



CO2 Hydrogenation: Na Doping Promotes CO and Hydrocarbon Formation over Ru/m-ZrO2 at Elevated Pressures in Gas Phase Media

 ,
,  , ,
, , 

Abstract
1. Introduction
2. Materials and Methods
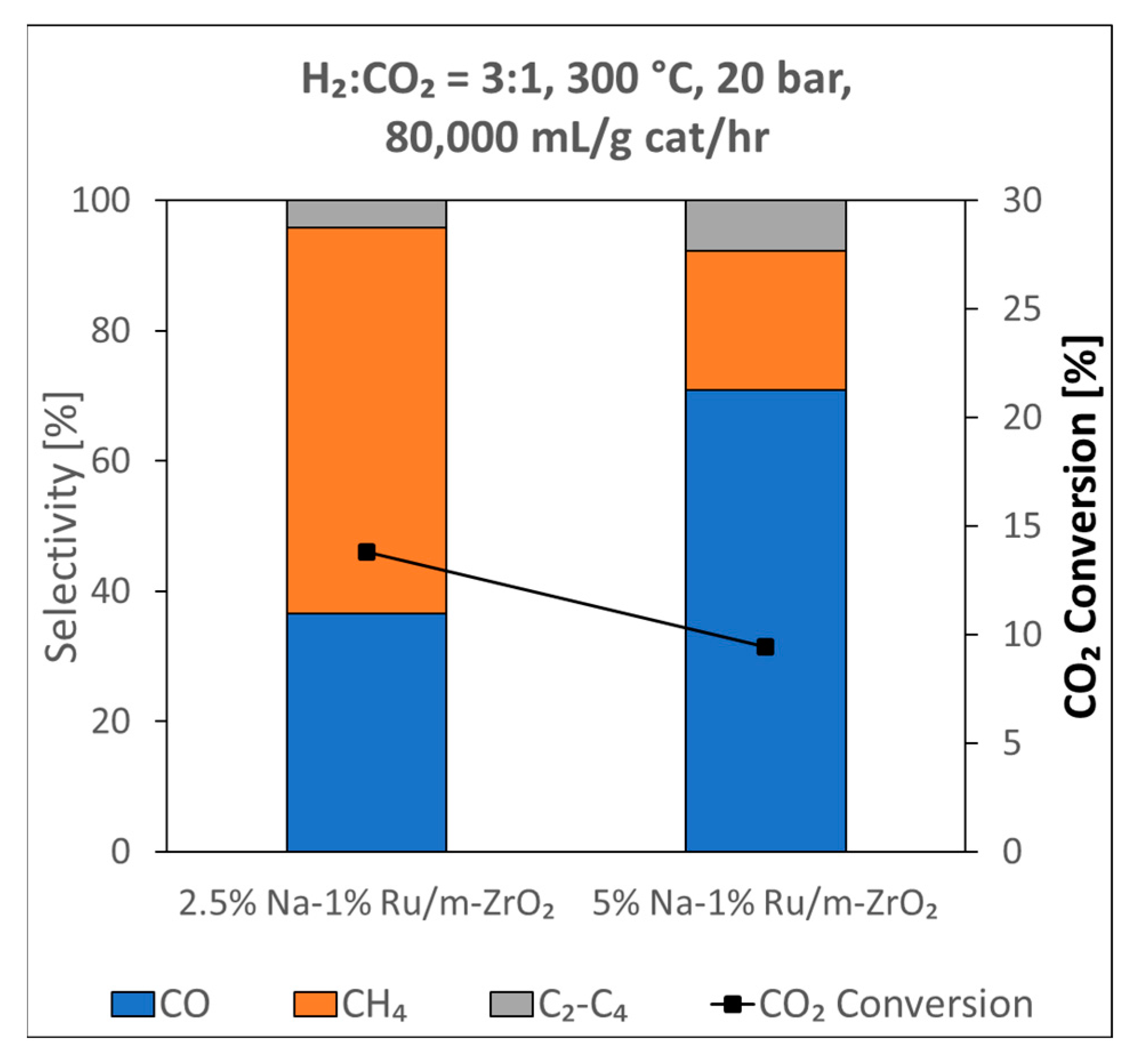
3. Results
3.1. Catalyst Characterization
3.1.1. BET Surface Area and Porosity
3.1.2. Chemisorption by H2-TPD
3.1.3. Catalyst Activation
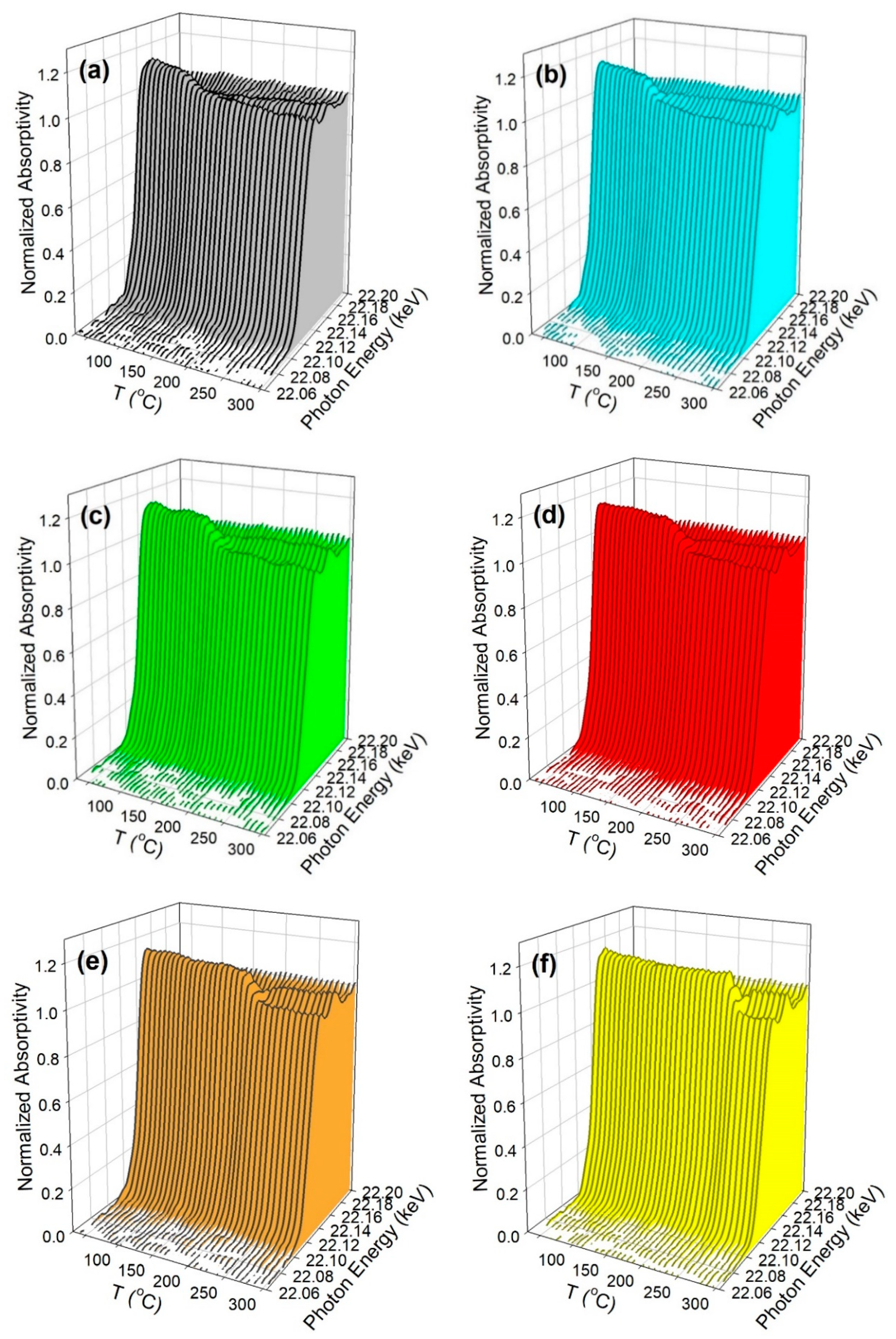
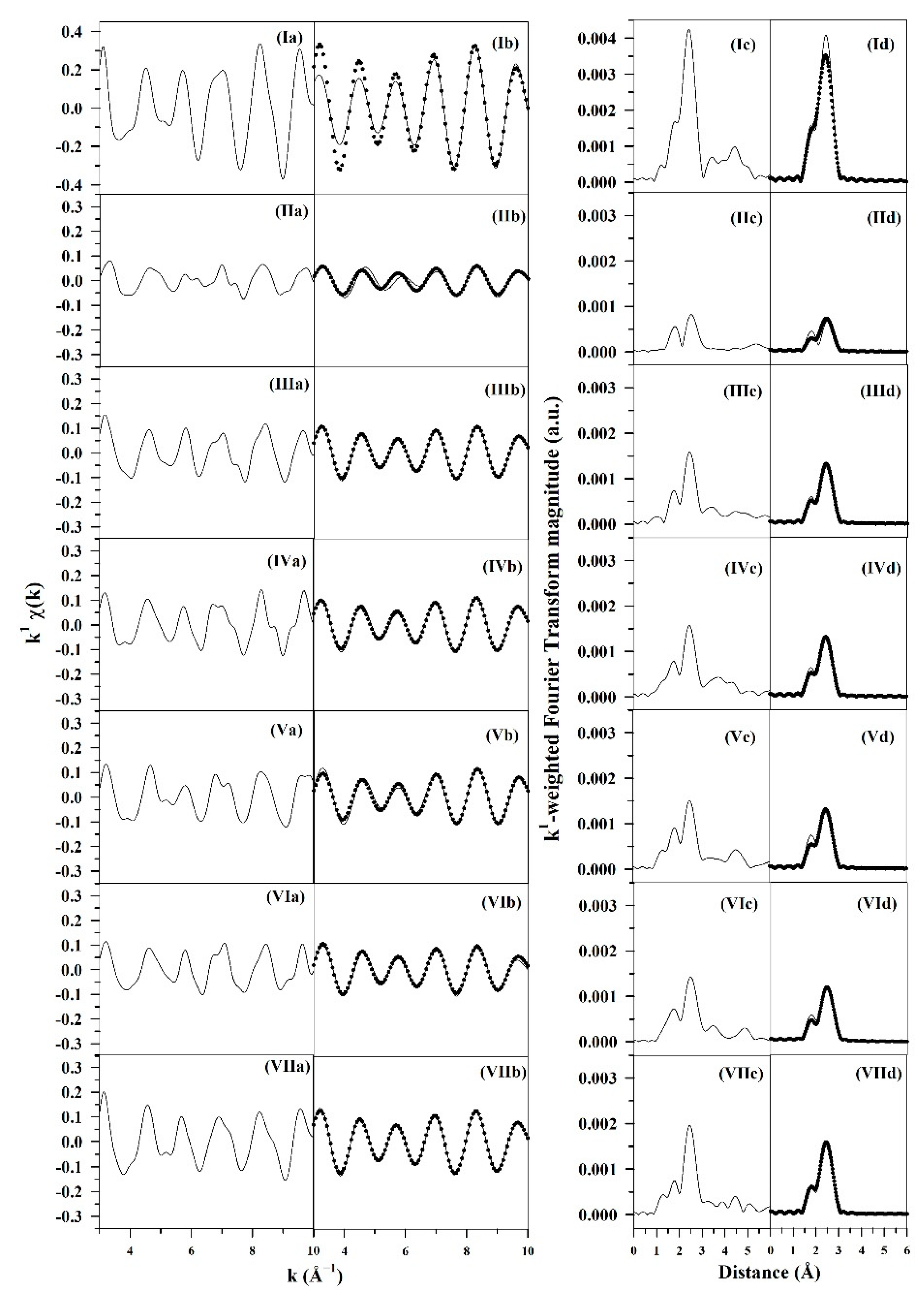
3.1.4. Synchrotron Methods
3.1.5. Thermal Desorption of CO2
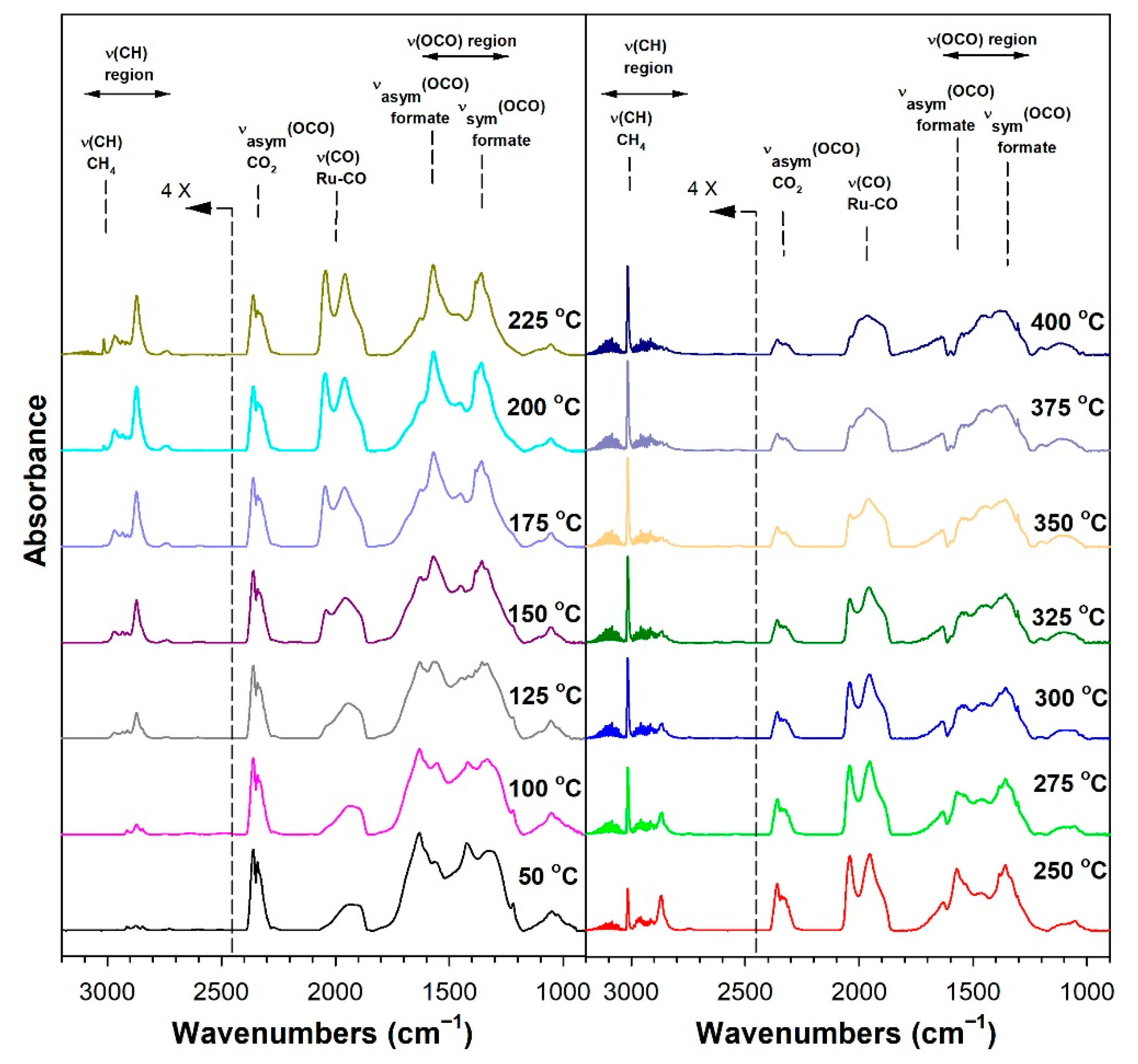
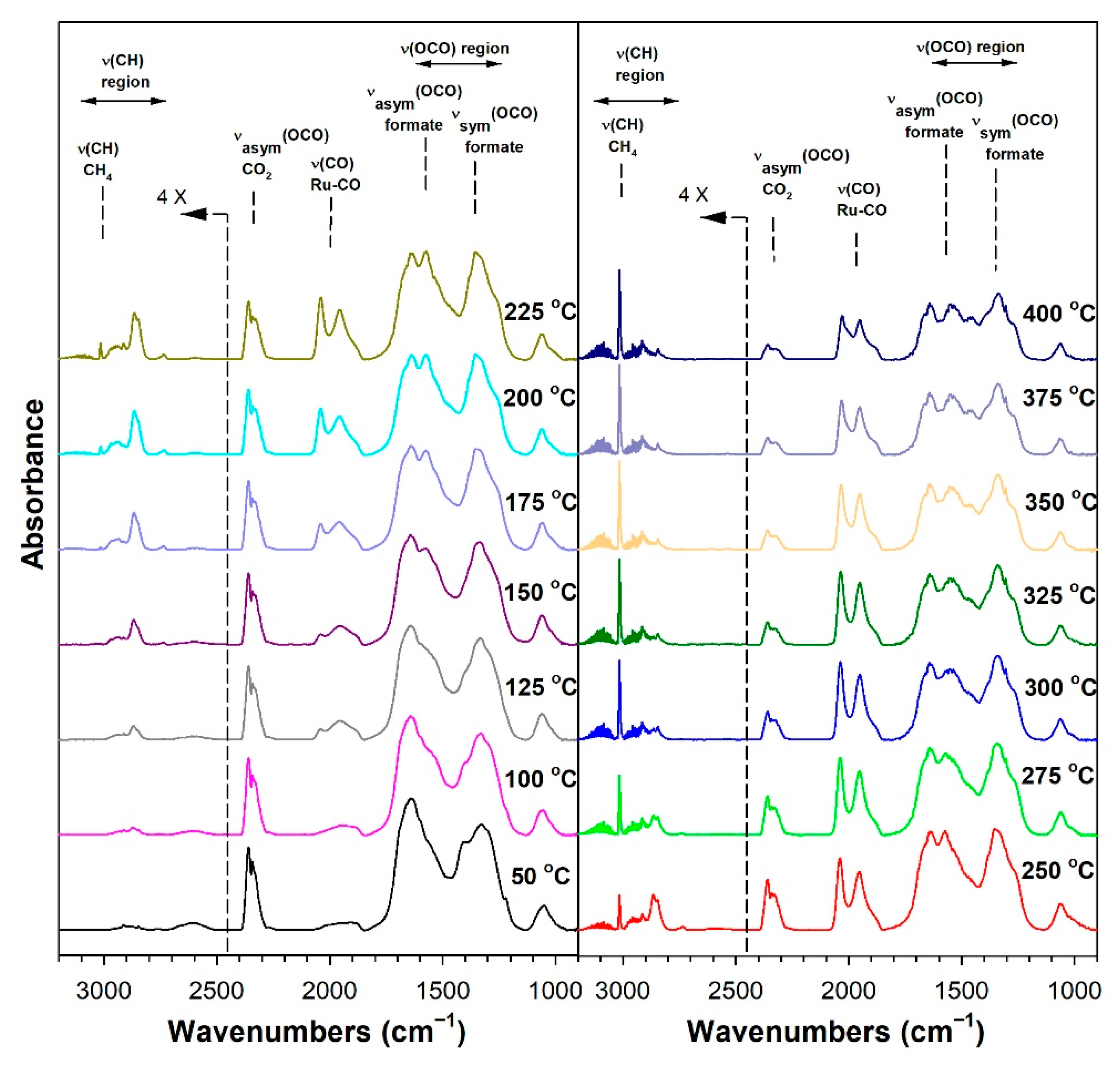
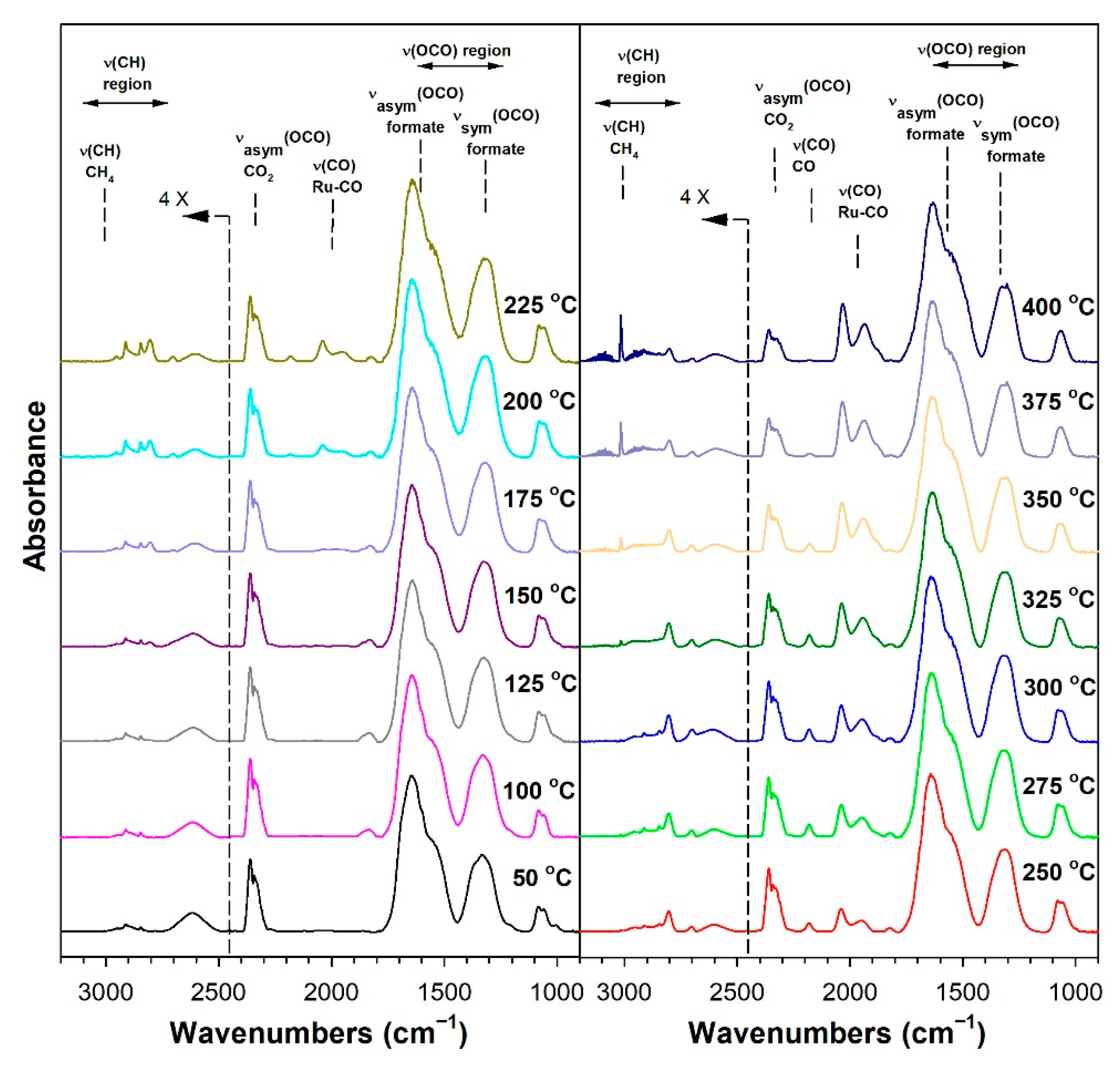
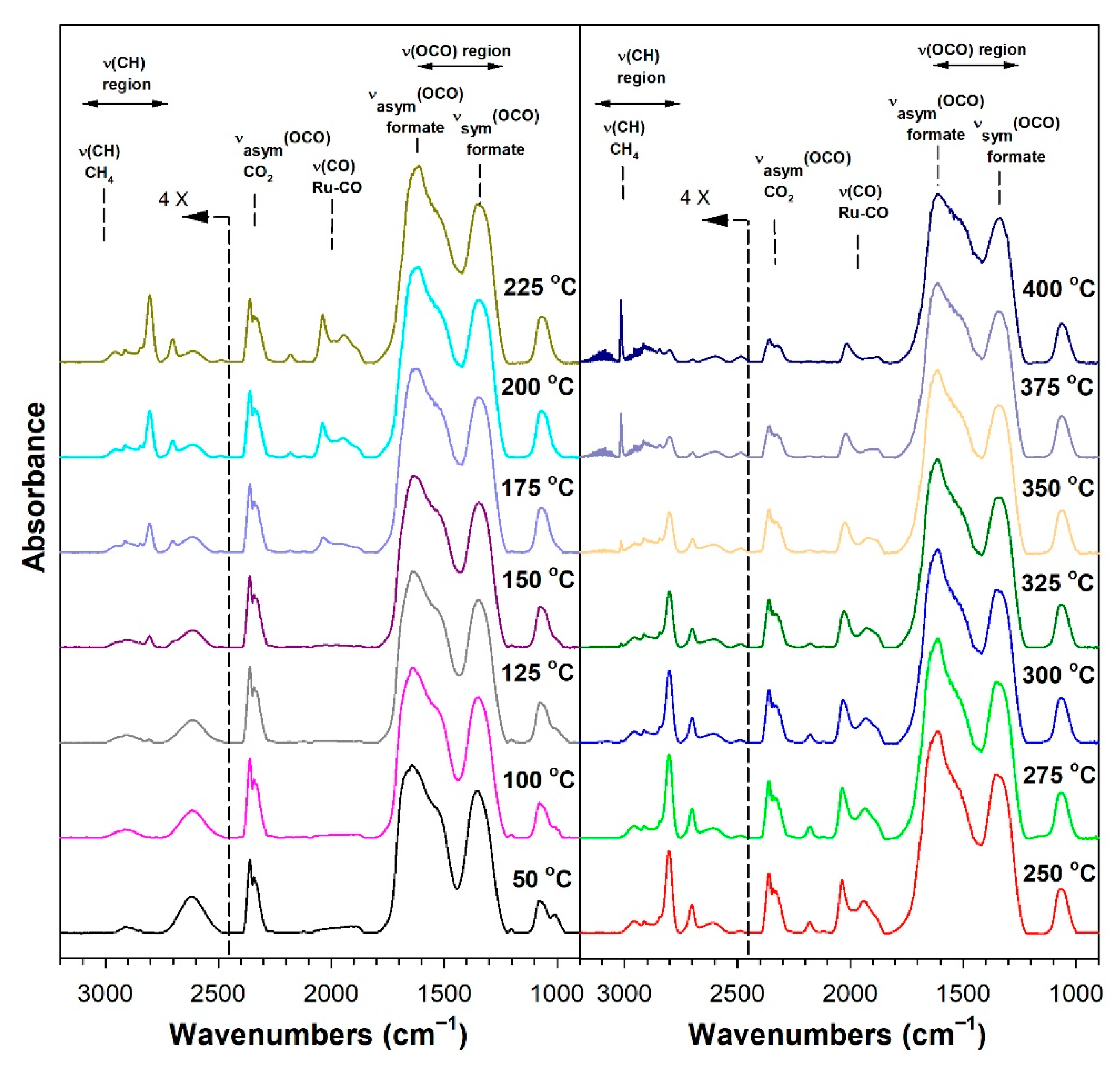
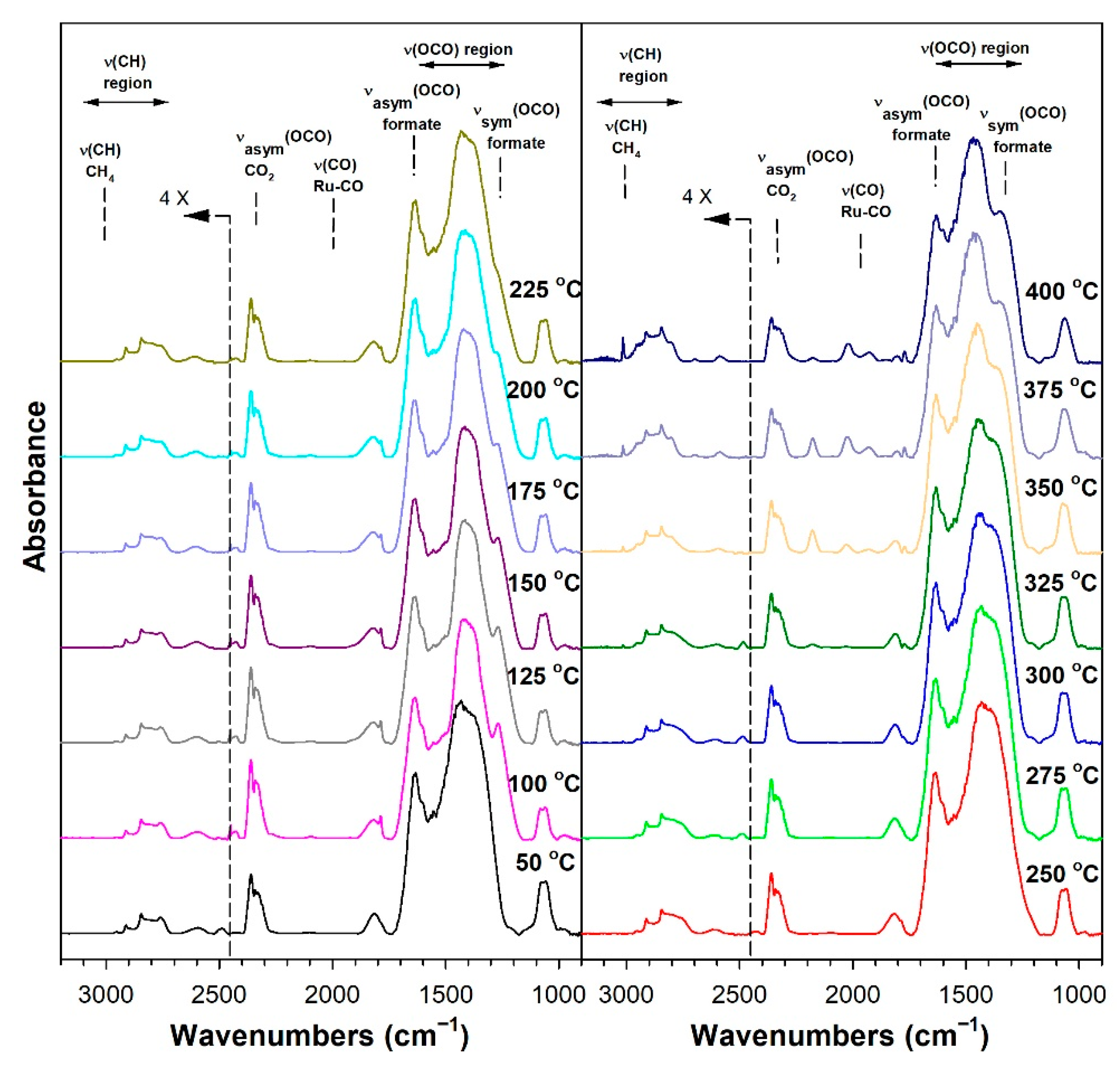
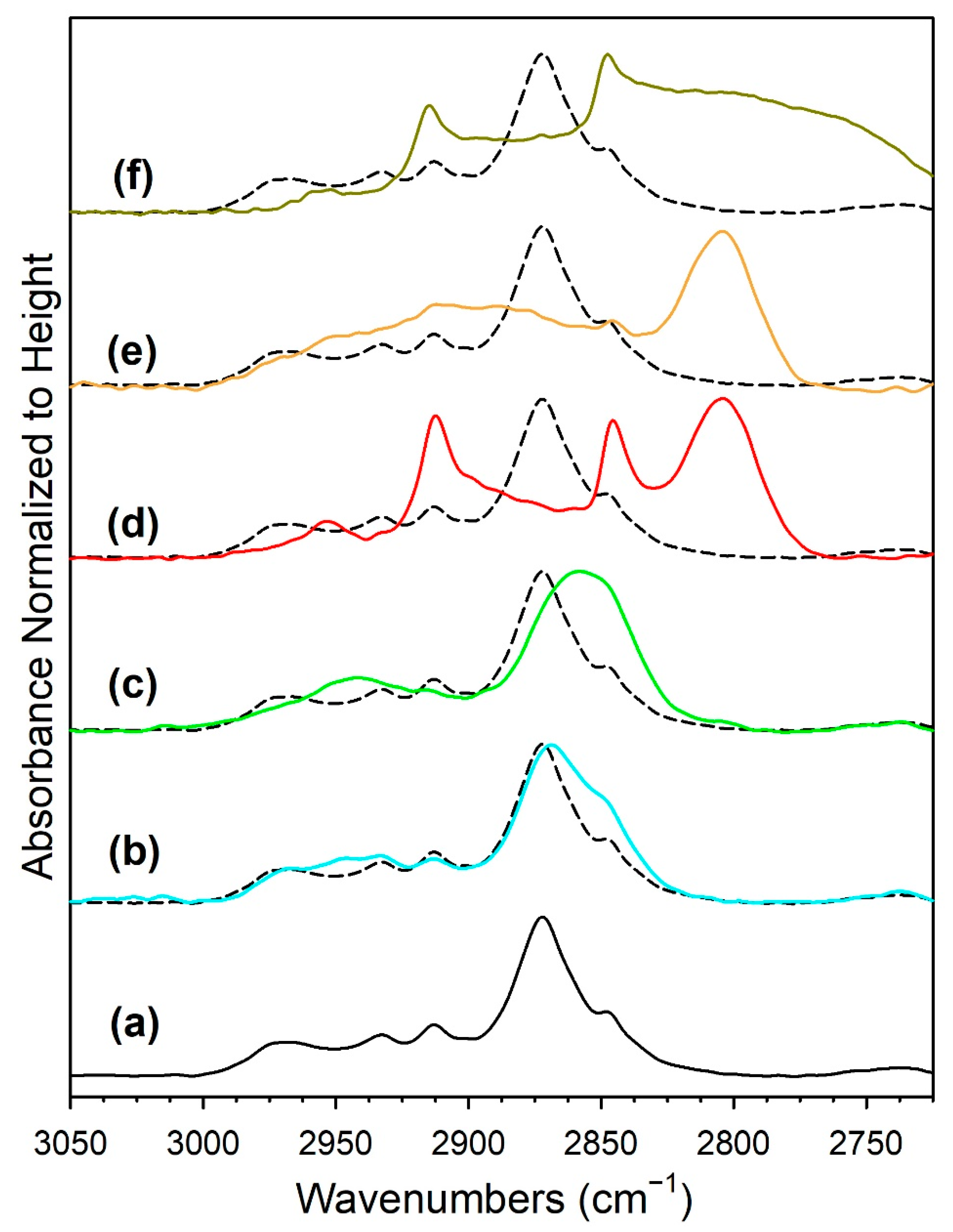
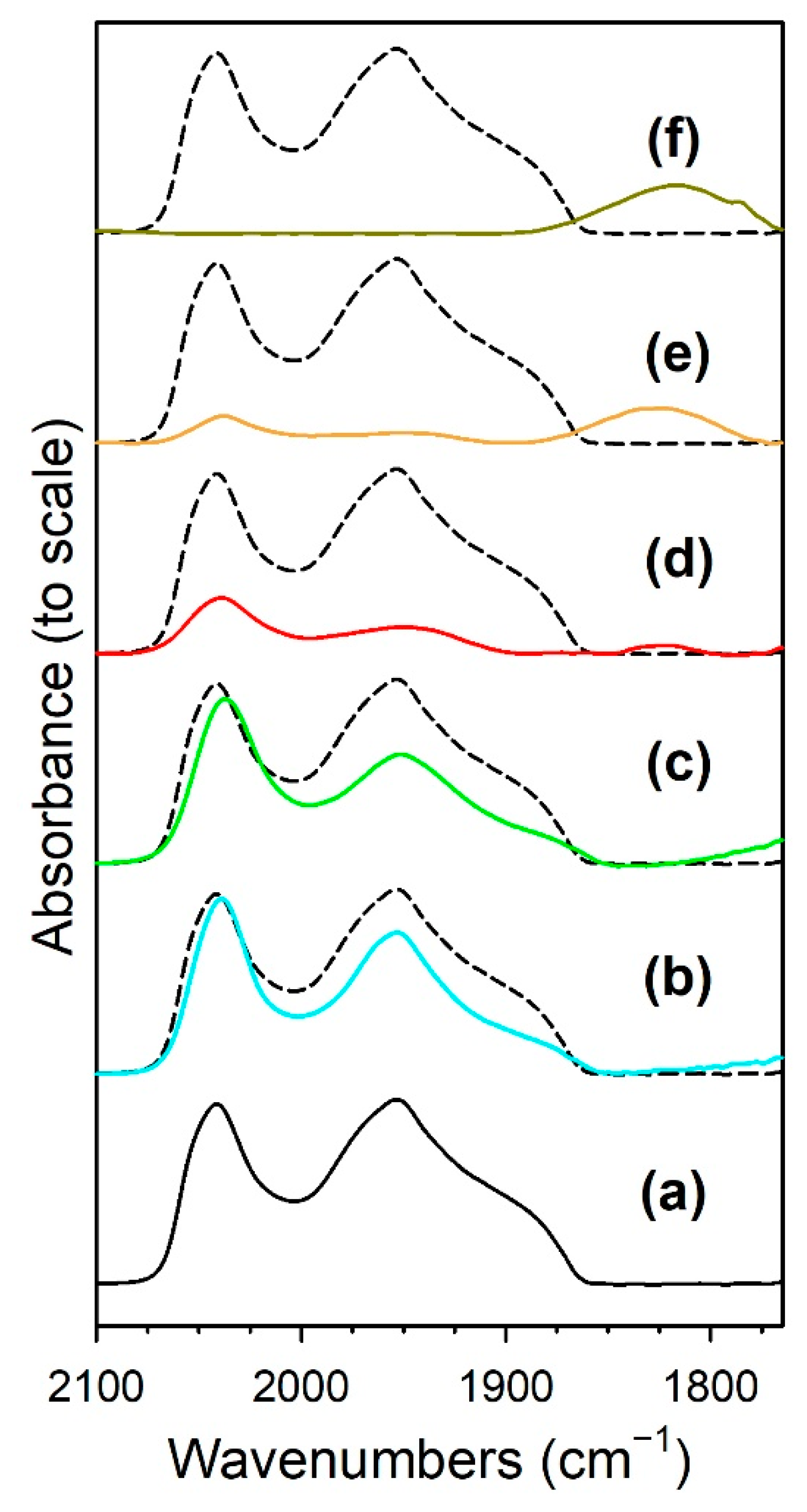
3.1.6. In Situ Infrared Spectroscopy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Description | N Ru-Ru Metal | R Ru-Ru (Å) Metal | e0 (eV) | σ2 (Å2) | R-Factor | Calc. # Atoms * | Calc. Diam. (nm) * |
|---|---|---|---|---|---|---|---|
| Ruthenium foil | 12 | 2.681 (0.0068) | −0.920 (1.457) | 0.00370 (0.000282) | 0.0095 | - | - |
| 0%Na-1%Ru/m-zirconia | 2.0 (0.81) | 2.674 (0.0178) | 1.442 (4.440) | 0.00294 (0.00254) | 0.016 | 4.7 ** | 0.31 |
| 0.5%Na-1%Ru/m-zirconia | 3.7 (0.47) | 2.665 (0.0053) | 0.592 (0.924) | 0.00334 (0.000938) | 0.0074 | 8.6 | 0.56 |
| 1%Na-1%Ru/m-zirconia | 3.4 (0.42) | 2.669 (0.0050) | 0.133 (0.915) | 0.00245 (0.000891) | 0.0063 | 7.9 | 0.52 |
| 1.8%Na-1%Ru/m-zirconia | 3.1 (0.64) | 2.666 (0.0082) | 1.484 (1.536) | 0.00160 (0.00147)) | 0.017 | 7.2 | 0.47 |
| 2.5%Na-1%Ru/m-zirconia | 3.7 (0.72) | 2.671 (0.0084) | 2.154 (1.372) | 0.00437 (0.00147) | 0.019 | 8.7 | 0.57 |
| 5%Na-1%Ru/m-zirconia | 4.5 (0.32) | 2.669 (0.0030) | −0.673 (0.523) | 0.00354 (0.000529) | 0.0023 | 10.6 | 0.69 |
| Catalyst | % T < 250 °C | % 250 °C < T < 400 °C | % T > 400 °C |
|---|---|---|---|
| 1%Ru/zirconia | 65 | 25 | 10 |
| 0.5%Na-1%Ru/m-zirconia | 50 | 25 | 25 |
| 1%Na-1%Ru/m-zirconia | 28 | 24 | 48 |
| 1.8%Na-1%Ru/m-zirconia | 20 | 18 | 62 |
| 2.5%Na-1%Ru/m-zirconia | 16 | 23 | 61 |
| 5%Na-1%Ru/m-zirconia | 10 | 21 | 69 |
| Catalyst | Band Position (cm−1) | ||||
|---|---|---|---|---|---|
| ν(C-H) Formate | ν(CO) Ru-CO | νasym (OCO) Formate | νsym (OCO) Formate | Δ(OCO) | |
| 1%Ru/m-ZrO2 | 2872, (2848) | 2042, (1970), 1955, (1900) | 1631, 1609, 1567, 1557 | (1417, 1386) 1358, 1335 | 199, 232 |
| w/0.5%Na | 2869, (2848) | 2040, (1965), 1954, (1900) | 1643, 1576, (1539) | (1384), 1337, (1266) | 239, 306 |
| w/1%Na | 2859, (2806) | 2038, 1952, (1900) | 1652, (1604–1450) | 1327 | 325 |
| w/1.8%Na | 2846, 2804 | 2039, 1950, 1825 | 1652 (1580–1450) | 1316 | 336 |
| w/2.5%Na | 2846, 2804 | 2038, 1950, 1827 | 1642 | (1365), 1315 | 327 |
| w/5%Na | 2846, 2834–2700 | 1818 | 1638 | 1435–1230 | 306 |









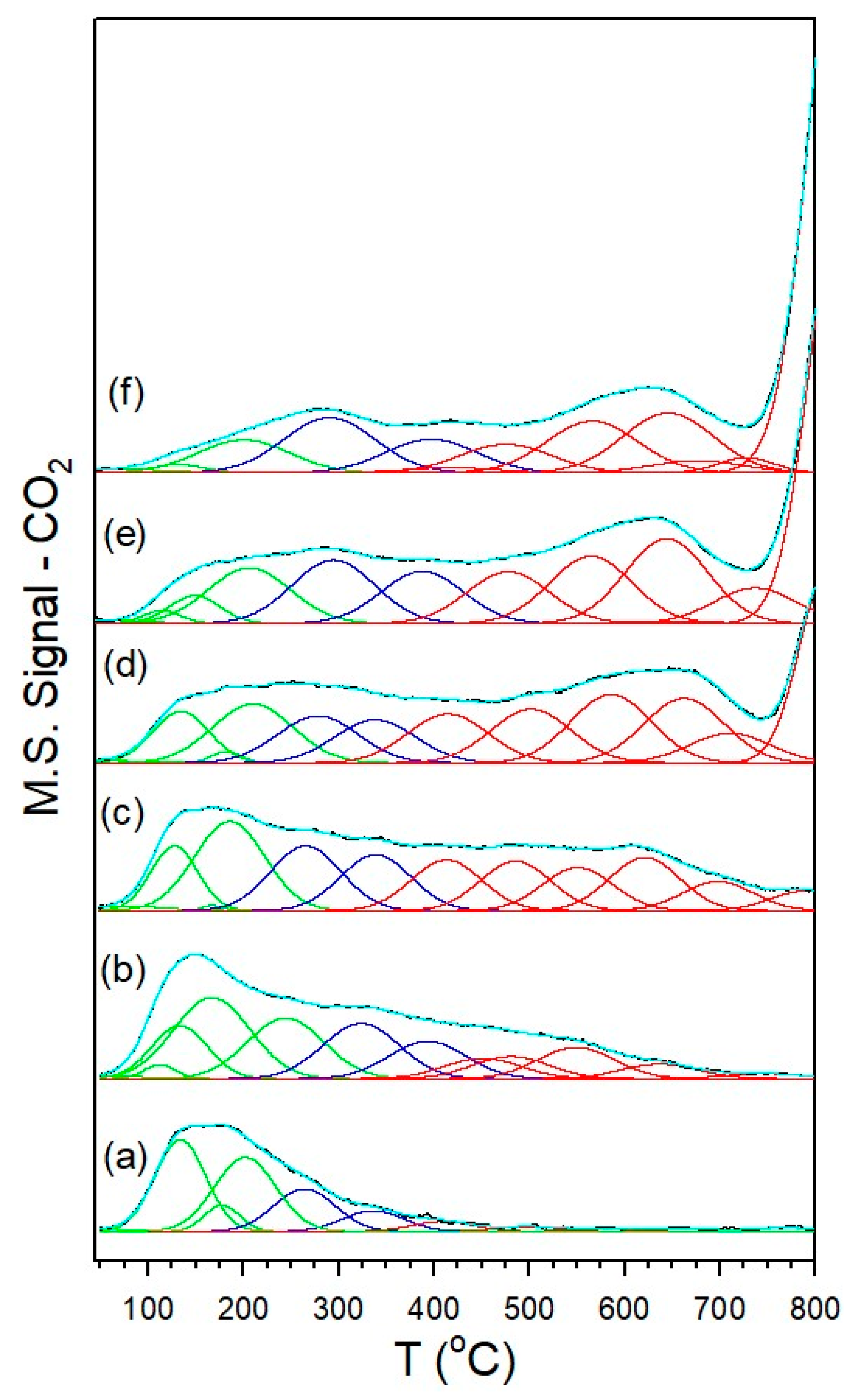
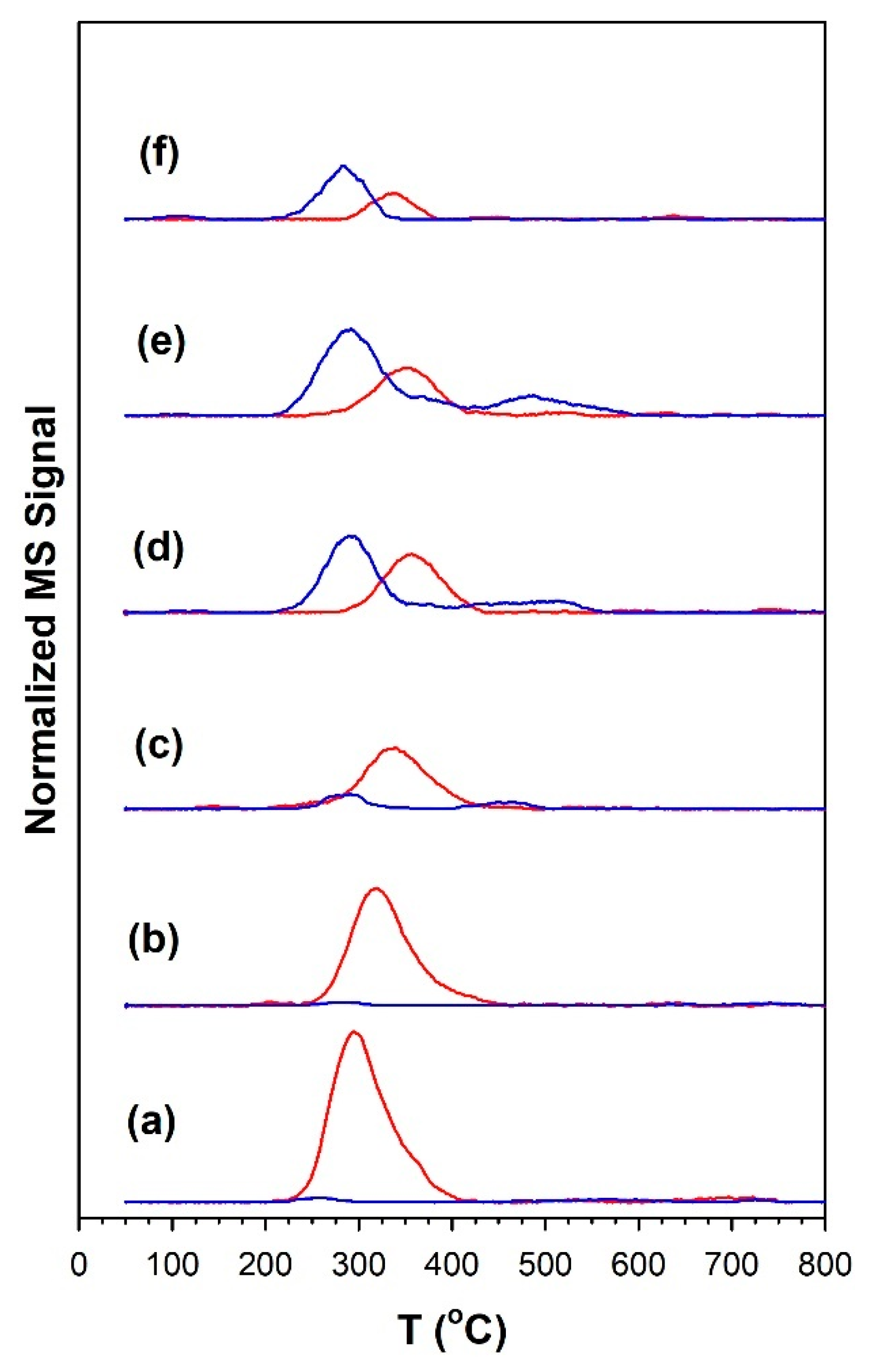
3.1.7. Temperature-Programmed Surface Reaction with Mass Spectrometry
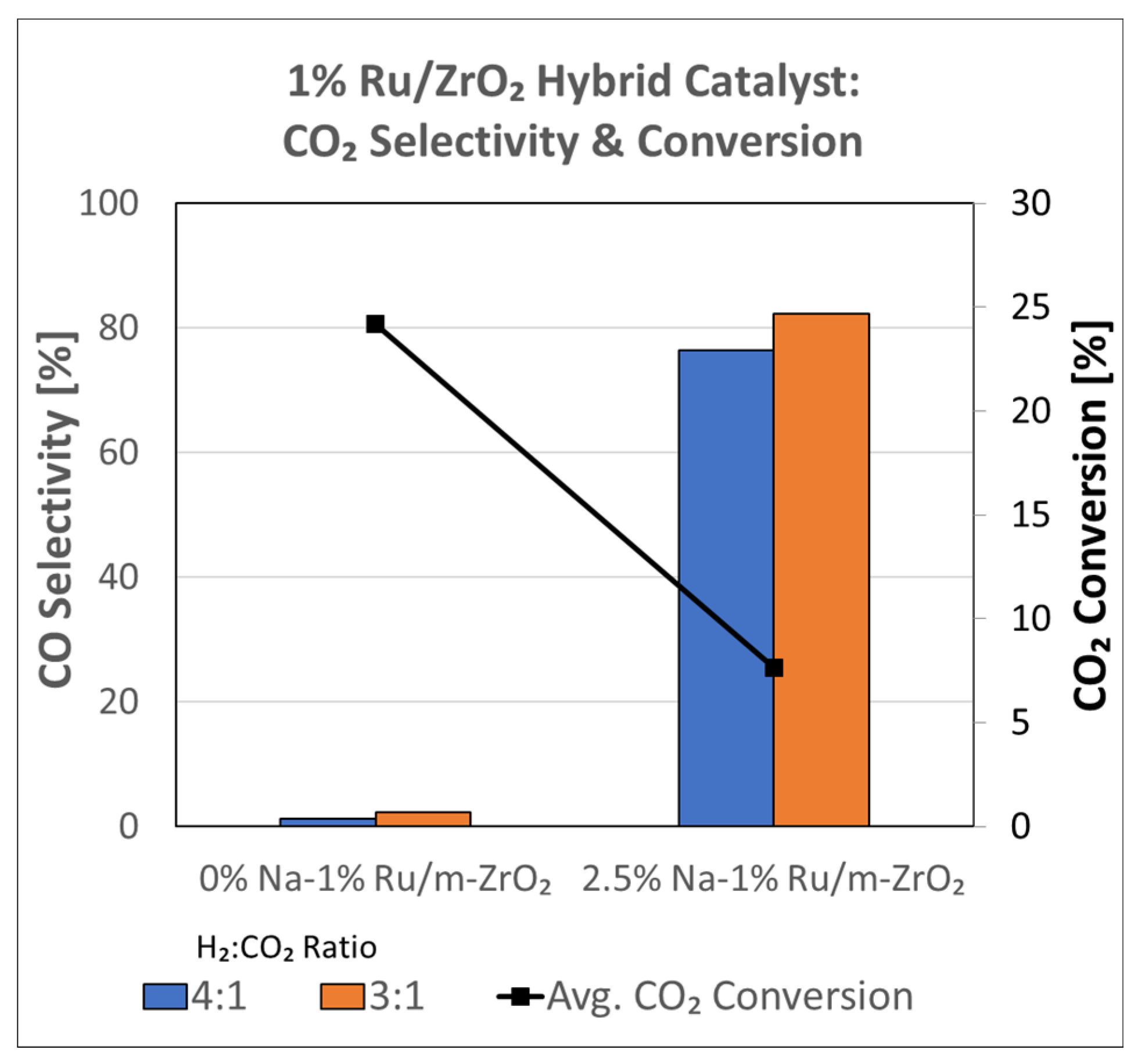
3.1.8. Catalytic Testing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alvez, L.M.N.C.; Almeida, M.P.; Ayala, M.; Watson, C.D.; Jacobs, G.; Rabelo-Neto, R.C.; Noronha, F.B.; Mattos, L.V. CO2 methanation over metal catalysts supported on ZrO2: Effect of the nature of the metallic phase on catalytic performance. Chem. Eng. Sci. 2021, 239, 116604. [Google Scholar] [CrossRef]
- Xie, Y.; Chen, J.; Wu, X.; Wen, J.; Zhao, R.; Li, Z.; Tian, G.; Zhang, Q.; Ning, P.; Hao, J. Frustrated Lewis pairs boosting low-temperature CO2 methanation performance over Ni/CeO2 nanocatalysts. ACS Catal. 2022, 12, 10587–10602. [Google Scholar] [CrossRef]
- Shafer, W.D.; Jacobs, G.; Graham, U.M.; Hamdeh, H.H.; Davis, B.H. Increased CO2 hydrogenation to liquid products using promoted iron catalysts. J. Catal. 2019, 369, 239–248. [Google Scholar] [CrossRef]
- Visconti, C.; Martinelli, M.; Falbo, L.; Fratalocchi, L.; Lietti, L. CO2 hydrogenation to hydrocarbons over Co and Fe-based Fischer-Tropsch catalysts. Catal. Today 2016, 277, 161–170. [Google Scholar] [CrossRef]
- Atsbha, T.A.; Yoon, T.; Seongho, P.; Lee, C.-J. A review on the catalytic conversion of CO2 using H2 for synthesis of CO, methanol, and hydrocarbons. J. CO2 Util. 2021, 44, 101413. [Google Scholar] [CrossRef]
- Santiago, R.G.; Coelho, J.A.; De Lucena, S.M.P.; Musse, A.P.S.; Portilho, M.D.F.; Rodriguez-Castellon, E.; De Azevedo, D.C.S.; Bastos-Neto, M. Synthesis of MeOH and DME from CO2 hydrogenation over commercial and modified catalysts. Front. Chem. 2022, 10, 903053. [Google Scholar] [CrossRef]
- Panahi, M.; Karimi, M.; Skogestad, S.; Hillestad, M.; Svendsen, H.F. Self-optimizing and control structure design for a CO2 capturing plant. Adv. Gas Proc. 2010, 2, 331–338. [Google Scholar] [CrossRef]
- Fugiel, A.; Burchart-Korol, D.; Czaplicka-Kolarz, K.; Smolinski, A. Environmental impact and damage categories caused by air pollution emissions from mining and quarrying sectors of European countries. J. Clean. Prod. 2017, 143, 159–168. [Google Scholar] [CrossRef]
- Soto, V.; Ulloa, C.; Garcia, X. A CFD design approach for industrial size tubular reactors for SNG production from biogas (CO2 Methanation). Energies 2021, 14, 6175. [Google Scholar] [CrossRef]
- Wang, T.; Wang, X.; Hou, C.; Liu, J. Quaternary functionalized mesoporous adsorbents for ultra-high kinetics of CO2 capture from air. Sci. Rep. 2020, 10, 21429. [Google Scholar] [CrossRef]
- Straatman, P.J.T.; van Sark, W.J.H.M. Indirect air CO2 capture and refinement based on OTEC seawater outgassing. iScience 2021, 24, 102754. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Li, J.; Dai, Z.; Akram, M.W. Modelling and analysis of a novel hydrogen production approach by full spectrum solar energy. Energy Conv. Manag. 2022, 263, 115694. [Google Scholar] [CrossRef]
- Goekcek, M. Hydrogen generation from small-scale wind-powered electrolysis system in different power matching modes. Int. J. Hydrogen Energy 2010, 35, 10050–10059. [Google Scholar] [CrossRef]
- Sahin, S.; Sahin, H.M. Generation-IV reactors and nuclear hydrogen production. Int. J. Hydrogen Energy 2021, 46, 28936–28948. [Google Scholar] [CrossRef]
- Guarieiro, L.L.N.; dos Anjos, J.P.; da Silva, L.A.; Santos, A.A.B.; Calixto, E.E.S.; Pessoa, F.L.P.; de Almeida, J.L.G.; Filho, M.A.; Marinho, F.S.; da Rochac, G.O.; et al. Technological perspectives and economic aspects of green hydrogen in the energetic transition: Challenges for chemistry. J. Braz. Chem. Soc. 2022, 33, 844–869. [Google Scholar] [CrossRef]
- Pahija, E.; Panaritis, C.; Rutherford, B.; Couillard, M.; Patarachao, B.; Shadbahr, J.; Bensebaa, F.; Patience, G.S.; Boffito, D.C. FeOX nanoparticle doping on Cu/Al2O3 catalysts for the reverse water gas shift. J. CO2 Util. 2022, 64, 102155. [Google Scholar] [CrossRef]
- Nielsen, A.S.; Ostadi, M.; Austboe, B.; Hillestad, M.; del Alamo, G.; Burheim, O. Enhancing the efficiency of power- and biomass-to-liquid fuel processes using fuel-assisted solid oxide electrolysis cells. Fuel 2021, 321, 123987. [Google Scholar] [CrossRef]
- Kianfar, E.; Mazaheri, H. Methanol to gasoline: A sustainable transport fuel. Adv. Chem. Res. 2020, 66, 207–236. [Google Scholar]
- Rabelo-Neto, R.C.; Almeida, M.P.; Silveira, E.B.; Ayala, M.; Watson, C.D.; Villarreal, J.; Cronauer, D.C.; Kropf, A.; Martinelli, M.; Noronha, F.B.; et al. CO2 hydrogenation: Selectivity control of CO versus CH4 achieved using Na doping over Ru/m-ZrO2 at low pressure. Appl. Catal. B Environ. 2022, 315, 121533. [Google Scholar] [CrossRef]
- Gnanamani, M.; Hamdeh, H.H.; Jacobs, G.; Shafer, W.D.; Hopps, S.D.; Thomas, G.A.; Davis, B.H. Hydrogenation of carbon dioxide over K-promoted FeCo bimetallic catalysts prepared from mixed metal oxalates. ChemCatChem 2017, 9, 1303–1312. [Google Scholar] [CrossRef]
- Numpilai, T.; Cheng, C.K.; Limtrakul, J.; Witoon, T. Recent advances in light olefins production from catalytic hydrogenation of carbon dioxide. Proc. Saf. Environ. Protect. 2021, 151, 401–427. [Google Scholar] [CrossRef]
- Pendyala, V.R.R.; Jacobs, G.; Graham, U.M.; Shafer, W.D.; Martinelli, M.; Kong, L.; Davis, B.H. Fischer-Tropsch synthesis: Influence of acid treatment and preparation method on carbon nanotube supported ruthenium catalysts. Ind. Eng. Chem. Res. 2017, 56, 6408–6418. [Google Scholar] [CrossRef]
- Porta, A.; Falbo, L.; Visconti, C.G.; Lietti, L.; Bassano, C.; Deiana, P. Synthesis of Ru-based catalysts for CO2 methanation and experimental assessment of intraporous transport limitations. Catal. Today 2020, 343, 38–47. [Google Scholar] [CrossRef]
- Jacobs, G.; Davis, B.H. Surface interfaces in low temperature water-gas shift: The metal-oxide synergy, the assistance of co-adsorbed water, and alkali doping. Int. J. Hydrogen Energy 2010, 35, 3522–3536. [Google Scholar] [CrossRef]
- Chenu, E.; Jacobs, G.; Crawford, A.C.; Keogh, R.A.; Patterson, P.M.; Sparks, D.E.; Davis, B.H. Water-gas shift: An examination of Pt promoted MgO and tetragonal and monoclinic ZrO2 by in-situ DRIFTS. Appl. Catal. B Environ. 2005, 59, 45–56. [Google Scholar] [CrossRef]
- Hagemeyer, A.; Carhart, R.E.; Yaccato, K.; Lesik, A.; Brooks, C.J.; Phillips, C.B. Platinum-Alkali/Alkaline-Earth Catalyst Formulations for Hydrogen Generation. U.S. Patent 7,744,849, 29 June 2010. [Google Scholar]
- Shido, T.; Iwasawa, Y. Reactant-promoted reaction mechanism for water-gas shift reaction of rhodium-doped ceria. J. Catal. 1993, 141, 71–78. [Google Scholar] [CrossRef]
- Jacobs, G.; Graham, U.M.; Chenu, E.; Patterson, P.M.; Dozier, A.; Davis, B.H. Low temperature water-gas shift: Impact of Pt promoter loading on the partial reduction of ceria, and consequences for catalyst design. J. Catal. 2005, 229, 499–512. [Google Scholar] [CrossRef]
- Pigos, J.M.; Brooks, C.J.; Jacobs, G.; Davis, B.H. Low temperature water-gas shift: Characterization of Pt-based ZrO2 catalyst promoted with Na discovered by combinatorial methods. Appl. Catal. A General 2007, 319, 47–57. [Google Scholar] [CrossRef]
- Pigos, J.M.; Brooks, C.J.; Jacobs, G.; Davis, B.H. Low temperature water-gas shift: The effect of alkali doping on the C-H bond of formate over Pt/ZrO2 catalysts. Appl. Catal. A Gen. 2007, 328, 14–26. [Google Scholar] [CrossRef]
- Martinelli, M.; Alhraki, N.; Castro, J.D.; Matamoros, M.E.; Jacobs, G. Effect of Na loading on Pt/ZrO2 catalysts for low temperature water-gas shift for the production and purification of hydrogen. In New Dimensions in Production and Utilization of Hydrogen; Nanda, S., Vo, D.-V., Tri, P.N., Eds.; Elsevier Books: Amsterdam, The Netherlands, 2020; pp. 143–160. ISBN 9780128195536. [Google Scholar] [CrossRef]
- Luukkanen, S.; Haukka, M.; Kallinen, M.; Pakkanen, T.A. The low-temperature water-gas shift reaction catalyzed by sodium-carbonate-activated ruthenium mono (bipyridine)/SiO2 complexes. Catal. Lett. 2000, 70, 123–125. [Google Scholar] [CrossRef]
- Basinska, A.; Domka, F. The influence of alkali metals on the activity of supported ruthenium catalysts for the water-gas shift reaction. Catal. Lett. 1997, 43, 59–61. [Google Scholar] [CrossRef]
- Pendyala, V.R.R.; Shafer, W.D.; Jacobs, G.; Graham, U.M.; Khalid, S.; Davis, B.H. Fischer-Tropsch synthesis: Effect of reducing agent for aqueous-phase synthesis over Ru nanoparticle and supported Ru catalysts. Catal. Lett. 2015, 145, 893–904. [Google Scholar] [CrossRef]
- Wang, C.; Zhao, H.; Wang, H.; Liu, L.; Xiao, C.; Ma, D. The effects of ionic additives on the aqueous-phase Fischer–Tropsch synthesis with a ruthenium nanoparticle catalyst. Catal. Today 2012, 183, 143–153. [Google Scholar] [CrossRef]
- Komaya, T.; Bell, A.T.; Wengsieh, Z.; Gronsky, R.; Engelke, F.; King, T.S.; Pruski, M. Effects of sodium on the structure and Fischer-Tropsch synthesis activity of Ru/TiO2. J. Catal. 1995, 152, 350–359. [Google Scholar] [CrossRef]
- Williams, F.J.; Lambert, R.M. A study of sodium promotion in Fischer–Tropsch synthesis: Electrochemical control of a ruthenium model catalyst. Catal. Lett. 2000, 70, 9–14. [Google Scholar] [CrossRef]
- VanderWiel, D.P.; Pruski, M.; King, T.S. A kinetic study on the adsorption and reaction of hydrogen over silica-supported ruthenium and silver–ruthenium catalysts during the hydrogenation of carbon monoxide. J. Catal. 1999, 188, 186–202. [Google Scholar] [CrossRef]
- Jacobs, G.; Ribeiro, M.C.; Ma, W.; Ji, Y.; Khalid, S.; Sumodjo, P.T.A.; Davis, B.H. Group 11 (Cu, Ag, Au) promotion of 15% Co/Al2O3 Fischer-Tropsch synthesis catalysts. Appl. Catal. A Gen. 2009, 361, 137–151. [Google Scholar] [CrossRef]
- Datye, A.K.; Schwank, J. Fischer-Tropsch synthesis on bimetallic ruthenium-gold catalysts. J. Catal. 1985, 93, 256–269. [Google Scholar] [CrossRef]
- McCue, A.J.; Aponaviciute, J.; Wells, R.P.K.; Anderson, J.A. Gold modified cobalt-based Fischer-Tropsch catalysts for conversion of synthesis gas to liquid fuels. Front. Chem. Sci. Eng. 2013, 7, 262–269. [Google Scholar] [CrossRef]
- Jacoby, M. X-ray absorption spectroscopy. Chem. Eng. News 2001, 79, 33–38. [Google Scholar] [CrossRef]
- Ressler, T. WinXAS: A Program for X-ray Absorption Spectroscopy Data Analysis under MS-Windows. J. Synchrotron Radiat. 1998, 5, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Ravel, B. ATOMS: Crystallography for the X-ray absorption spectroscopist. J. Synchrotron Rad. 2001, 8, 314–316. [Google Scholar] [CrossRef] [PubMed]
- Newville, M.; Ravel, B.; Haskel, D.; Rehr, J.J.; Stern, E.A.; Yacoby, Y. Analysis of multiple-scattering XAFS data using theoretical standards. Phys. B Condens. Matter 1995, 208, 154–156. [Google Scholar] [CrossRef]
- Fisher, I.A.; Bell, A.T. In situ infrared study of methanol synthesis from H2/CO over Cu/SiO2 and Cu/ZrO2/SiO2. J. Catal. 1998, 178, 153–173. [Google Scholar] [CrossRef]
- Kondo, J.; Sakata, Y.; Domen, K.; Maruya, K.; Onishi, T. Infrared study of hydrogen adsorbed on ZrO2. J. Chem. Soc. Faraday Trans. 1990, 86, 397–401. [Google Scholar] [CrossRef]
- Laachir, A.; Perrichon, V.; Badri, A.; Lamotte, J.; Catherine, E.; Lavalley, J.C.; El Fallah, J.; Hilaire, L.; Le Normand, F.; Quemere, E.; et al. Reduction of CeO2 by Hydrogen. Magnetic Susceptibility and Fourier-Transform IR, UV and X-ray Photoelectron Spectroscopy Measurements. J. Chem. Soc. Faraday Trans. 1991, 87, 1601–1609. [Google Scholar] [CrossRef]
- McHugh, B.J.; Larsen, G.; Haller, G.L. Characterization of platinum particle interaction with L-zeolite by X-ray absorption spectroscopy: Binding energy shifts, X-ray absorption near-edge structure, and extended X-ray absorption fine structure. J. Phys. Chem. 1990, 94, 8621–8624. [Google Scholar] [CrossRef]
- Bunluesin, T.; Gorte, R.J.; Graham, G.W. Studies of the water-gas-shift reaction on ceria-supported Pt, Pd, and Rh: Implications for oxygen-storage properties. Appl. Catal. B Environ. 1998, 15, 107–114. [Google Scholar] [CrossRef]
- Marinkovic, N.S.; Sasaki, K.; Adzic, R. Nanoparticle size evaluation of catalysts by EXAFS: Advantages and limitations. Zast. Mater. 2016, 57, 101–109. [Google Scholar] [CrossRef]
- Binet, C.; Daturi, M.; Lavalley, J.C. IR study of polycrystalline ceria properties in oxidized and reduced states. Catal. Today 1999, 50, 207–225. [Google Scholar] [CrossRef]
- Görling, A.; Ackermann, L.; Lauber, J.; Knappe, P.; Rösch, N. On the coadsorption of CO and alkali atoms at transition metal surfaces: A LCGTO-LDF cluster study. Surf. Sci. 1993, 286, 26–45. [Google Scholar] [CrossRef]
- Blyholder, G. Molecular orbital view of chemisorbed carbon monoxide. J. Phys. Chem. 1964, 68, 2772–2777. [Google Scholar] [CrossRef]
- Martinelli, M.; Castro, J.D.; Alhraki, N.; Matamoros, M.E.; Kropf, A.J.; Cronauer, D.C.; Jacobs, G. Effect of sodium loading on Pt/ZrO2 during ethanol steam reforming. Appl. Catal. A Gen. 2021, 610, 117947. [Google Scholar] [CrossRef]








| Catalyst | Main Peaks, T (°C) |
|---|---|
| 1%Ru/m-zirconia | 187 |
| Same with: 0.5%Na | 218 |
| 1%Na | 222, 234 |
| 1.8%Na | 234, 272 |
| 2.5%Na | 235, 327 |
| 5%Na | 238, 349, 390, 402 |
| Catalyst | T (°C) at Which the ν(CH) of CH4 Was Detected |
|---|---|
| 1%Ru/m-zirconia | 150 |
| same with 0.5 wt.% Na | 175 |
| 1 wt.% Na | 175 |
| 1.8 wt.% Na | 250 |
| 2.5 wt.% Na | 275 |
| 5 wt.% Na | 300 |
| Catalyst | Tmax (°C) at Which CO Was Detected | Tmax (°C) at Which CH4 Was Detected | CO/CH4 Signal Ratio |
|---|---|---|---|
| 1%Ru/m-ZrO2 | 251 | 296 | <0.05 |
| w/0.5%Na | 280 | 317 | <0.05 |
| w/1%Na | 291 | 339 | 0.33 |
| w/1.8%Na | 291 | 355 | 1.5 |
| w/2.5%Na | 291 | 355 | 2.3 |
| w/5%Na | 283 | 339 | 2.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seuser, G.; Staffel, R.; Hocaoglu, Y.; Upton, G.F.; Garcia, E.S.; Cronauer, D.C.; Kropf, A.J.; Martinelli, M.; Jacobs, G. CO2 Hydrogenation: Na Doping Promotes CO and Hydrocarbon Formation over Ru/m-ZrO2 at Elevated Pressures in Gas Phase Media. Nanomaterials 2023, 13, 1155. https://doi.org/10.3390/nano13071155
Seuser G, Staffel R, Hocaoglu Y, Upton GF, Garcia ES, Cronauer DC, Kropf AJ, Martinelli M, Jacobs G. CO2 Hydrogenation: Na Doping Promotes CO and Hydrocarbon Formation over Ru/m-ZrO2 at Elevated Pressures in Gas Phase Media. Nanomaterials. 2023; 13(7):1155. https://doi.org/10.3390/nano13071155
Chicago/Turabian StyleSeuser, Grant, Raechel Staffel, Yagmur Hocaoglu, Gabriel F. Upton, Elijah S. Garcia, Donald C. Cronauer, A. Jeremy Kropf, Michela Martinelli, and Gary Jacobs. 2023. "CO2 Hydrogenation: Na Doping Promotes CO and Hydrocarbon Formation over Ru/m-ZrO2 at Elevated Pressures in Gas Phase Media" Nanomaterials 13, no. 7: 1155. https://doi.org/10.3390/nano13071155
APA StyleSeuser, G., Staffel, R., Hocaoglu, Y., Upton, G. F., Garcia, E. S., Cronauer, D. C., Kropf, A. J., Martinelli, M., & Jacobs, G. (2023). CO2 Hydrogenation: Na Doping Promotes CO and Hydrocarbon Formation over Ru/m-ZrO2 at Elevated Pressures in Gas Phase Media. Nanomaterials, 13(7), 1155. https://doi.org/10.3390/nano13071155