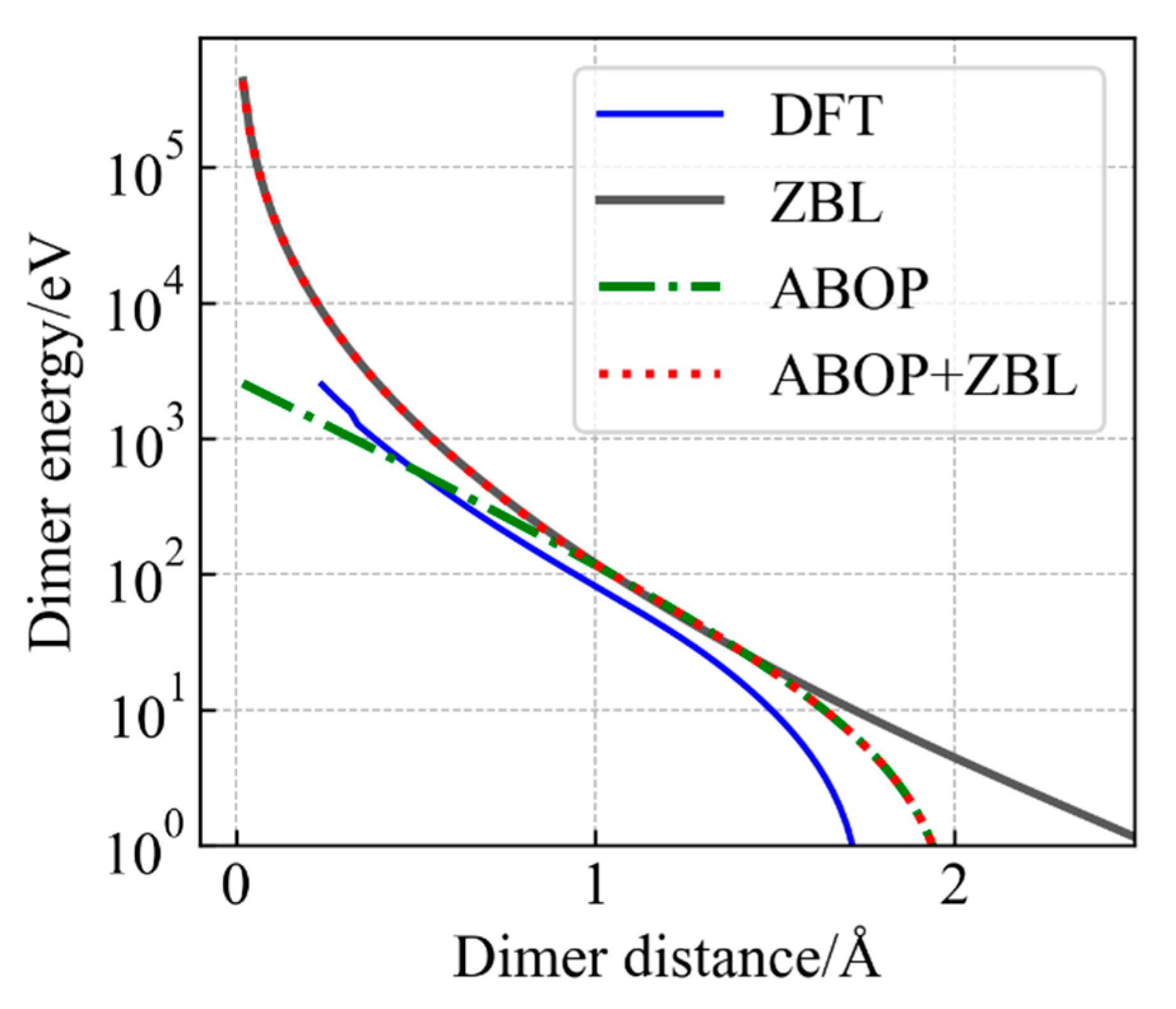
3.1. Accuracy of Modified Potential
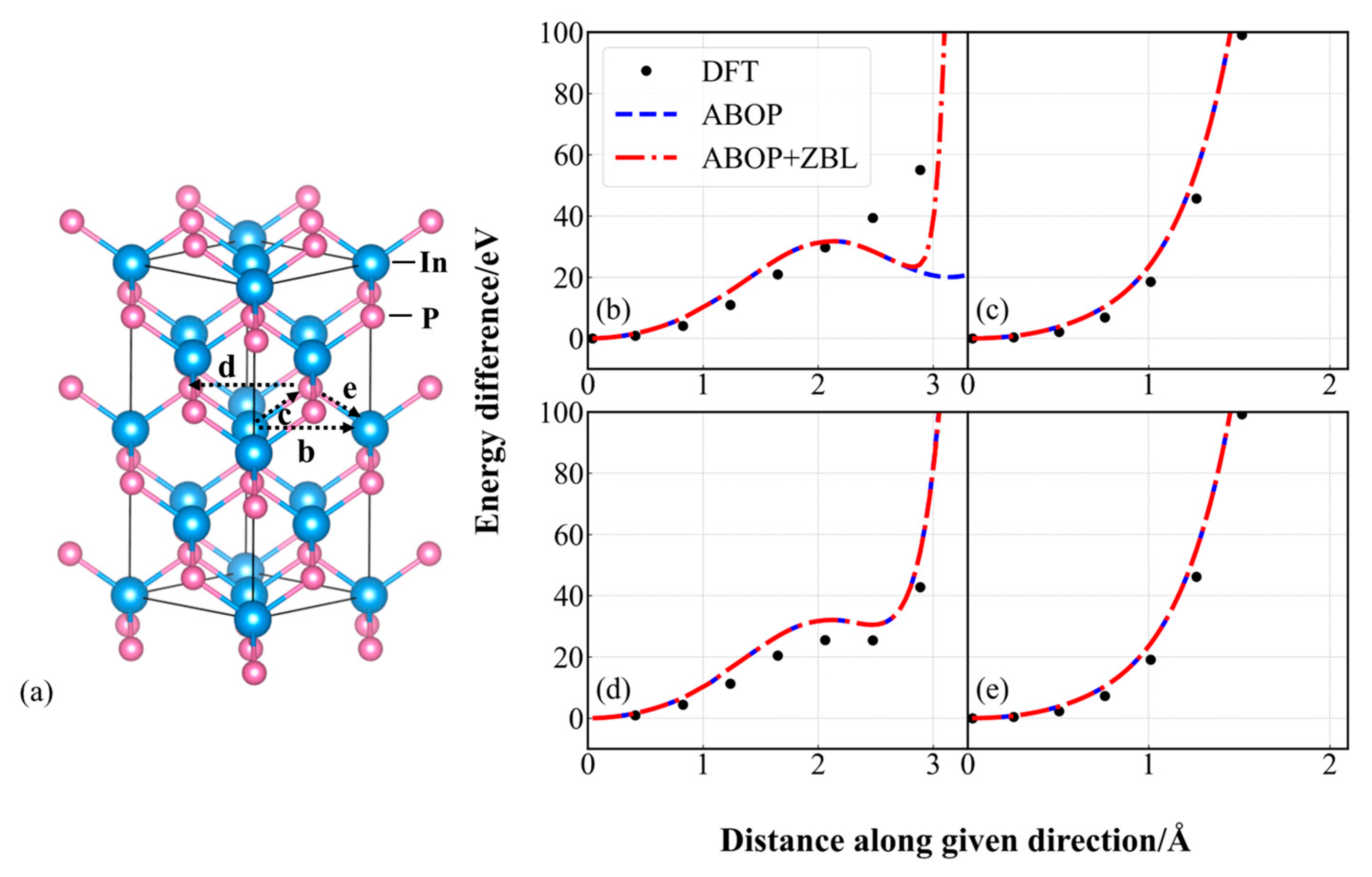
3.2. Quasi-Static Calculations
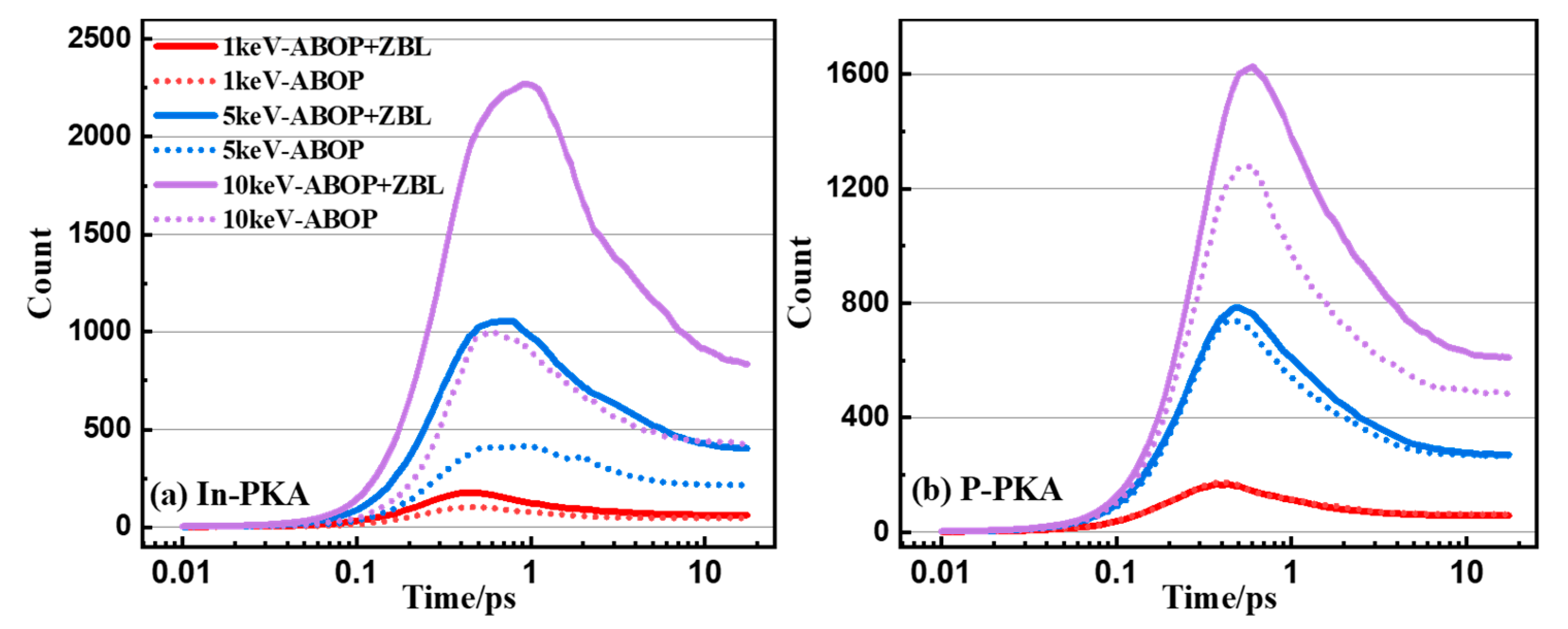
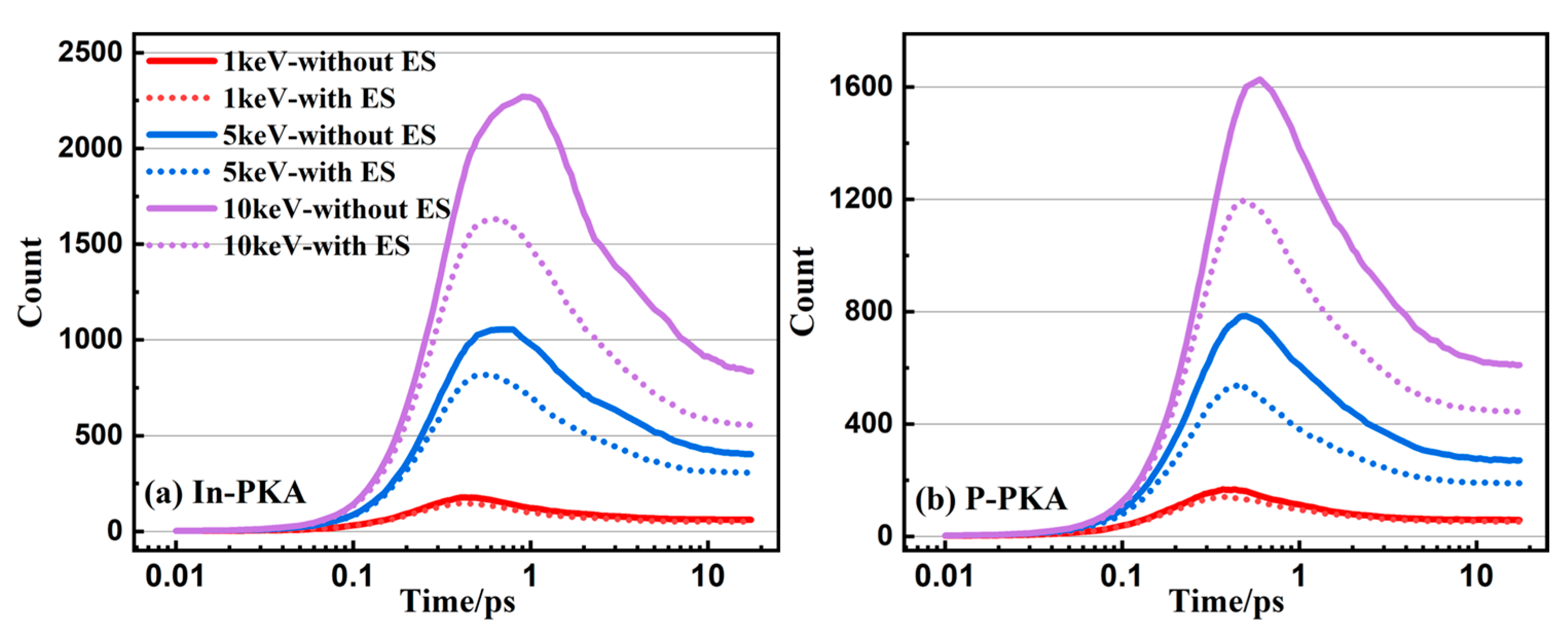
3.3. Point Defects
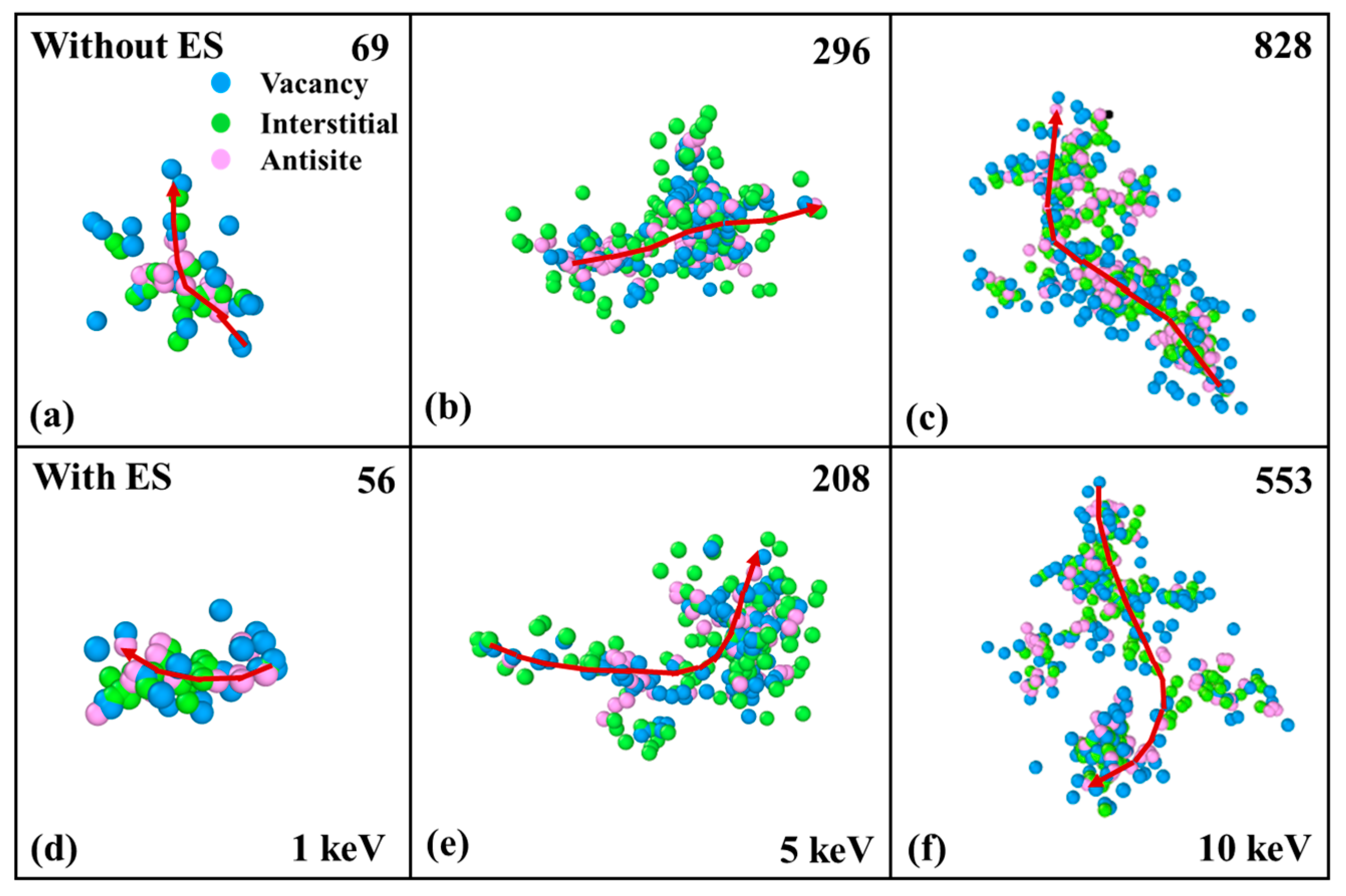
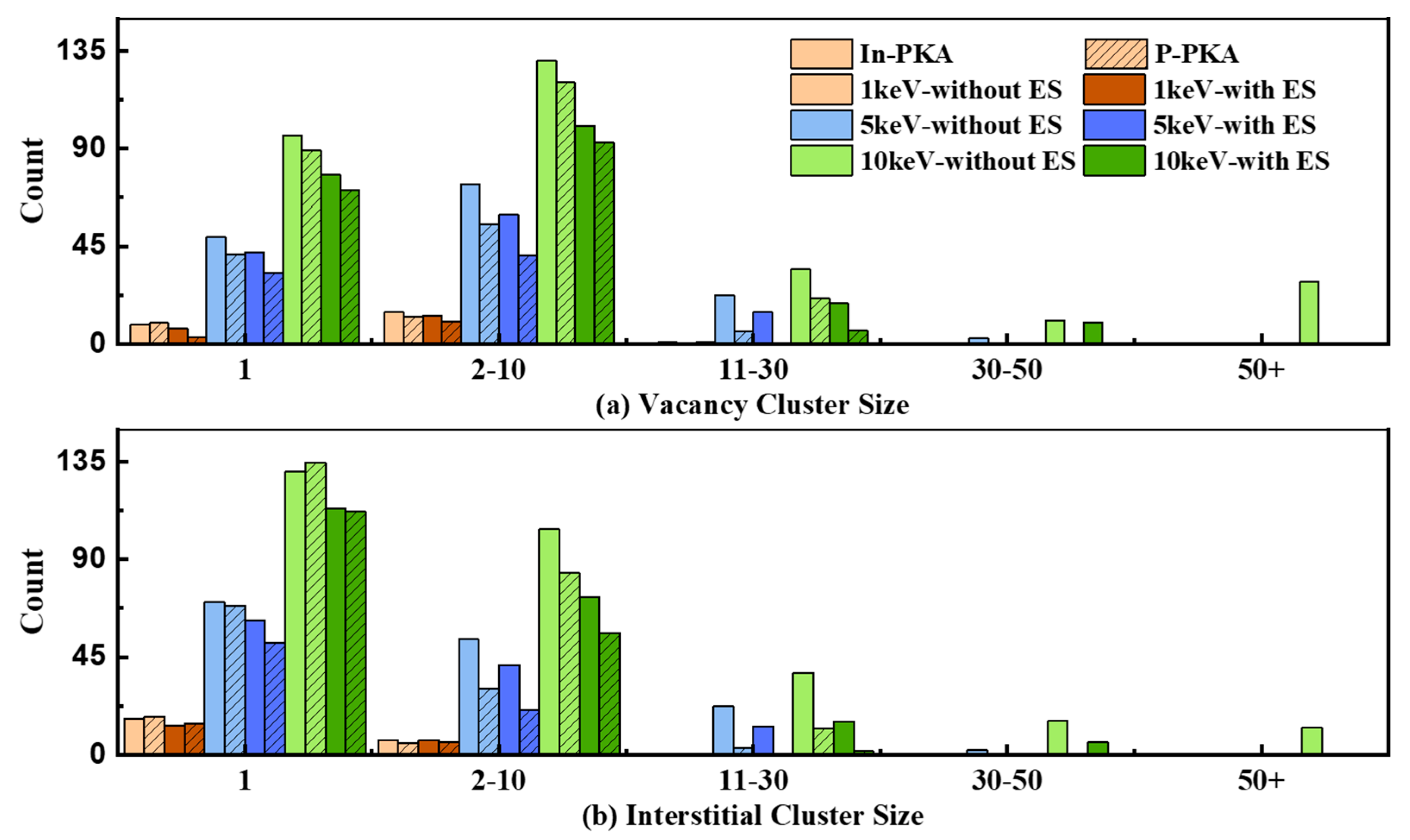
3.4. Cluster Defects
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yamaguchi, M.; Ando, K. Mechanism for Radiation Resistance of InP Solar Cells. J. Appl. Phys. 1988, 63, 5555–5562. [Google Scholar] [CrossRef]
- Keyes, B.M.; Ahrenkiel, R.K.; Shaw, G.J.; Summers, G.P. Minority-Carrier Lifetime Damage Coefficient of Irradiated InP. J. Appl. Phys. 1997, 82, 2156–2163. [Google Scholar] [CrossRef]
- Shatalov, A.; Subramanian, S.; Dentai, A. Neutron Irradiation Effects in InP/InGaAs Single Heterojunction Bipolar Transistors. IEEE Trans. Nucl. Sci. 2000, 47, 2551–2556. [Google Scholar] [CrossRef]
- Vuppala, S.; Li, C.; Zwicknagl, P.; Subramanian, S. Neutron, Proton, and Electron Irradiation Effects in InGaP/GaAs Single Heterojunction Bipolar Transistors. IEEE Trans. Nucl. Sci. 2003, 50, 1846–1851. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Takamoto, T.; Taylor, S.J.; Walters, R.J.; Summers, G.P.; Flood, D.J.; Ohmori, M. Correlations for Damage in Diffused-Junction InP Solar Cells Induced by Electron and Proton Irradiation. J. Appl. Phys. 1997, 81, 6013–6018. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, Y.; Lu, H.; Zhang, Y.; Zhang, J.; Ren, X. Investigation of Proton Irradiation Effects on InP/InGaAs Double Heterojunction Bipolar Transistors. Solid-State Electron. 2015, 109, 52–57. [Google Scholar] [CrossRef]
- Zhao, M.-L.; Lu, H.-L.; Zhang, Y.-M.; Zhang, Y.-M.; Zhao, X.-H. Effect of Deep Level Traps on the I-V and C-V Characteristics of InP/InGaAs Heterojunction. In Proceedings of the 2018 18th International Workshop on Junction Technology (IWJT), Shanghai, China, 8–9 March 2018; pp. 1–4. [Google Scholar]
- Sun, S.-X.; Chang, M.-M.; Li, M.-K.; Ma, L.-H.; Zhong, Y.-H.; Li, Y.-X.; Ding, P.; Jin, Z.; Wei, Z.-C. Effect of Defects Properties on InP-Based High Electron Mobility Transistors. Chin. Phys. B 2019, 28, 078501. [Google Scholar] [CrossRef]
- Chen, X.; Li, L.; Ren, H. Mechanisms for Radiation Resistance of InP Photovoltaic Cells: A First Principle Study. Sol. RRL 2023, 7, 2201042. [Google Scholar] [CrossRef]
- Liu, J.; Song, Y.; Xu, X.; Li, W.; Yang, J.; Li, X. Identification of Intrinsic Defects and Hydrogen Passivation in InP Using Hybrid Functional. J. Appl. Phys. 2023, 134, 115702. [Google Scholar] [CrossRef]
- Tapster, P.R. A DLTS Study of Electron Irradiated InP. J. Cryst. Growth 1983, 64, 200–205. [Google Scholar] [CrossRef]
- Tian, S.; He, C.; He, H.; Liao, W.; Bai, Y.; Li, Y. Insight of Displacement Cascade Evolution in Gallium Arsenide through Molecular Dynamics Simulations. Comput. Mater. Sci. 2022, 202, 111016. [Google Scholar] [CrossRef]
- Jia, T.; Wang, Z.; Tang, M.; Xue, Y.; Huang, G.; Nie, X.; Lai, S.; Ma, W.; He, B.; Gou, S. Simulation Study on the Defect Generation, Accumulation Mechanism and Mechanical Response of GaAs Nanowires under Heavy-Ion Irradiation. Nanomaterials 2022, 12, 611. [Google Scholar] [CrossRef]
- He, H.; He, C.; Zhang, J.; Liao, W.; Zang, H.; Li, Y.; Liu, W. Primary Damage of 10 keV Ga PKA in Bulk GaN Material under Different Temperatures. Nucl. Eng. Technol. 2020, 52, 1537–1544. [Google Scholar] [CrossRef]
- Liu, Y.; Xiong, Z.; Ouyang, X. Molecular Dynamics Study on the Mechanism of Gallium Nitride Radiation Damage by Alpha Particles. Materials 2023, 16, 4224. [Google Scholar] [CrossRef] [PubMed]
- Samolyuk, G.D.; Osetsky, Y.N.; Stoller, R.E. Molecular Dynamics Modeling of Atomic Displacement Cascades in 3C–SiC: Comparison of Interatomic Potentials. J. Nucl. Mater. 2015, 465, 83–88. [Google Scholar] [CrossRef]
- Liao, W.; He, C.; He, H. Molecular Dynamics Simulation of Displacement Damage in 6H-SiC. Radiat. Eff. Defects Solids 2019, 174, 729–740. [Google Scholar] [CrossRef]
- Chen, N.; Huang, D.; Heller, E.R.; Cardimona, D.A.; Gao, F. Atomistic Simulation of Displacement Damage and Effective Nonionizing Energy Loss in InAs. Phys. Rev. Mater. 2021, 5, 033603. [Google Scholar] [CrossRef]
- Branicio, P.S.; Rino, J.P.; Gan, C.K.; Tsuzuki, H. Interaction Potential for Indium Phosphide: A Molecular Dynamics and First-Principles Study of the Elastic Constants, Generalized Stacking Fault and Surface Energies. J. Phys. Condens. Matter 2009, 21, 095002. [Google Scholar] [CrossRef]
- Vashishta, P.; Kalia, R.K.; Nakano, A.; Rino, J.P. Interaction Potential for Silicon Carbide: A Molecular Dynamics Study of Elastic Constants and Vibrational Density of States for Crystalline and Amorphous Silicon Carbide. J. Appl. Phys. 2007, 101, 103515. [Google Scholar] [CrossRef]
- Chrobak, D.; Majtyka-Piłat, A.; Ziółkowski, G.; Chrobak, A. Interatomic Potential for InP. Materials 2022, 15, 4960. [Google Scholar] [CrossRef]
- Li, J.; Shi, T.; Sun, Y.; Cai, X.; Gao, R.; Peng, Q.; Lu, P.; Lu, C. Description of Short-Range Interactions of Carbon-Based Materials with a Combined AIREBO and ZBL Potential. Nanomaterials 2024, 14, 1423. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-W.; Stewart, J.A.; Dingreville, R.; Foiles, S.M.; Schleife, A. Multiscale Simulations of Electron and Ion Dynamics in Self-Irradiated Silicon. Phys. Rev. B 2020, 102, 024107. [Google Scholar] [CrossRef]
- Zarkadoula, E.; Samolyuk, G.; Zhang, Y.; Weber, W.J. Electronic Stopping in Molecular Dynamics Simulations of Cascades in 3C–SiC. J. Nucl. Mater. 2020, 540, 152371. [Google Scholar] [CrossRef]
- Liao, W.; He, H.; Li, Y.; Liu, W.; Zang, H.; Wei, J.; He, C. Effects of Electronic Energy Deposition on Pre-Existing Defects in 6H–SiC. Nucl. Eng. Technol. 2021, 53, 2357–2363. [Google Scholar] [CrossRef]
- Kempner, M.; Sestito, J.M.; Wang, Y.; Zarkadoula, E. Molecular Dynamics Simulations of Cascade Events in AlN. Results Mater. 2023, 17, 100383. [Google Scholar] [CrossRef]
- Stewart, J.A.; Brookman, G.; Price, P.; Franco, M.; Ji, W.; Hattar, K.; Dingreville, R. Characterizing Single Isolated Radiation-Damage Events from Molecular Dynamics via Virtual Diffraction Methods. J. Appl. Phys. 2018, 123, 165902. [Google Scholar] [CrossRef]
- Zarkadoula, E.; Duffy, D.M.; Nordlund, K.; Seaton, M.A.; Todorov, I.T.; Weber, W.J.; Trachenko, K. Electronic Effects in High-Energy Radiation Damage in Tungsten. J. Phys. Condens. Matter 2015, 27, 135401. [Google Scholar] [CrossRef]
- Zarkadoula, E.; Daraszewicz, S.L.; Duffy, D.M.; Seaton, M.A.; Todorov, I.T.; Nordlund, K.; Dove, M.T.; Trachenko, K. Electronic Effects in High-Energy Radiation Damage in Iron. J. Phys. Condens. Matter 2014, 26, 085401. [Google Scholar] [CrossRef]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; In ’T Veld, P.J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS—A Flexible Simulation Tool for Particle-Based Materials Modeling at the Atomic, Meso, and Continuum Scales. Comput. Phys. Commun. 2022, 271, 108171. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and Analysis of Atomistic Simulation Data with OVITO–the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2010, 18, 015012. [Google Scholar] [CrossRef]
- Ziegler, J.F.; Ziegler, M.D.; Biersack, J.P. SRIM—The Stopping and Range of Ions in Matter (2010). Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2010, 268, 1818–1823. [Google Scholar] [CrossRef]
- Nord, J.; Albe, K.; Erhart, P.; Nordlund, K. Modelling of Compound Semiconductors: Analytical Bond-Order Potential for Gallium, Nitrogen and Gallium Nitride. J. Phys. Condens. Matter 2003, 15, 5649–5662. [Google Scholar] [CrossRef]
- Albe, K.; Nordlund, K.; Nord, J.; Kuronen, A. Modeling of Compound Semiconductors: Analytical Bond-Order Potential for Ga, As, and GaAs. Phys. Rev. B 2002, 66, 035205. [Google Scholar] [CrossRef]
- Chen, N.; Rasch, E.; Huang, D.; Heller, E.R.; Gao, F. Atomic-Scale Simulation for Pseudometallic Defect-Generation Kinetics and Effective NIEL in GaN. IEEE Trans. Nucl. Sci. 2018, 65, 1108–1118. [Google Scholar] [CrossRef]
- Biersack, J.P.; Ziegler, J.F. Refined Universal Potentials in Atomic Collisions. Nucl. Instrum. Methods Phys. Res. 1982, 194, 93–100. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Mishra, R.; Restrepo, O.D.; Kumar, A.; Windl, W. Native Point Defects in Binary InP Semiconductors. J. Mater. Sci. 2012, 47, 7482–7497. [Google Scholar] [CrossRef]
- He, H.; Zhao, J.; Byggmästar, J.; He, R.; Nordlund, K.; He, C.; Djurabekova, F. Threshold Displacement Energy Map of Frenkel Pair Generation in β-Ga2O3 from Machine-Learning-Driven Molecular Dynamics Simulations. Acta Mater. 2024, 276, 120087. [Google Scholar] [CrossRef]
- Zhao, J.; Byggmästar, J.; He, H.; Nordlund, K.; Djurabekova, F.; Hua, M. Complex Ga2O3 Polymorphs Explored by Accurate and General-Purpose Machine-Learning Interatomic Potentials. Npj Comput. Mater. 2023, 9, 159. [Google Scholar] [CrossRef]
- Gao, F.; Weber, W.J.; Devanathan, R. Atomic-Scale Simulation of Displacement Cascades and Amorphization in b-SiC. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2001, 180, 176–186. [Google Scholar] [CrossRef]
- Aleksandrov, I.A.; Zhuravlev, K.S. Migration Barriers for Diffusion of As and P Atoms in InP and InAs via Vacancies and Interstitial Atoms. Acta Mater. 2024, 270, 119854. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PKA Energy (EPKA)/keV | Simulation Box Size (a0 = 5.869 Å) |
|---|---|
| 1 | (21,600 atoms) |
| 5 | (1,000,000 atoms) |
| 10 | (8,000,000 atoms) |
| Parameter | Experiment [19] | ABOP [21] | ABOP + ZBL |
|---|---|---|---|
| 5.87 | 5.83 | 5.83 | |
| −3.34/−3.48 | −3.794 | −3.794 | |
| B/GPa | 71.1/72.5 | 69.1 | 69.1 |
| C11/GPa | 101.1/102.2 | 94.21 | 94.21 |
| C12/GPa | 56.1/57.6 | 56.5 | 56.5 |
| C44/GPa | 45.6/46 | 44.13 | 44.13 |
| Defect | DFT [38] | ABOP [21] | ABOP + ZBL | DFT [38] | ABOP [21] | ABOP + ZBL |
|---|---|---|---|---|---|---|
| In-rich | P-rich | |||||
| −1.52, −6.94 | −1.66, −5.93 | −3.96, −4.5 | −4.1, −3.49 | |||
| 5.26 | 5.21 | 5.24 | 2.83 | 2.77 | 2.8 | |
| 0.98 | 0.93 | 0.97 | 3.42 | 3.37 | 3.41 | |
| 1.62 | 1.66 | 1.54 | 4.06 | 4.10 | 3.98 | |
| 4.19 | 5.81 | 5.57 | 1.76 | 3.37 | 3.13 | |
| 4.88 | 4.88 | 5.13 | - | - | - | |
| - | - | - | 4.88 | 4.88 | 4.89 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, Y.; Liao, W.; Chen, Z.; Li, W.; Liu, W.; He, H.; He, C. Molecular Dynamic Simulation of Primary Damage with Electronic Stopping in Indium Phosphide. Nanomaterials 2024, 14, 1738. https://doi.org/10.3390/nano14211738
Bai Y, Liao W, Chen Z, Li W, Liu W, He H, He C. Molecular Dynamic Simulation of Primary Damage with Electronic Stopping in Indium Phosphide. Nanomaterials. 2024; 14(21):1738. https://doi.org/10.3390/nano14211738
Chicago/Turabian StyleBai, Yurong, Wenlong Liao, Zhongcun Chen, Wei Li, Wenbo Liu, Huan He, and Chaohui He. 2024. "Molecular Dynamic Simulation of Primary Damage with Electronic Stopping in Indium Phosphide" Nanomaterials 14, no. 21: 1738. https://doi.org/10.3390/nano14211738
APA StyleBai, Y., Liao, W., Chen, Z., Li, W., Liu, W., He, H., & He, C. (2024). Molecular Dynamic Simulation of Primary Damage with Electronic Stopping in Indium Phosphide. Nanomaterials, 14(21), 1738. https://doi.org/10.3390/nano14211738