
High Thermoelectric Performance of a Novel γ-PbSnX2 (X = S, Se, Te) Monolayer: Predicted Using First Principles



Abstract
1. Introduction
2. Computational Method
3. Results
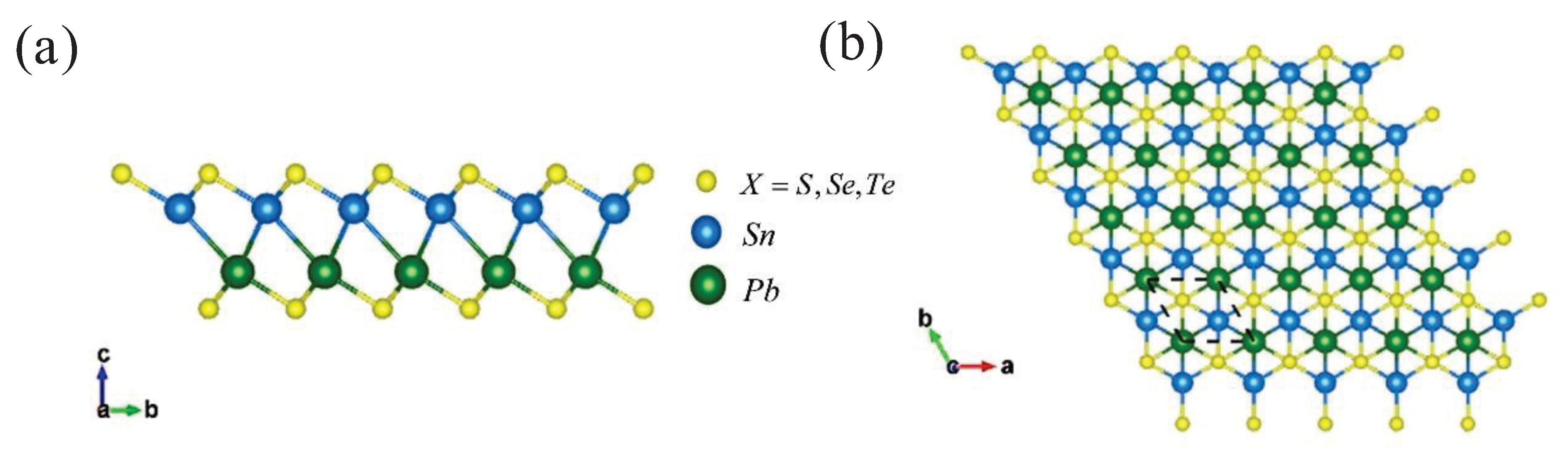
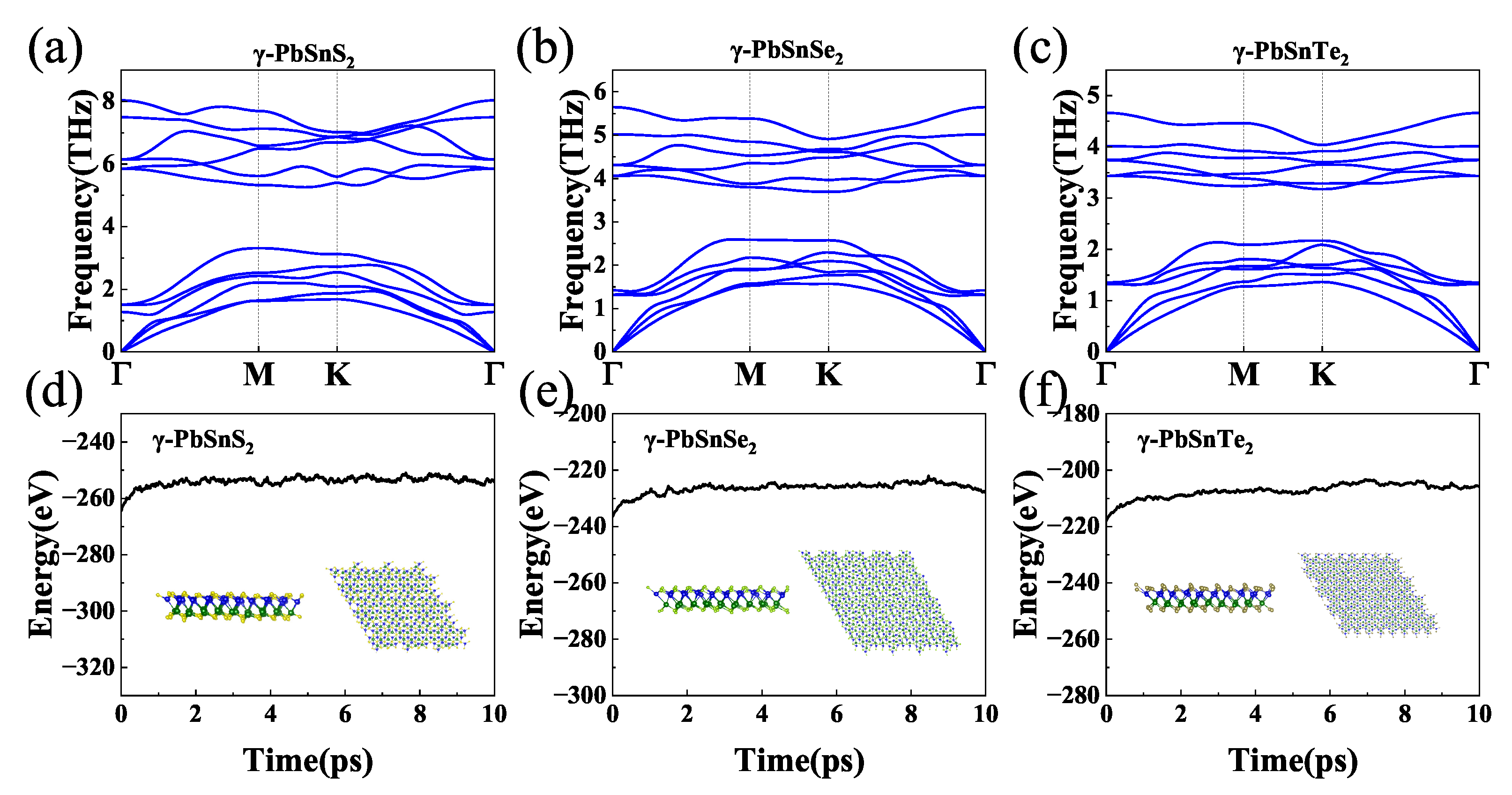
3.1. Structure and Stability
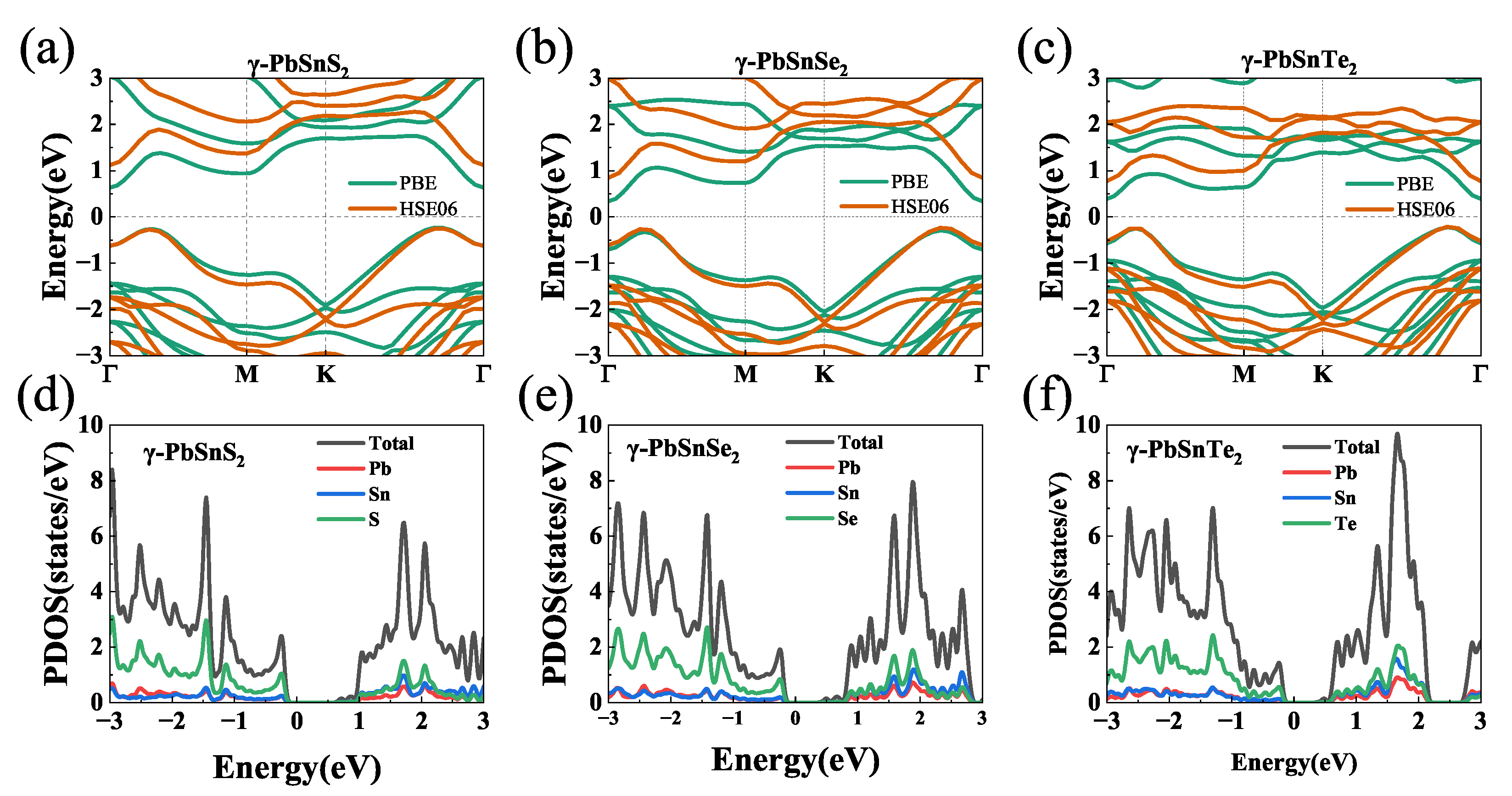
3.2. Electronic Band Structure
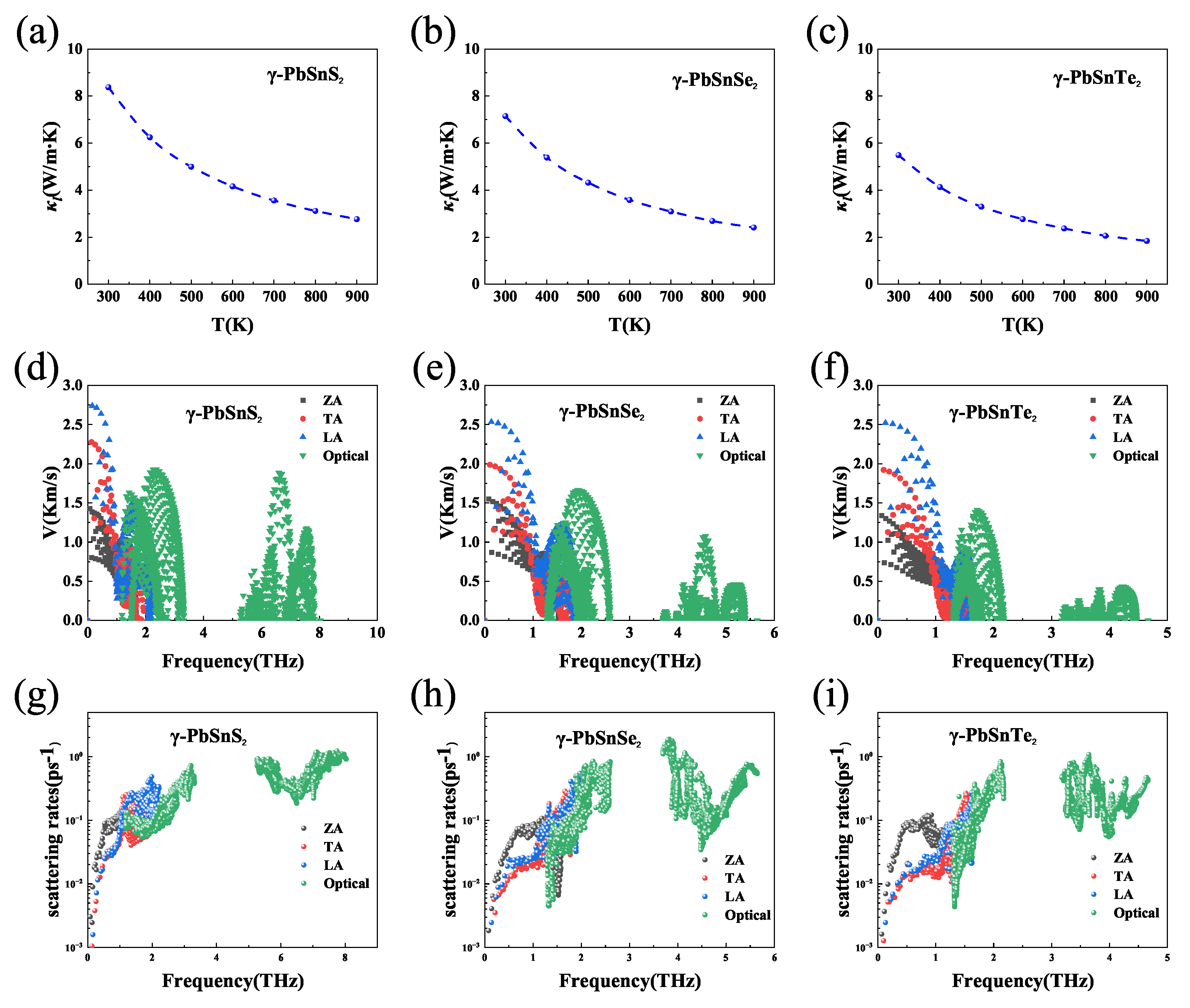
3.3. Carrier Mobility and Relaxation Time
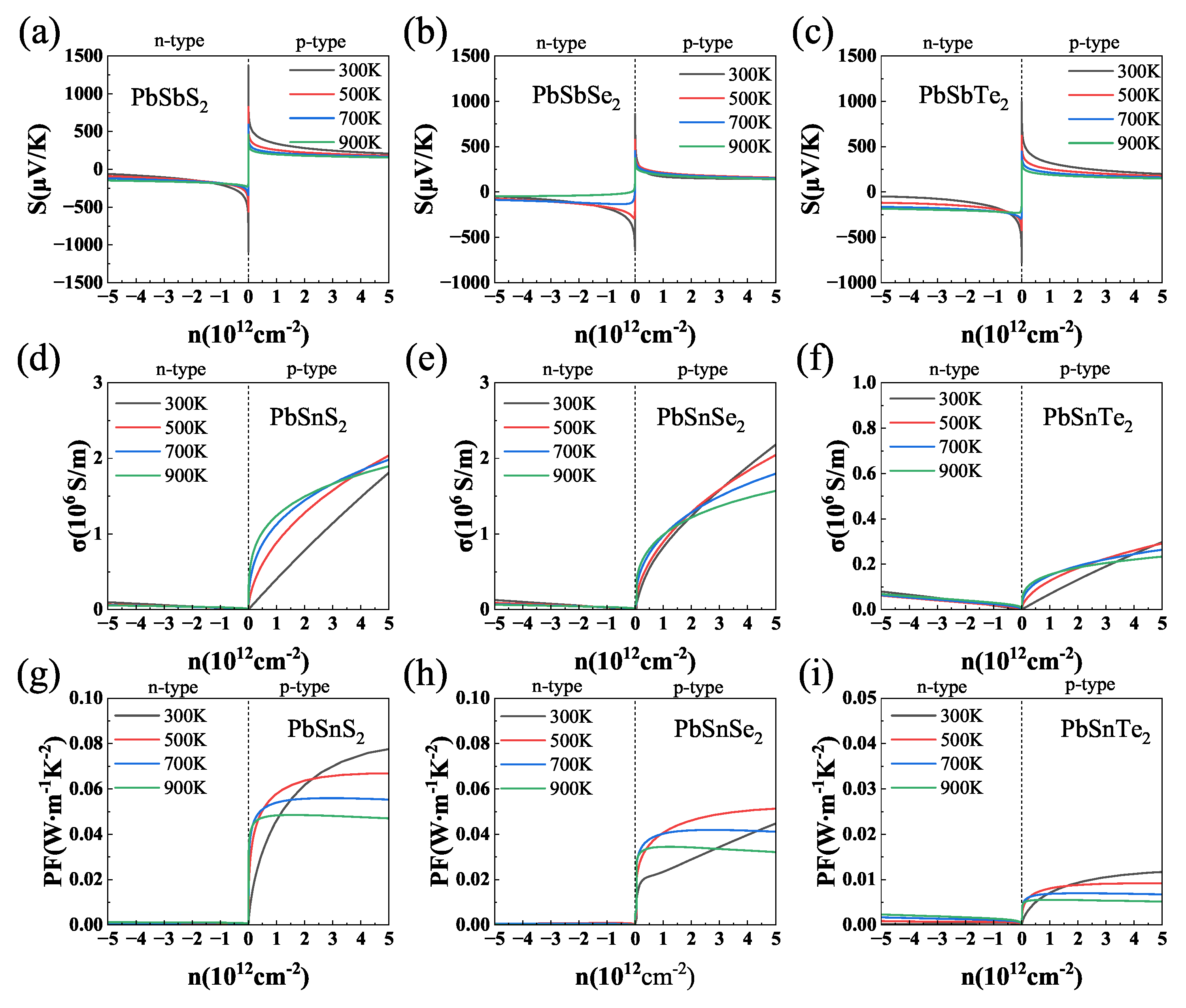
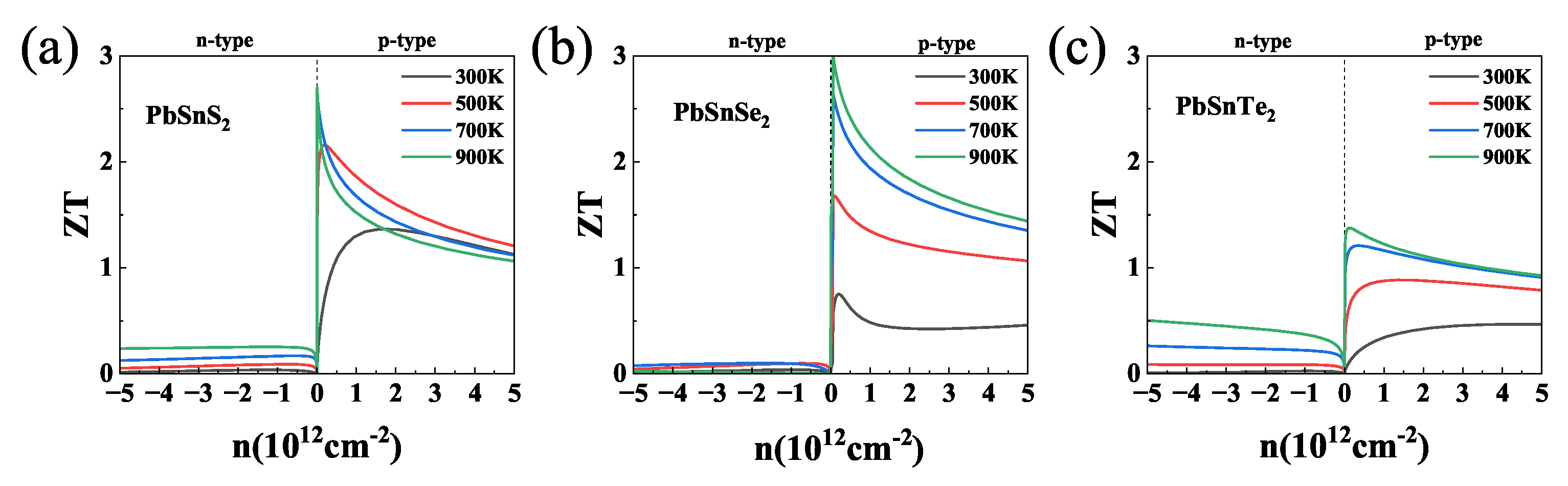
3.4. Thermoelectric Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yang, L.; Chen, Z.G.; Dargusch, M.S.; Zou, J. High Performance Thermoelectric Materials: Progress and Their Applications. Adv. Energy Mater. 2018, 8, 1701797. [Google Scholar] [CrossRef]
- Chu, S.; Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 2012, 488, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.H.; Fan, X.; Singh, D.J.; Zheng, W.T. Thermoelectric properties of monolayer GeAsSe and SnSbTe. J. Mater. Chem. C 2020, 8, 9763–9774. [Google Scholar] [CrossRef]
- Wu, D.; Cao, X.H.; Chen, S.Z.; Tang, L.M.; Feng, Y.X.; Chen, K.Q.; Zhou, W.X. Pure spin current generated in thermally driven molecular magnetic junctions: A promising mechanism for thermoelectric conversion. J. Mater. Chem. A 2019, 7, 19037–19044. [Google Scholar] [CrossRef]
- Zeng, Y.J.; Wu, D.; Cao, X.H.; Zhou, W.X.; Tang, L.M.; Chen, K.Q. Nanoscale Organic Thermoelectric Materials: Measurement, Theoretical Models, and Optimization Strategies. Adv. Funct. Mater. 2020, 30, 1903873. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, L.D. Thermoelectric materials: Energy conversion between heat and electricity. J. Mater. 2015, 1, 92–105. [Google Scholar] [CrossRef]
- Jonson, M.; Mahan, G.D. Mott’s formula for the thermopower and the Wiedemann-Franz law. Phys. Rev. B 1980, 21, 4223–4229. [Google Scholar] [CrossRef]
- Slack, G.A.; Rowe, D. CRC Handbook of Thermoelectrics; CRC Press: Boca Raton, FL, USA, 1995. [Google Scholar]
- Ding, Z.K.; Zeng, Y.J.; Pan, H.; Luo, N.; Zeng, J.; Tang, L.M.; Chen, K.Q. Edge states of topological acoustic phonons in graphene zigzag nanoribbons. Phys. Rev. B 2022, 106, L121401. [Google Scholar] [CrossRef]
- Pan, H.; Ding, Z.K.; Zeng, B.W.; Luo, N.N.; Zeng, J.; Tang, L.M.; Chen, K.Q. Ab initio Boltzmann approach to coupled magnon-phonon thermal transport in ferromagnetic crystals. Phys. Rev. B 2023, 107, 104303. [Google Scholar] [CrossRef]
- Pan, H.; Ding, Z.K.; Zeng, Y.J.; Li, Q.Q.; Tang, L.M.; Chen, K.Q. Tuning quantum heat transport in magnetic nanostructures by spin-phonon interaction. Europhys. Lett. 2022, 138, 36001. [Google Scholar] [CrossRef]
- Pan, H.; Tang, L.M.; Chen, K.Q. Quantum mechanical modeling of magnon-phonon scattering heat transport across three-dimensional ferromagnetic/nonmagnetic interfaces. Phys. Rev. B 2022, 105, 064401. [Google Scholar] [CrossRef]
- Zeng, B.; Ding, Z.K.; Pan, H.; Luo, N.; Zeng, J.; Tang, L.M.; Chen, K.Q. Strong strain-dependent phonon hydrodynamic window in bilayer graphene. Appl. Phys. Lett. 2022, 121, 252202. [Google Scholar] [CrossRef]
- Dong, B.; Wang, Z.; Hung, N.T.; Oganov, A.R.; Yang, T.; Saito, R.; Zhang, Z. New two-dimensional phase of tin chalcogenides: Candidates for high-performance thermoelectric materials. Phys. Rev. Mater. 2019, 3, 013405. [Google Scholar] [CrossRef]
- Guo, S.D. Biaxial strain tuned thermoelectric properties in monolayer PtSe2. J. Mater. Chem. C 2016, 4, 9366–9374. [Google Scholar] [CrossRef]
- Lin, C.M.; Chen, W.C.; Chen, C.C. First-principles study of strain effect on the thermoelectric properties of LaP and LaAs. Phys. Chem. Chem. Phys. 2021, 23, 18189–18196. [Google Scholar] [CrossRef]
- Miao, T.; Yu, D.; Xing, L.; Li, D.; Jiao, L.; Ma, W.; Zhang, X. Current Rectification in a Structure: ReSe2/Au Contacts on Both Sides of ReSe2. Nanoscale Res. Lett. 2019, 14, 1. [Google Scholar] [CrossRef]
- Wu, C.W.; Ren, X.; Xie, G.; Zhou, W.X.; Zhang, G.; Chen, K.Q. Enhanced High-Temperature Thermoelectric Performance by Strain Engineering in BiOCl. Phys. Rev. Appl. 2022, 18, 014053. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, Z.; Nan, P.; Xiong, F.; Lin, S.; Zhang, X.; Chen, Y.; Chen, L.; Ge, B.; Pei, Y. Lattice Strain Advances Thermoelectrics. Joule 2019, 3, 1276–1288. [Google Scholar] [CrossRef]
- Zhou, W.X.; Wu, D.; Xie, G.; Chen, K.Q.; Zhang, G. α-Ag2S: A Ductile Thermoelectric Material with High ZT. ACS Omega 2020, 5, 5796–5804. [Google Scholar] [CrossRef]
- Gu, B.C.; Li, Z.; Liu, J.D.; Zhang, H.J.; Ye, B.J. Effect of vacancies on thermoelectric properties of β-CuAgSe studied by positron annihilation. Appl. Phys. Lett. 2019, 115, 192106. [Google Scholar] [CrossRef]
- Han, D.; Yang, X.; Du, M.; Xin, G.; Zhang, J.; Wang, X.; Cheng, L. Improved thermoelectric properties of WS2–WSe2 phononic crystals: Insights from first-principles calculations. Nanoscale 2021, 13, 7176–7192. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.X.; Chen, X.K.; Yu, X.; Deng, Y.X.; Zhang, Y.; Zhou, W.X.; Jia, P.Z. Intrinsic thermoelectric properties in biphenylene nanoribbons and effect of lattice defects. Comput. Mater. Sci. 2023, 220, 112041. [Google Scholar] [CrossRef]
- Cao, X.H.; Wu, D.; Feng, Y.X.; Zhou, W.X.; Tang, L.M.; Chen, K.Q. Effect of electrophilic substitution and destructive quantum interference on the thermoelectric performance in molecular devices. J. Phys. Condens. Matter 2019, 31, 345303. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.X.; Chen, S.Z.; Hong, J.; Jia, P.Z.; Zhang, Y.; Yu, X.; Chen, K.Q. Perfect spin-filtering effect in molecular junctions based on half-metallic penta-hexa-graphene nanoribbons. J. Phys. Condens. Matter 2022, 34, 285302. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.J.; Wu, D.; Cao, X.H.; Feng, Y.X.; Tang, L.M.; Chen, K.Q. Significantly enhanced thermoelectric performance of molecular junctions by the twist angle dependent phonon interference effect. J. Mater. Chem. A 2020, 8, 11884–11891. [Google Scholar] [CrossRef]
- Gibson, Q.D.; Zhao, T.; Daniels, L.M.; Walker, H.C.; Daou, R.; Hebert, S.; Zanella, M.; Dyer, M.S.; Claridge, J.B.; Slater, B.; et al. Low thermal conductivity in a modular inorganic material with bonding anisotropy and mismatch. Science 2021, 373, 1017–1022. [Google Scholar] [CrossRef]
- Wang, J.; Cao, X.H.; Zeng, Y.J.; Luo, N.N.; Tang, L.M.; Chen, K.Q. Excellent thermoelectric properties of monolayer MoS2-MoSe2 aperiodic superlattices. Appl. Surf. Sci. 2023, 612, 155914. [Google Scholar] [CrossRef]
- Xie, Z.X.; Zhang, Y.; Yu, X.; Li, K.M.; Chen, Q. Ballistic thermal conductance by phonons through superlattice quantum-waveguides. J. Appl. Phys. 2014, 115, 104309. [Google Scholar] [CrossRef]
- Jia, P.Z.; Xie, J.P.; Chen, X.K.; Zhang, Y.; Yu, X.; Zeng, Y.J.; Xie, Z.X.; Deng, Y.X.; Zhou, W.X. Recent progress of two-dimensional heterostructures for thermoelectric applications. J. Phys. Condens. Matter 2023, 35, 073001. [Google Scholar] [CrossRef]
- Jia, P.Z.; Zeng, Y.J.; Wu, D.; Pan, H.; Cao, X.H.; Zhou, W.X.; Xie, Z.X.; Zhang, J.X.; Chen, K.Q. Excellent thermoelectric performance induced by interface effect in MoS2/MoSe2 van der Waals heterostructure. J. Phys. Condens. Matter 2020, 32, 055302. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, Y.W. Thermoelectric properties of two-dimensional transition metal dichalcogenides. J. Mater. Chem. C 2017, 5, 7684–7698. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, Y.; Rui, K.; Hng, H.H.; Hippalgaonkar, K.; Xu, J.; Sun, W.; Zhu, J.; Yan, Q.; Huang, W. 2D Black Phosphorus for Energy Storage and Thermoelectric Applications. Small 2017, 13, 1700661. [Google Scholar] [CrossRef]
- Hicks, L.D.; Dresselhaus, M.S. Thermoelectric figure of merit of a one-dimensional conductor. Phys. Rev. B 1993, 47, 16631–16634. [Google Scholar] [CrossRef]
- Shafique, A.; Shin, Y.H. Thermoelectric and phonon transport properties of two-dimensional IV–VI compounds. Sci. Rep. 2017, 7, 506. [Google Scholar] [CrossRef]
- Babaei, H.; Khodadadi, J.M.; Sinha, S. Large theoretical thermoelectric power factor of suspended single-layer MoS2. Appl. Phys. Lett. 2014, 105, 193901. [Google Scholar] [CrossRef]
- Chang, C.; Wu, M.; He, D.; Pei, Y.; Wu, C.F.; Wu, X.; Yu, H.; Zhu, F.; Wang, K.; Chen, Y.; et al. 3D charge and 2D phonon transports leading to high out-of-plane ZT in n-type SnSe crystals. Science 2018, 360, 778–783. [Google Scholar] [CrossRef]
- Zhao, L.D.; Lo, S.H.; Zhang, Y.; Sun, H.; Tan, G.; Uher, C.; Wolverton, C.; Dravid, V.P.; Kanatzidis, M.G. Ultralow thermal conductivity and high thermoelectric figure of merit in SnSe crystals. Nature 2014, 508, 373–377. [Google Scholar] [CrossRef]
- Gupta, R.; Kakkar, S.; Dongre, B.; Carrete, J.; Bera, C. Enhancement in the Thermoelectric Performance of SnS Monolayer by Strain Engineering. ACS Appl. Energy Mater. 2023, 6, 3944–3952. [Google Scholar] [CrossRef]
- Lu, A.Y.; Zhu, H.; Xiao, J.; Chuu, C.P.; Han, Y.; Chiu, M.H.; Cheng, C.C.; Yang, C.W.; Wei, K.H.; Yang, Y.; et al. Janus monolayers of transition metal dichalcogenides. Nat. Nanotechnol. 2017, 12, 744–749. [Google Scholar] [CrossRef]
- Sa, B.; Sun, Z.; Wu, B. The development of two dimensional group IV chalcogenides, blocks for van der Waals heterostructures. Nanoscale 2016, 8, 1169–1178. [Google Scholar] [CrossRef]
- Jia, P.Z.; Xie, Z.X.; Deng, Y.X.; Zhang, Y.; Tang, L.M.; Zhou, W.X.; Chen, K.Q. High thermoelectric performance induced by strong anharmonic effects in monolayer (PbX)2 (X = S, Se, Te). Appl. Phys. Lett. 2022, 121, 043901. [Google Scholar] [CrossRef]
- Eivari, H.A.; Sohbatzadeh, Z.; Mele, P.; Assadi, M.H.N. Low thermal conductivity: Fundamentals and theoretical aspects in thermoelectric applications. Mater. Today Energy 2021, 21, 100744. [Google Scholar] [CrossRef]
- Gorai, P.; Stevanović, V.; Toberer, E.S. Computationally guided discovery of thermoelectric materials. Nat. Rev. Mater. 2017, 2, 17053. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, R.E.; Scuseria, G.E.; Frisch, M.J. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem. Phys. 1998, 109, 8218–8224. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561, PRB. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269, PRB. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775, PRB. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Madsen, G.K.H.; Singh, D.J. BoltzTraP. A code for calculating band-structure dependent quantities. Comput. Phys. Commun. 2006, 175, 67–71. [Google Scholar] [CrossRef]
- Li, W.; Carrete, J.; Katcho, N.A.; Mingo, N. ShengBTE: A solver of the Boltzmann transport equation for phonons. Comput. Phys. Commun. 2014, 185, 1747–1758. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104, PRB. [Google Scholar] [CrossRef]
- Bruzzone, S.; Fiori, G. Ab-initio simulations of deformation potentials and electron mobility in chemically modified graphene and two-dimensional hexagonal boron-nitride. Appl. Phys. Lett. 2011, 99, 222108. [Google Scholar] [CrossRef]
- Fiori, G.; Iannaccone, G. Multiscale Modeling for Graphene-Based Nanoscale Transistors. Proc. IEEE 2013, 101, 1653–1669. [Google Scholar] [CrossRef]
- Lang, H.; Zhang, S.; Liu, Z. Mobility anisotropy of two-dimensional semiconductors. Phys. Rev. B 2016, 94, 235306. [Google Scholar] [CrossRef]
- Wang, V.; Xu, N.; Liu, J.C.; Tang, G.; Geng, W.T. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Rawat, A.; Jena, N.; Dimple; De Sarkar, A. A comprehensive study on carrier mobility and artificial photosynthetic properties in group VI B transition metal dichalcogenide monolayers. J. Mater. Chem. A 2018, 6, 8693–8704. [Google Scholar] [CrossRef]
- Shafique, A.; Samad, A.; Shin, Y.H. Ultra low lattice thermal conductivity and high carrier mobility of monolayer SnS2 and SnSe2: A first principles study. Phys. Chem. Chem. Phys. 2017, 19, 20677–20683. [Google Scholar] [CrossRef] [PubMed]
- Hung, N.T.; Hasdeo, E.H.; Nugraha, A.R.; Dresselhaus, M.S.; Saito, R. Quantum Effects in the Thermoelectric Power Factor of Low-Dimensional Semiconductors. Phys. Rev. Lett. 2016, 117, 036602. [Google Scholar] [CrossRef] [PubMed]
- Hung, N.T.; Nugraha, A.R.T.; Yang, T.; Saito, R. Confinement Effect in Thermoelectric Properties of Two–Dimensional Materials. MRS Adv. 2020, 5, 469–479. [Google Scholar] [CrossRef]
- Li, Y.; Wu, M.N.; Ding, T.; Ma, K.; Liu, F.S.; Ao, W.Q.; Li, J.Q. Promising thermoelectric properties and anisotropic electrical and thermal transport of monolayer SnTe. Appl. Phys. Lett. 2019, 114, 083901. [Google Scholar] [CrossRef]
- Kumar, S.; Schwingenschlögl, U. Thermoelectric Response of Bulk and Monolayer MoSe2 and WSe2. Chem. Mater. 2015, 27, 1278–1284. [Google Scholar] [CrossRef]
- Patel, A.; Singh, D.; Sonvane, Y.; Thakor, P.B.; Ahuja, R. High Thermoelectric Performance in Two-Dimensional Janus Monolayer Material WS-X (X = Se and Te). Acs Appl. Mater. Interfaces 2020, 12, 46212–46219. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | a (Å) | (Å) | (Å) | (Å) | h (Å) | (N/m) | (N/m) | (N/m) |
|---|---|---|---|---|---|---|---|---|
| PbSnS2 | 3.96 | 2.64 | 2.68 | 3.58 | 5.49 | 39.5 | 12.9 | 13.2 |
| PbSnSe2 | 4.11 | 2.77 | 2.81 | 3.51 | 5.52 | 42.7 | 14.5 | 14.1 |
| PbSnTe2 | 4.37 | 2.97 | 3.01 | 3.46 | 5.59 | 43.5 | 12.7 | 15.4 |
| Material | Structure | Gap-Type | Eg (PBE) | Eg (HSE06) |
|---|---|---|---|---|
| PbSnS2 | hexagonal (2D) | Indirect | 0.86 eV | 1.37 eV |
| PbSnSe2 | hexagonal (2D) | Indirect | 0.63 eV | 1.08 eV |
| PbSnTe2 | hexagonal (2D) | Indirect | 0.61 eV | 0.98 eV |
| Material | Carrier | (N/m) | (eV) | (×103 cm2 V−1 s−1) | (ps) | |
|---|---|---|---|---|---|---|
| PbSnS2 | e | 39.5 | 0.22 | 4.92 | 0.476 | 0.059 |
| h | 0.50 | 1.02 | 4.04 | 1.14 | ||
| PbSnSe2 | e | 42.75 | 0.18 | 5.52 | 0.654 | 0.067 |
| h | 0.357 | 2.66 | 1.421 | 0.288 | ||
| PbSnTe2 | e | 43.55 | 0.23 | 6.52 | 0.256 | 0.033 |
| h | 0.596 | 4.14 | 0.245 | 0.083 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, C.; Duan, Z.; Luo, N.; Zeng, J.; Ren, W.; Tang, L.; Chen, K. High Thermoelectric Performance of a Novel γ-PbSnX2 (X = S, Se, Te) Monolayer: Predicted Using First Principles. Nanomaterials 2023, 13, 1519. https://doi.org/10.3390/nano13091519
Ding C, Duan Z, Luo N, Zeng J, Ren W, Tang L, Chen K. High Thermoelectric Performance of a Novel γ-PbSnX2 (X = S, Se, Te) Monolayer: Predicted Using First Principles. Nanomaterials. 2023; 13(9):1519. https://doi.org/10.3390/nano13091519
Chicago/Turabian StyleDing, Changhao, Zhifu Duan, Nannan Luo, Jiang Zeng, Wei Ren, Liming Tang, and Keqiu Chen. 2023. "High Thermoelectric Performance of a Novel γ-PbSnX2 (X = S, Se, Te) Monolayer: Predicted Using First Principles" Nanomaterials 13, no. 9: 1519. https://doi.org/10.3390/nano13091519
APA StyleDing, C., Duan, Z., Luo, N., Zeng, J., Ren, W., Tang, L., & Chen, K. (2023). High Thermoelectric Performance of a Novel γ-PbSnX2 (X = S, Se, Te) Monolayer: Predicted Using First Principles. Nanomaterials, 13(9), 1519. https://doi.org/10.3390/nano13091519