
Functionalized Gold Nanoparticles and Halogen Bonding Interactions Involving Fentanyl and Fentanyl Derivatives
 ,
,
Abstract
1. Introduction
2. Experimental Details
2.1. Materials and Instrumentation
2.2. Computational Methodology
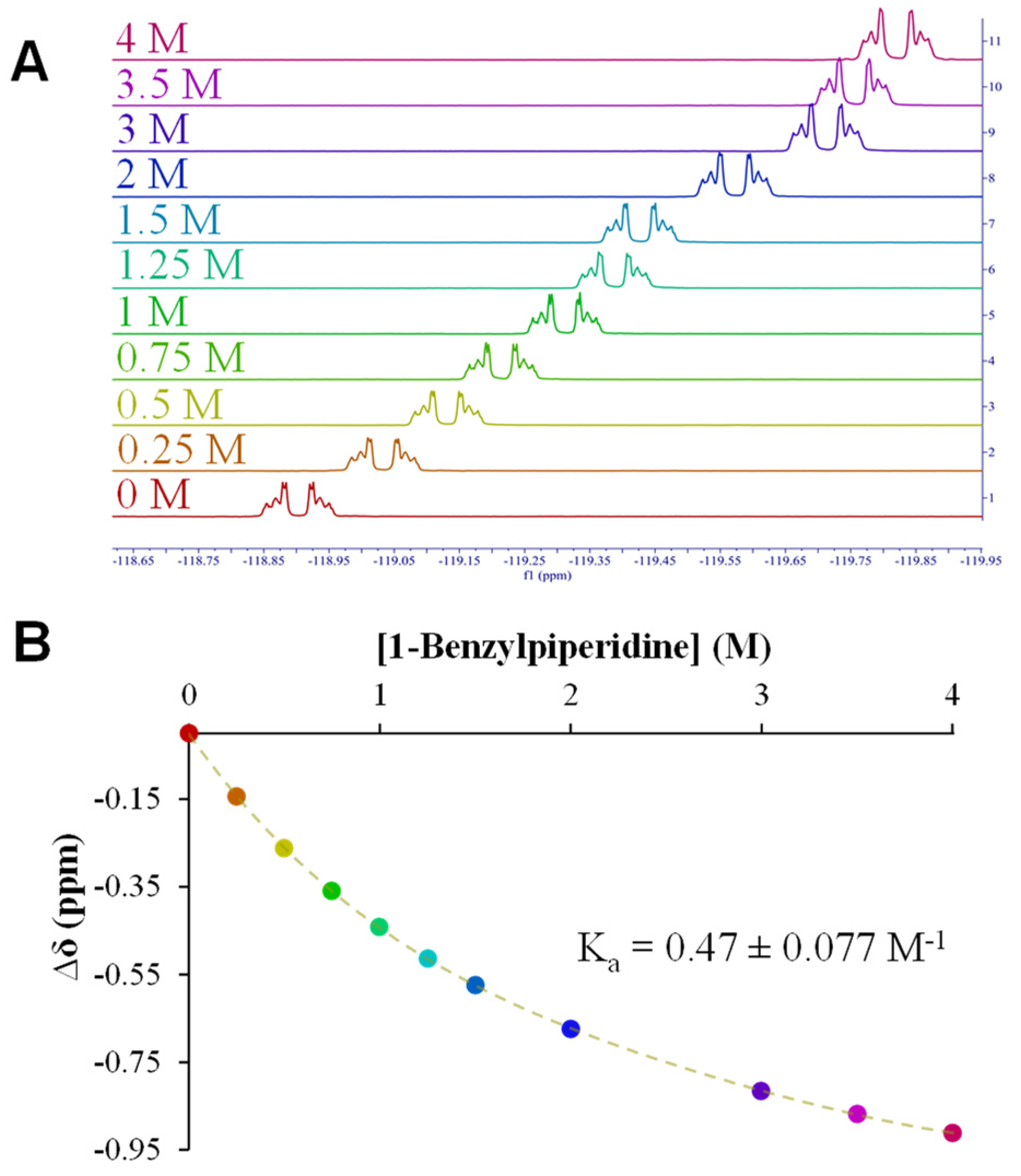
2.3. NMR Titrations
2.4. Electrochemical Measurements
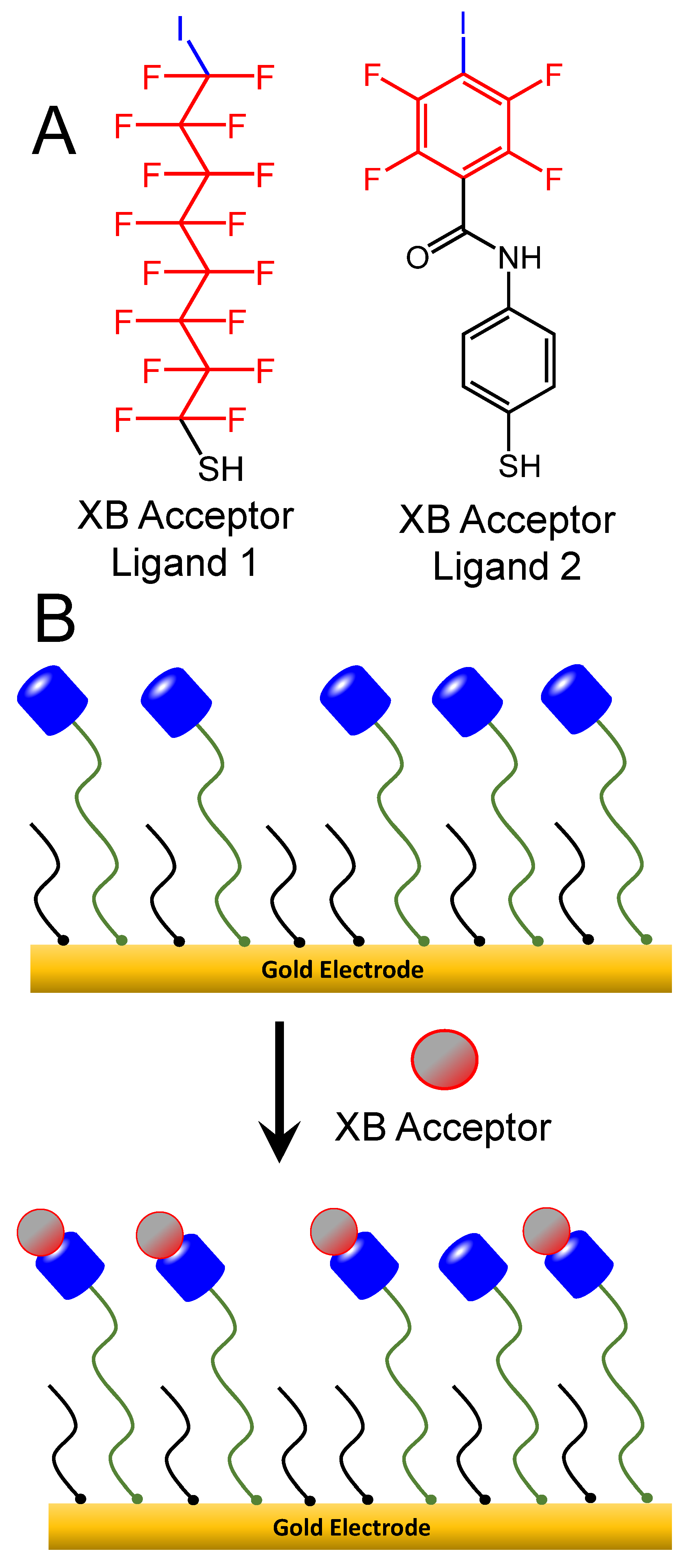
2.5. Synthesis, Functionalization, and Characterization of XB Donor SAMs, Au-NPs, and Au-NP Films
2.6. UV–Vis Spectroscopy and TEM Imaging
3. Results and Discussion
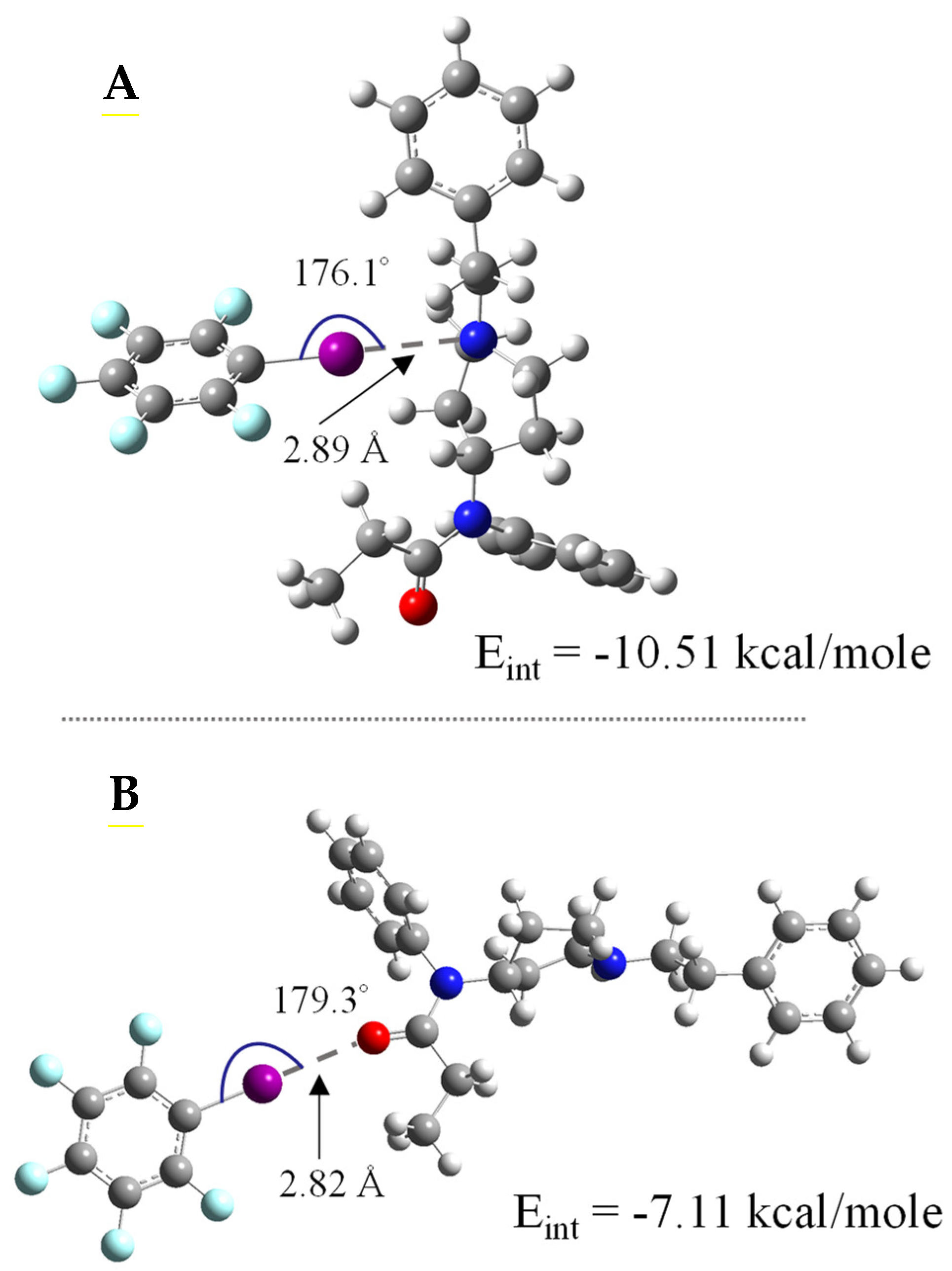
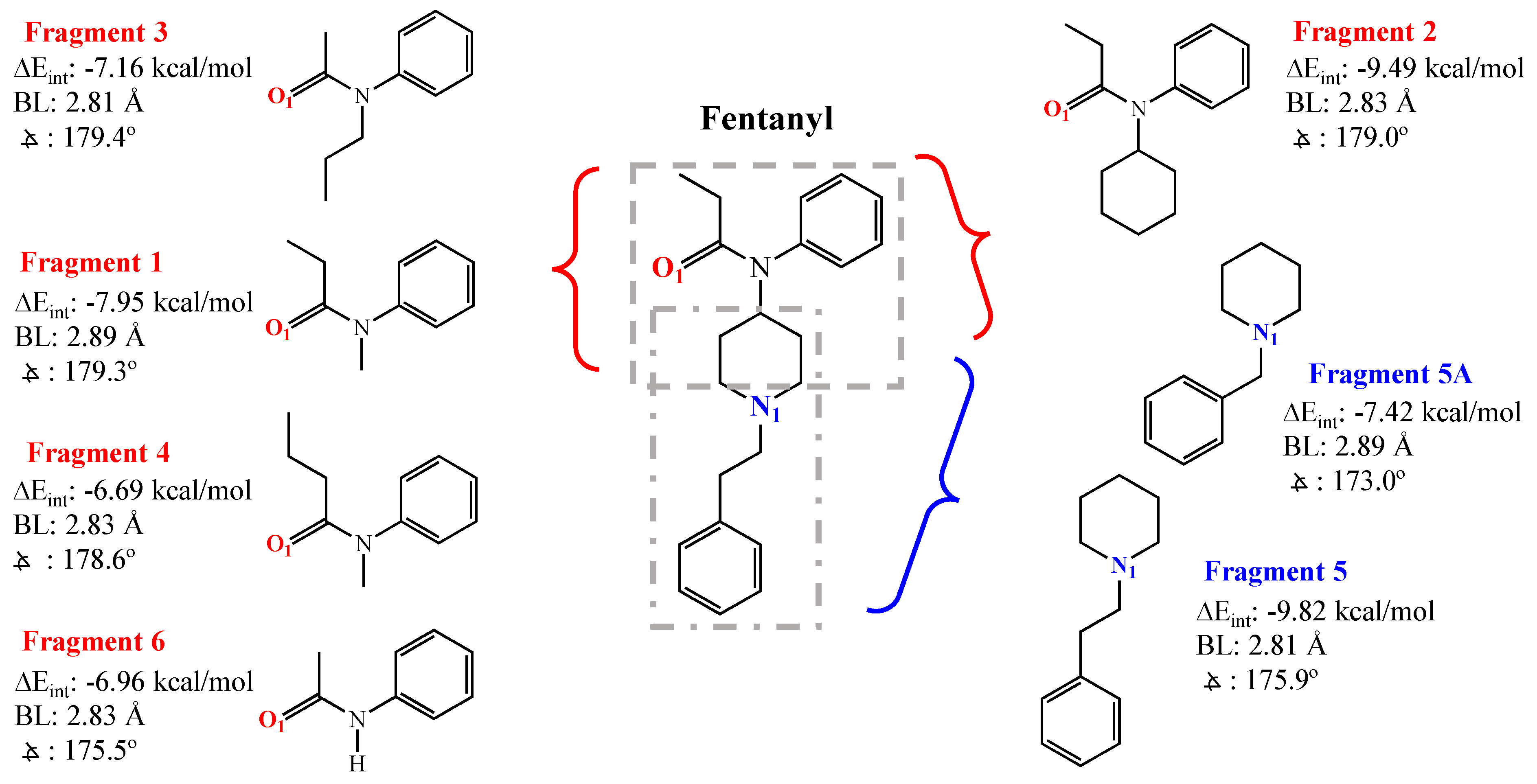
3.1. DFT Calculations for Fentanyl and Fentanyl Derivatives
3.2. NMR Titrations: Fentanyl Fragments
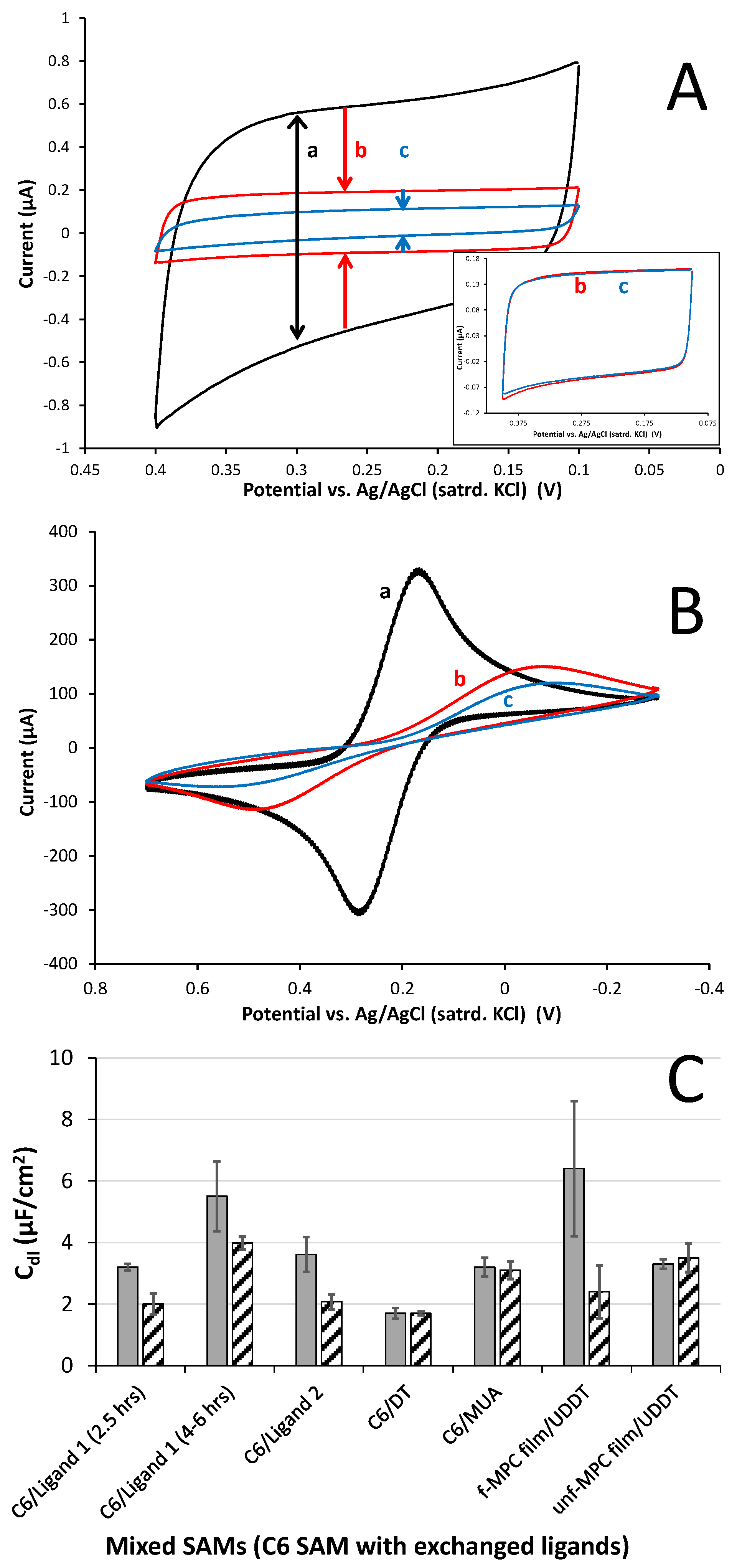
3.3. Electrochemical Support of XB Interactions Using XB Donor Self-Assembled Monolayers
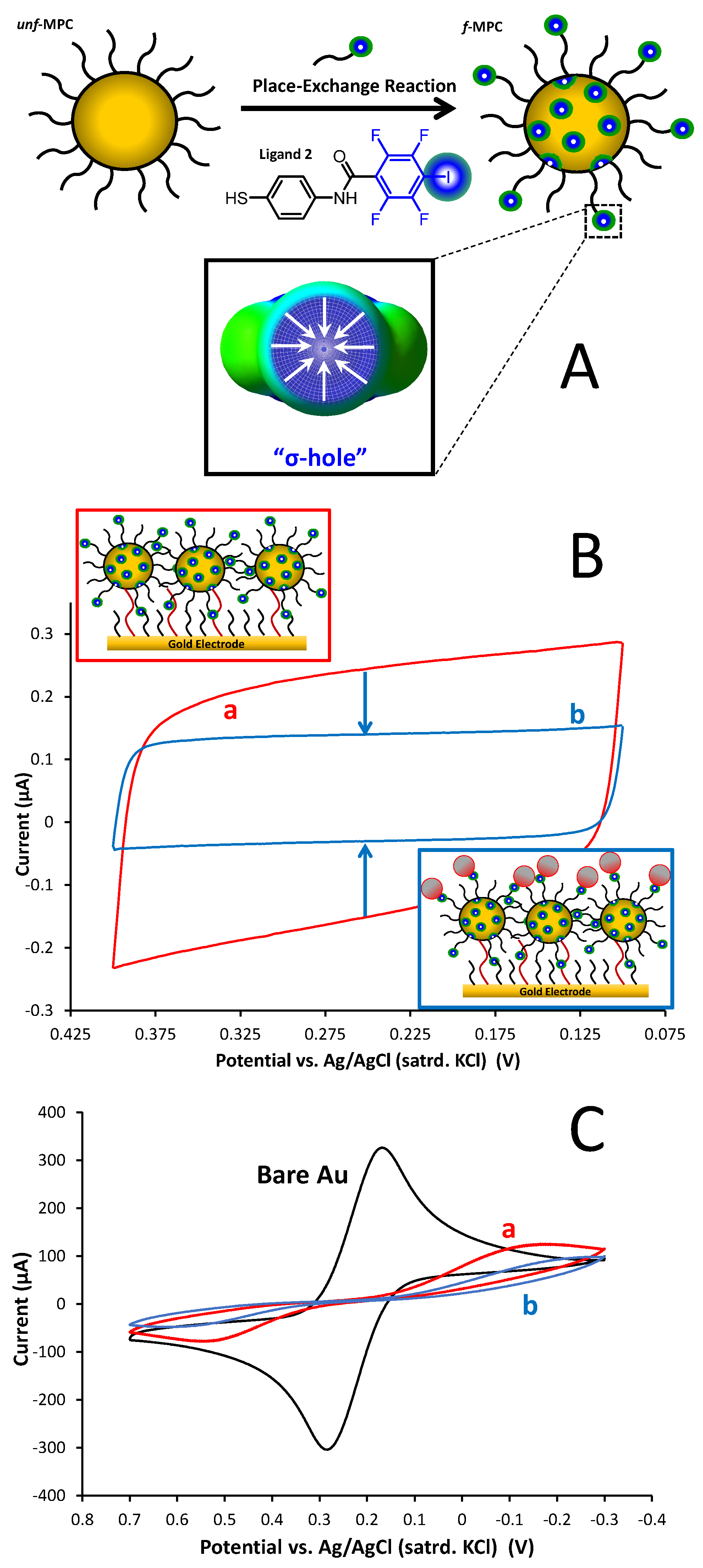
3.4. XB Interactions at Interfaces Using XB Donor-Functionalized Gold Nanoparticles
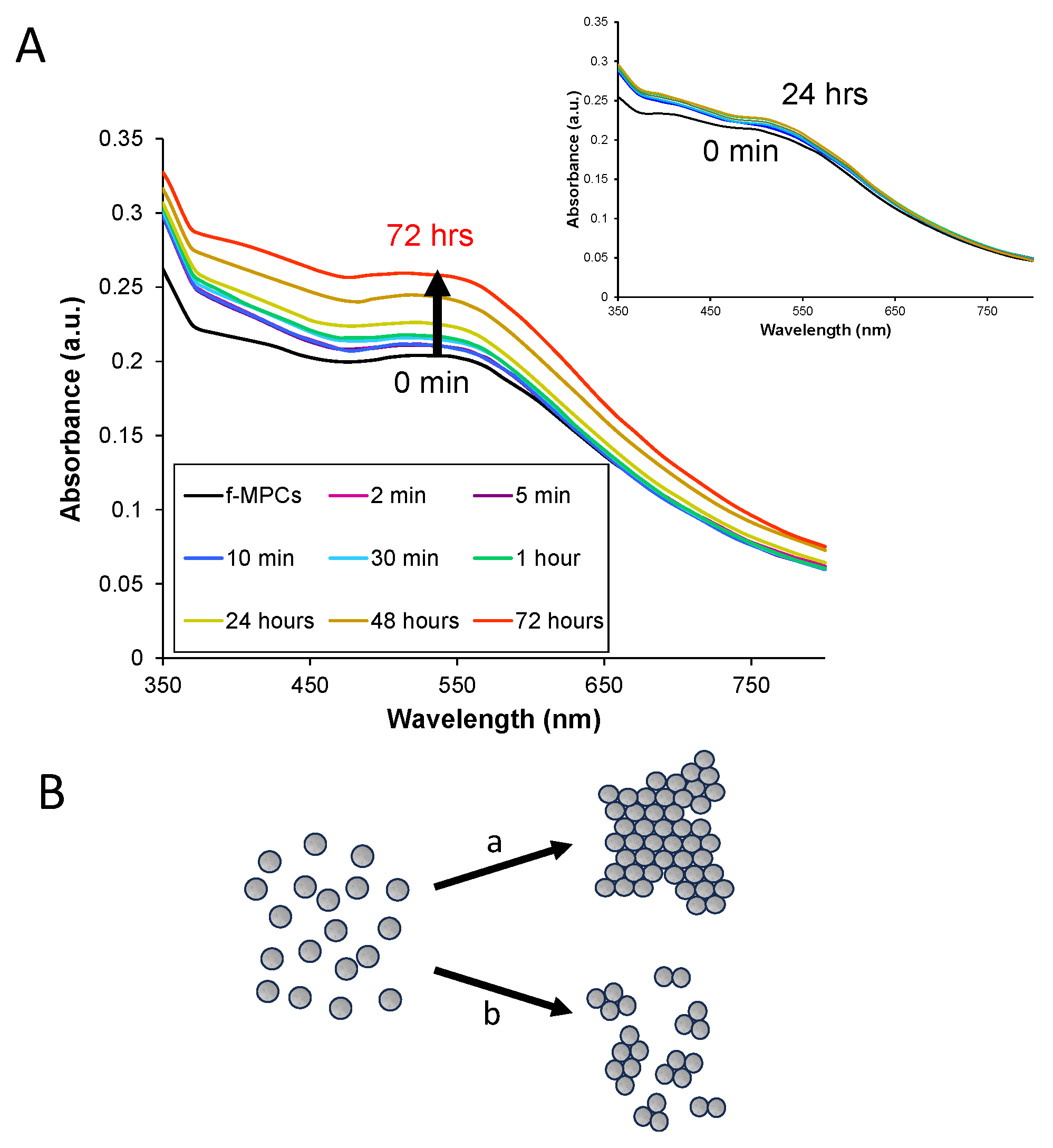
3.5. XB Interactions in Solution Using XB Donor-Functionalized Gold Nanoparticles
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vadivelu, N.; Kai, A.M.; Kodumudi, V.; Sramcik, J.; Kaye, A.D. The Opioid Crisis: A Comprehensive Overview. Curr. Pain Headache Rep. 2018, 22, 16. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, S.G.; Gordon, A.I. A crisis of opioids and the limits of prescription control: United States. Addiction 2019, 114, 169–180. [Google Scholar] [CrossRef]
- Fakayode, S.O.; Brady, P.N.; Grant, C.; Fernand Narcisse, V.; Rosado Flores, P.; Lisse, C.H.; Bwambok, D.K. Electrochemical Sensors, Biosensors, and Optical Sensors for the Detection of Opioids and Their Analogs: Pharmaceutical, Clinical, and Forensic Applications. Chemosensors 2024, 12, 58. [Google Scholar] [CrossRef]
- National Center for Drug Abuse Statistics. Drug Overdose Death Rates. Available online: https://drugabusestatistics.org/drug-overdose-deaths (accessed on 11 January 2024).
- Centers for Disease Control and Prevention. Drug Overdose Deaths. Available online: https://www.cdc.gov/drugoverdose/deaths/index.html (accessed on 11 January 2024).
- Han, Y.; Yan, W.; Zheng, Y.B.; Khan, M.Z.; Yuan, K.; Lu, L. The rising crisis of illicit fentanyl use, overdose, and potential therapeutic strategies. Transl. Psychiatry 2019, 9, 282. [Google Scholar] [CrossRef] [PubMed]
- Niles, J.K.; Gudin, J.; Radcliff, J.; Kaufman, H.W. The Opioid Epidemic Within the COVID-19 Pandemic: Drug Testing in 2020. Popul. Health Manag. 2021, 24, S43–S51. [Google Scholar] [CrossRef] [PubMed]
- Galarneau, L.R.; Hilburt, J.; O’Neill, Z.R.; Buxton, J.A.; Scheuermeyer, F.X.; Dong, K.; Kaczorowski, J.; Orkin, A.M.; Barbic, S.P.; Bath, M.; et al. Experiences of people with opioid use disorder during the COVID-19 pandemic: A qualitative study. PLoS ONE 2021, 16, e0255396. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.Q.; Kaelber, D.C.; Xu, R.; Volkow, N.D. COVID-19 risk and outcomes in patients with substance use disorders: Analyses from electronic health records in the United States. Mol. Psychiatr. 2021, 26, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Glasscott, M.W.; Vannoy, K.J.; Fernando, P.U.A.I.; Kosgei, G.K.; Moores, L.C.; Dick, J.E. Electrochemical sensors for the detection of fentanyl and its analogs: Foundations and recent advances. TrAC Trends Anal. Chem. 2020, 132, 116037. [Google Scholar] [CrossRef]
- Zhu, D.T.; Friedman, J.; Bourgois, P.; Montero, F.; Tamang, S. The emerging fentanyl-xylazine syndemic in the USA: Challenges and future directions. Lancet 2023, 402, 1949–1952. [Google Scholar] [CrossRef]
- Wu, P.E.; Austin, E. Xylazine in the illicit opioid supply. Can. Med. Assoc. J. 2024, 196, E133. [Google Scholar] [CrossRef]
- Marchei, E.; Pacifici, R.; Mannocchi, G.; Marinelli, E.; Busardò, F.P.; Pichini, S. New synthetic opioids in biological and non-biological matrices: A review of current analytical methods. TrAC Trends Anal. Chem. 2018, 102, 1–15. [Google Scholar] [CrossRef]
- Fakayode, S.O.; Lisse, C.; Medawala, W.; Brady, P.N.; Bwambok, D.K.; Anum, D.; Alonge, T.; Taylor, M.E.; Baker, G.A.; Mehari, T.F.; et al. Fluorescent chemical sensors: Applications in analytical, environmental, forensic, pharmaceutical, biological, and biomedical sample measurement, and clinical diagnosis. Appl. Spectrosc. Rev. 2024, 59, 1–89. [Google Scholar] [CrossRef]
- Rosendo, L.M.; Antunes, M.; Simao, A.Y.; Brinca, A.T.; Catarro, G.; Pelixo, R.; Martinho, J.; Pires, B.; Soares, S.; Cascalheira, J.F.; et al. Sensors in the Detection of Abused Substances in Forensic Contexts: A Comprehensive Review. Micromachines 2023, 14, 2249. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.H.; Shen, F.C.; Mishra, R.K.; Wang, Z.F.; Zhao, X.L.; Zhu, Z.G. Advances of Drugs Electroanalysis Based on Direct Electrochemical Redox on Electrodes: A Review. Crit. Rev. Anal. Chem. 2022, 54, 269–314. [Google Scholar] [CrossRef] [PubMed]
- Fulton, A.C.; Vaughan, S.R.; DeGreeff, L.E. Non-contact detection of fentanyl by a field-portable ion mobility spectrometer. Drug Test. Anal. 2022, 14, 1451–1459. [Google Scholar] [CrossRef] [PubMed]
- Angelini, D.J.; Biggs, T.D.; Prugh, A.M.; Smith, J.A.; Hanburger, J.A.; Llano, B.; Avelar, R.; Ellis, A.; Lusk, B.; Naanaa, A.; et al. Detection of fentanyl and derivatives using a lateral flow immunoassay. Forensic Chem. 2021, 23, 100309. [Google Scholar] [CrossRef]
- Gilbert, N.; Antonides, L.H.; Schofield, C.J.; Costello, A.; Kilkelly, B.; Cain, A.R.; Dalziel, P.R.V.; Horner, K.; Mewis, R.E.; Sutcliffe, O.B. Hitting the Jackpot—Development of gas chromatography-mass spectrometry (GC-MS) and other rapid screening methods for the analysis of 18 fentanyl-derived synthetic opioids. Drug Test. Anal. 2020, 12, 798–811. [Google Scholar] [CrossRef]
- Smith, C.D.; Giordano, B.C.; Collins, G.E. Assessment of opioid surrogates for colorimetric testing (Part I). Forensic Chem. 2022, 27, 100398. [Google Scholar] [CrossRef]
- Mishra, R.K.; Krishnakumar, A.; Zareei, A.; Heredia-Rivera, U.; Rahimi, R. Electrochemical sensor for rapid detection of fentanyl using laser-induced porous carbon-electrodes. Microchim. Acta 2022, 189, 198. [Google Scholar] [CrossRef]
- Lin, Y.; Sun, J.; Tang, M.; Zhang, G.; Yu, L.; Zhao, X.; Ai, R.; Yu, H.; Shao, B.; He, Y. Synergistic Recognition-Triggered Charge Transfer Enables Rapid Visual Colorimetric Detection of Fentanyl. Anal. Chem. 2021, 93, 6544–6550. [Google Scholar] [CrossRef]
- Mishra, R.K.; Goud, K.Y.; Li, Z.; Moonla, C.; Mohamed, M.A.; Tehrani, F.; Teymourian, H.; Wang, J. Continuous Opioid Monitoring along with Nerve Agents on a Wearable Microneedle Sensor Array. J. Am. Chem. Soc. 2020, 142, 5991–5995. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kumar, P.; Pournara, A.; Vellingiri, K.; Kim, K.-H. Nanomaterials for the sensing of narcotics: Challenges and opportunities. TrAC Trends Anal. Chem. 2018, 106, 84–115. [Google Scholar] [CrossRef]
- Razlansari, M.; Ulucan-Karnak, F.; Kahrizi, M.; Mirinejad, S.; Sargazi, S.; Mishra, S.; Rahdar, A.; Díez-Pascual, A.M. Nanobiosensors for detection of opioids: A review of latest advancements. Eur. J. Pharm. Biopharm. 2022, 179, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, H.; Gooch, J.; Frascione, N. Nanomaterials for optical biosensors in forensic analysis. Talanta 2023, 253, 123945. [Google Scholar] [CrossRef]
- Jun, D.N.; Sammis, G.; Rezazadeh-Azar, P.; Ginoux, E.; Bizzotto, D. Development of a Graphene-Oxide-Deposited Carbon Electrode for the Rapid and Low-Level Detection of Fentanyl and Derivatives. Anal. Chem. 2022, 94, 12706–12714. [Google Scholar] [CrossRef] [PubMed]
- González-Hernández, J.; Moya-Alvarado, G.; Alvarado-Gámez, A.L.; Urcuyo, R.; Barquero-Quirós, M.; Arcos-Martínez, M.J. Electrochemical biosensor for quantitative determination of fentanyl based on immobilized cytochrome on multi-walled carbon nanotubes modified screen-printed carbon electrodes. Microchim. Acta 2022, 189, 483. [Google Scholar] [CrossRef]
- Shao, W.T.; Zeng, Z.D.; Star, A. An Ultrasensitive Norfentanyl Sensor Based on a Carbon Nanotube-Based Field-Effect Transistor for the Detection of Fentanyl Exposure. ACS Appl. Mater. Interfaces 2023, 15, 37784–37793. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Sohouli, E.; Mousavi, F. An Electrochemical Sensor for Fentanyl Detection Based on Multi-Walled Carbon Nanotubes as Electrocatalyst and the Electrooxidation Mechanism. J. Anal. Chem. 2020, 75, 1209–1217. [Google Scholar] [CrossRef]
- Yan, K.; Wang, L.C.; Zhu, Z.H.; Duan, S.Q.; Hua, Z.D.; Xu, P.; Xu, H.; Hu, C.; Wang, Y.M.; Di, B. Cucurbituril-protected dual-readout gold nanoclusters for sensitive fentanyl detection. Analyst 2023, 148, 1253–1258. [Google Scholar] [CrossRef]
- Costa, P.J. The halogen bond: Nature and applications. Phys. Sci. Rev. 2017, 2, 20170136. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Dang, Q.M.; Simpson, J.H.; Parish, C.A.; Leopold, M.C. Evaluating Halogen-Bond Strength as a Function of Molecular Structure Using Nuclear Magnetic Resonance Spectroscopy and Computational Analysis. J. Phys. Chem. A 2021, 125, 9377–9393. [Google Scholar] [CrossRef] [PubMed]
- Weis, J.G.; Ravnsbaek, J.B.; Mirica, K.A.; Swager, T.M. Employing Halogen Bonding Interactions in Chemiresistive Gas Sensors. Acs Sens. 2016, 1, 115–119. [Google Scholar] [CrossRef]
- Hein, R.; Beer, P.D. Halogen bonding and chalcogen bonding mediated sensing. Chem. Sci. 2022, 13, 7098–7125. [Google Scholar] [CrossRef] [PubMed]
- Jaini, A.K.A.; Hughes, L.B.; Kitimet, M.M.; Ulep, K.J.; Leopold, M.C.; Parish, C.A. Halogen Bonding Interactions for Aromatic and Nonaromatic Explosive Detection. ACS Sens. 2019, 4, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Dang, Q.M.; Gilmore, S.T.; Lalwani, K.; Conk, R.J.; Simpson, J.H.; Leopold, M.C. Monolayer-Protected Gold Nanoparticles Functionalized with Halogen Bonding Capability─An Avenue for Molecular Detection Schemes. Langmuir 2022, 38, 4747–4762. [Google Scholar] [CrossRef] [PubMed]
- Sherard, M.M.; Dang, Q.M.; Reiff, S.C.; Simpson, J.H.; Leopold, M.C. On-Site Detection of Neonicotinoid Pesticides Using Functionalized Gold Nanoparticles and Halogen Bonding. ACS Appl. Nano Mater. 2023, 6, 8367–8381. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Greene, T.; Parish, C.A. Manuscript in preparation. TBD 2024.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian 16; Gaussian Inc.: Wallington, CT, USA, 2016. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Kendall, R.A., Jr.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian-Basis Sets for Use in Correlated Molecular Calculations. 1. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Peterson, K.A.; Shepler, B.C.; Figgen, D.; Stoll, H. On the spectroscopic and thermochemical properties of ClO, BrO, IO, and their anions. J. Phys. Chem. A 2006, 110, 13877–13883. [Google Scholar] [CrossRef] [PubMed]
- Stoll, H.; Metz, B.; Dolg, M. Relativistic energy-consistent pseudopotentials--recent developments. J. Comput. Chem. 2002, 23, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Xu, Z.; Zhu, W. Interaction Nature and Computational Methods for Halogen Bonding: A Perspective. J. Chem. Inf. Model. 2020, 60, 2683–2696. [Google Scholar] [CrossRef] [PubMed]
- Leopold, M.C.; Bowden, E.F. Influence of gold substrate topography on the voltammetry of cytochrome c adsorbed on carboxylic acid terminated self-assembled monolayers. Langmuir 2002, 18, 2239–2245. [Google Scholar] [CrossRef]
- Kasmi, A.E.; Wallace, J.M.; Bowden, E.F.; Binet, S.M.; Linderman, R.J. Controlling Interfacial Electron-Transfer Kinetics of Cytochrome c with Mixed Self-Assembled Monolayers. Sect. Title Gen. Biochem. 1998, 120, 225–226. [Google Scholar] [CrossRef]
- Leopold, M.C.; Doan, T.T.; Mullaney, M.J.; Loftus, A.F.; Kidd, C.M. Electrochemical characterization of self-assembled monolayers on gold substrates derived from thermal decomposition of monolayer-protected cluster films. J. Appl. Electrochem. 2015, 45, 1069–1084. [Google Scholar] [CrossRef]
- Kittredge, K.W.; Fox, M.A.; Whitesell, J.K. Effect of alkyl chain length on the fluorescence of 9-alkylfluorenyl thiols as self-assembled monolayers on gold. J. Phys. Chem. B 2001, 105, 10594–10599. [Google Scholar] [CrossRef]
- Walczak, M.M.; Popenoe, D.D.; Deinhammer, R.S.; Lamp, B.D.; Chung, C.; Porter, M.D. Reductive desorption of alkanethiolate monolayers at gold: A measure of surface coverage. Sect. Title Surf. Chem. Colloids 1991, 7, 2687–2693. [Google Scholar] [CrossRef]
- Brust, M.; Fink, J.; Bethell, D.; Schiffrin, D.J.; Kiely, C. Synthesis and Reactions of Functionalized Gold Nanoparticles. J. Chem. Soc. Chem. Comm. 1995, 16, 1655–1656. [Google Scholar] [CrossRef]
- Oberdörster, G. Safety assessment for nanotechnology and nanomedicine: Concepts of nanotoxicology. J. Intern. Med. 2010, 267, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Hostetler, M.J.; Wingate, J.E.; Zhong, C.J.; Harris, J.E.; Vachet, R.W.; Clark, M.R.; Londono, J.D.; Green, S.J.; Stokes, J.J.; Wignall, G.D.; et al. Alkanethiolate Gold Cluster Molecules with Core Diameters from 1.5 to 5.2 nm: Core and Monolayer Properties as a Function of Core Size. Langmuir 1998, 14, 17–30. [Google Scholar] [CrossRef]
- Ingram, R.S.; Hostetler, M.J.; Murray, R.W. Poly-hetero-omega-functionalized alkanethiolate-stabilized gold cluster compounds. J. Am. Chem. Soc. 1997, 119, 9175–9178. [Google Scholar] [CrossRef]
- Loftus, A.F.; Reighard, K.P.; Kapourales, S.A.; Leopold, M.C. Monolayer-protected nanoparticle film assemblies as platforms for controlling interfacial and adsorption properties in protein monolayer electrochemistry. J. Am. Chem. Soc. 2008, 130, 1649–1661. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.J.; Stewart, J.; Donald, K.J.; Parish, C.A. Halogen Bonding in DNA Base Pairs. J. Am. Chem. Soc. 2012, 134, 5165–5172. [Google Scholar] [CrossRef] [PubMed]
- Ciancaleoni, G. Characterization of Halogen Bonded Adducts in Solution by Advanced NMR Techniques. Magnetochemistry 2017, 3, 30. [Google Scholar] [CrossRef]
- Sarwar, M.G.; Dragisic, B.; Salsberg, L.J.; Gouliaras, C.; Taylor, M.S. Thermodynamics of Halogen Bonding in Solution: Substituent, Structural, and Solvent Effects. J. Am. Chem. Soc. 2010, 132, 1646–1653. [Google Scholar] [CrossRef] [PubMed]
- Thordarson, P. Determining association constants from titration experiments in supramolecular chemistry. Chem. Soc. Rev. 2011, 40, 1305–1323. [Google Scholar]
- Brett, C.M.A.; Brett, A.M.O. Electrochemistry Principles, Methods, and Applications; Oxford Univeristy Press: New York, NY, USA, 1993. [Google Scholar]
- Labban, N.; Wayu, M.B.; Steele, C.M.; Munoz, T.S.; Pollock, J.A.; Case, W.S.; Leopold, M.C. First Generation Amperometric Biosensing of Galactose with Xerogel-Carbon Nanotube Layer-By-Layer Assemblies. Nanomaterials 2019, 9, 42. [Google Scholar] [CrossRef]
- Wayu, M.B.; Pannell, M.J.; Leopold, M.C. Layered Xerogel Films Incorporating Monolayer-Protected Cluster Networks on Platinum-Black-Modified Electrodes for Enhanced Sensitivity in First-Generation Uric Acid Biosensing. Chemelectrochem 2016, 3, 1245–1252. [Google Scholar] [CrossRef]
- Vargo, M.L.; Gulka, C.P.; Gerig, J.K.; Manieri, C.M.; Dattelbaum, J.D.; Marks, C.B.; Lawrence, N.T.; Trawick, M.L.; Leopold, M.C. Distance Dependence of Electron Transfer Kinetics for Azurin Protein Adsorbed to Monolayer Protected Nanoparticle Film Assemblies. Langmuir 2010, 26, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Vilela, D.; Gonzalez, M.C.; Escarpa, A. Sensing colorimetric approaches based on gold and silver nanoparticles aggregation: Chemical creativity behind the assay. A review. Anal. Chim. Acta 2012, 751, 24–43. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, N.; Nagase, H.; Loftsson, T.; Endo, T.; Takahashi, C.; Kawashima, Y.; Ueda, H.; Yamamoto, H. Crystallographic and theoretical studies of an inclusion complex of β-cyclodextrin with fentanyl. Int. J. Pharm. 2017, 531, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Mayer, B.; Valdez, C.; Kennedy, D. Forensic analytical applications of designer cyclodextrin-modified magnetic nanoparticles for extraction of fentanyl and its analogues. J. Am. Chem. Soc. 2016, 252, 1155. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fentanyl Derivatives | X••B Interaction | ΔEint (kcal/mol) | X••B Distance (Å) | R–X••B Angle (θ) | |
|---|---|---|---|---|---|
| A | Fentanyl | N1 | −10.51 | 2.89 | 176.1 |
| O1 | −7.11 | 2.82 | 179.3 | ||
| B | a-Mefentanyl | N1 | −10.54 | 2.90 | 175.9 |
| O1 | −7.12 | 2.82 | 179.3 | ||
| C | Crotonylfentanyl | N1 | −9.29 | 2.85 | 177.0 |
| O1 | −7.14 | 2.83 | 179.3 | ||
| D | Cyclopropylfentanyl | N1 | −9.04 | 2.86 | 177.3 |
| O1 | −7.22 | 2.81 | 178.7 | ||
| E | Mefentanyl | N1 | −8.62 | 2.86 | 175.9 |
| O1 | −7.70 | 2.84 | 172.6 | ||
| F | Methoxyacetylfentanyl | N1 | −9.12 | 2.85 | 176.9 |
| O1 | −9.02 | 2.83 | 176.7 | ||
| O2 | −11.16 | 2.96 | 164.5 | ||
| G | p-Fluorobutyrylfentanyl | N1 | −10.81 | 2.90 | 176.0 |
| O1 | −7.13 | 2.83 | 179.7 | ||
| H | Ohmefentanyl | N1 | −12.52 | 2.97 | 168.7 |
| O1 | −10.07 | 2.83 | 178.6 | ||
| O2 | −9.24 | 2.92 | 170.2 | ||
| I | Phenaridine | N1 | −9.61 | 2.96 | 175.7 |
| O1 | −10.45 | 2.82 | 178.8 | ||
| J | Valerylfentanyl | N1 | −9.59 | 2.86 | 174.2 |
| O1 | −8.63 | 2.84 | 173.6 | ||
| K | p-Methoxybutyrylfentanyl | N1 | −10.23 | 2.98 | 162.7 |
| O1 | −7.48 | 2.82 | 179.3 | ||
| O2 | −4.46 | 2.92 | 175.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sherard, M.M.; Kaplan, J.S.; Simpson, J.H.; Kittredge, K.W.; Leopold, M.C. Functionalized Gold Nanoparticles and Halogen Bonding Interactions Involving Fentanyl and Fentanyl Derivatives. Nanomaterials 2024, 14, 917. https://doi.org/10.3390/nano14110917
Sherard MM, Kaplan JS, Simpson JH, Kittredge KW, Leopold MC. Functionalized Gold Nanoparticles and Halogen Bonding Interactions Involving Fentanyl and Fentanyl Derivatives. Nanomaterials. 2024; 14(11):917. https://doi.org/10.3390/nano14110917
Chicago/Turabian StyleSherard, Molly M., Jamie S. Kaplan, Jeffrey H. Simpson, Kevin W. Kittredge, and Michael C. Leopold. 2024. "Functionalized Gold Nanoparticles and Halogen Bonding Interactions Involving Fentanyl and Fentanyl Derivatives" Nanomaterials 14, no. 11: 917. https://doi.org/10.3390/nano14110917
APA StyleSherard, M. M., Kaplan, J. S., Simpson, J. H., Kittredge, K. W., & Leopold, M. C. (2024). Functionalized Gold Nanoparticles and Halogen Bonding Interactions Involving Fentanyl and Fentanyl Derivatives. Nanomaterials, 14(11), 917. https://doi.org/10.3390/nano14110917