




Synthesis of 3D Porous Cu Nanostructures on Ag Thin Film Using Dynamic Hydrogen Bubble Template for Electrochemical Conversion of CO2 to Ethanol



Abstract
:1. Introduction
2. Experiments and Methods
2.1. Electrode and Electrolyte Preparation
2.2. Electrochemical Measurements
2.3. Product Characterization
3. Results and Discussion
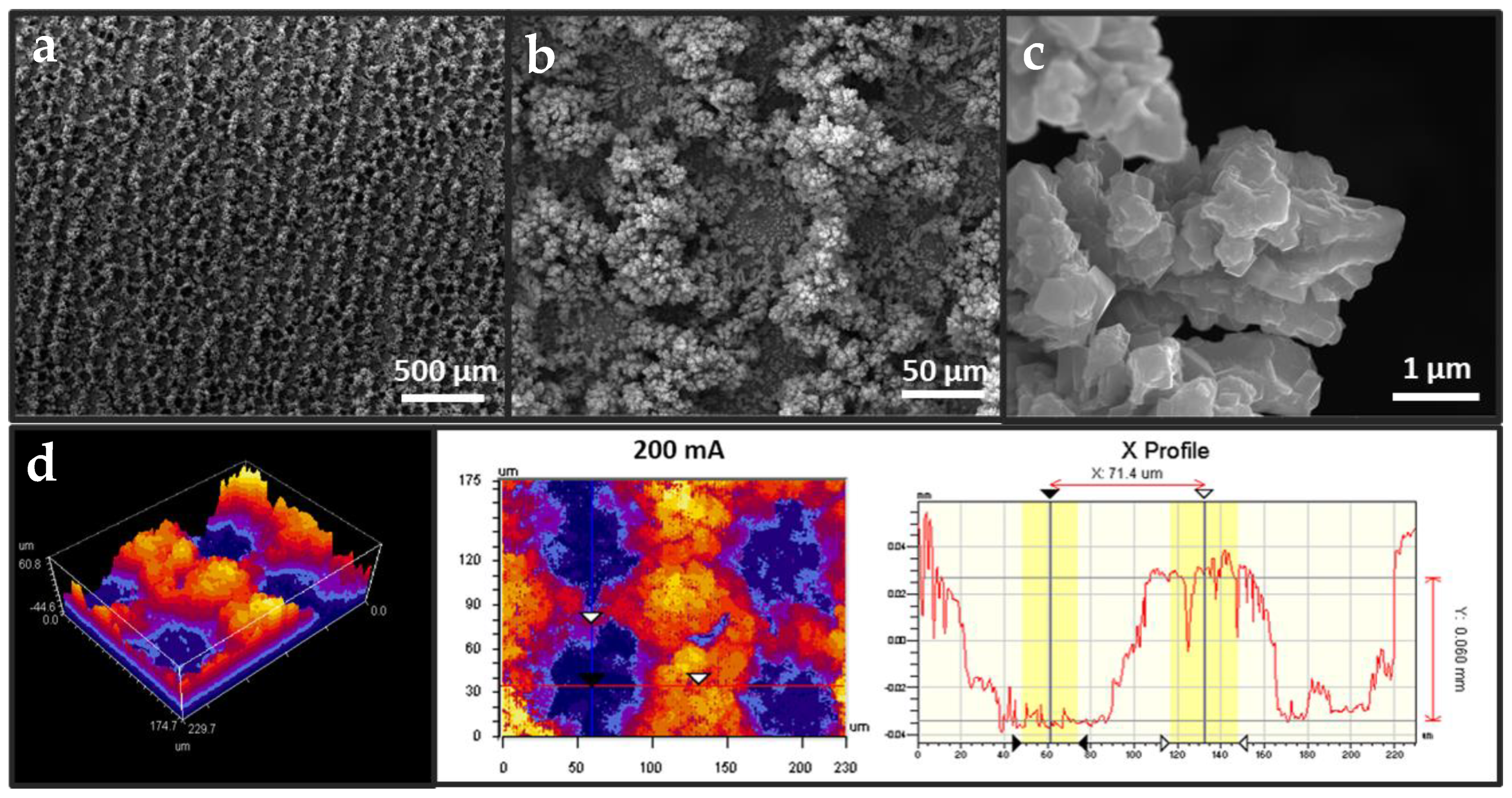
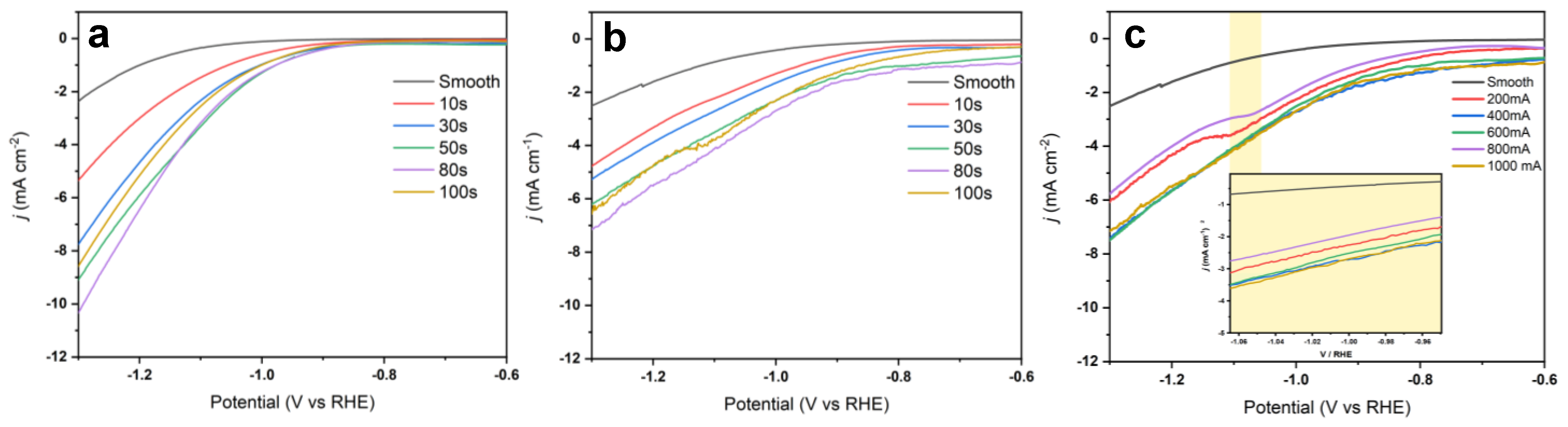
3.1. Fabrication of Cu Foam under Different Deposition Times
3.2. Fabrication of Cu Foam under Different Deposition Currents
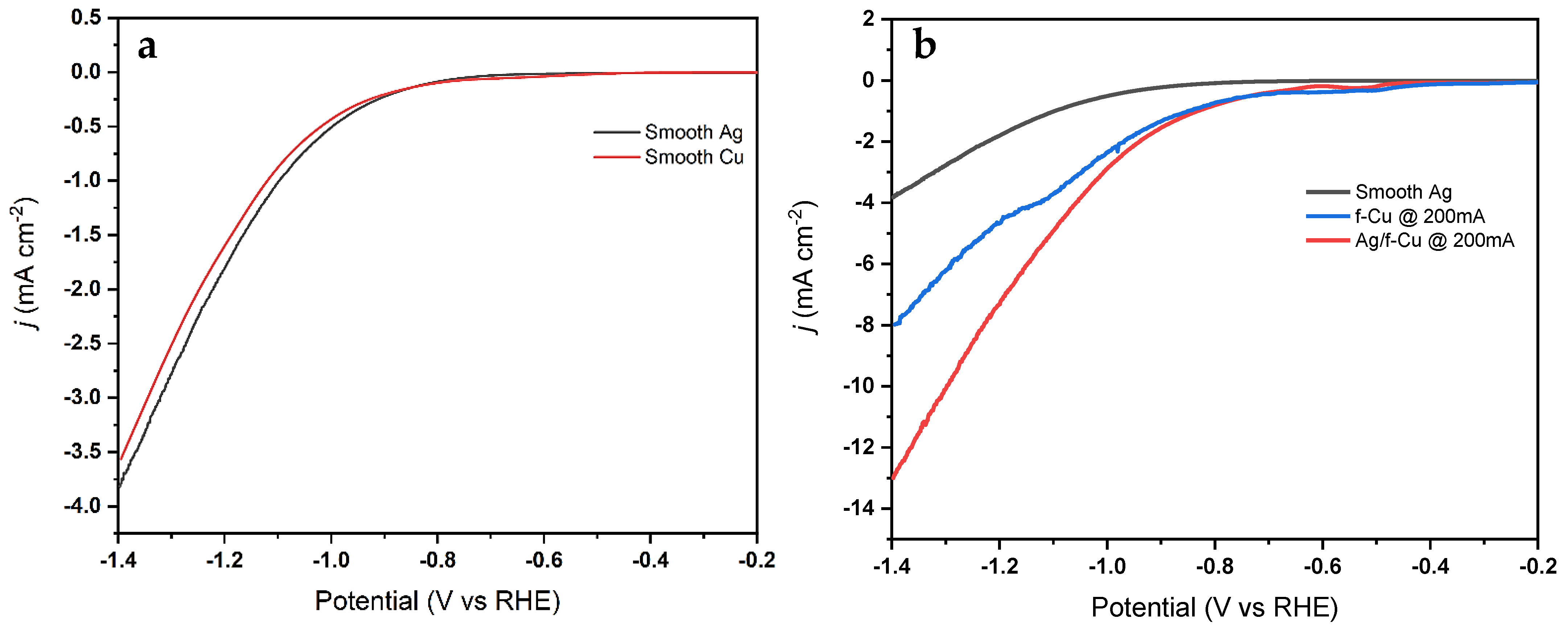
3.3. Synthesis of Ag-Modified Cu Foam
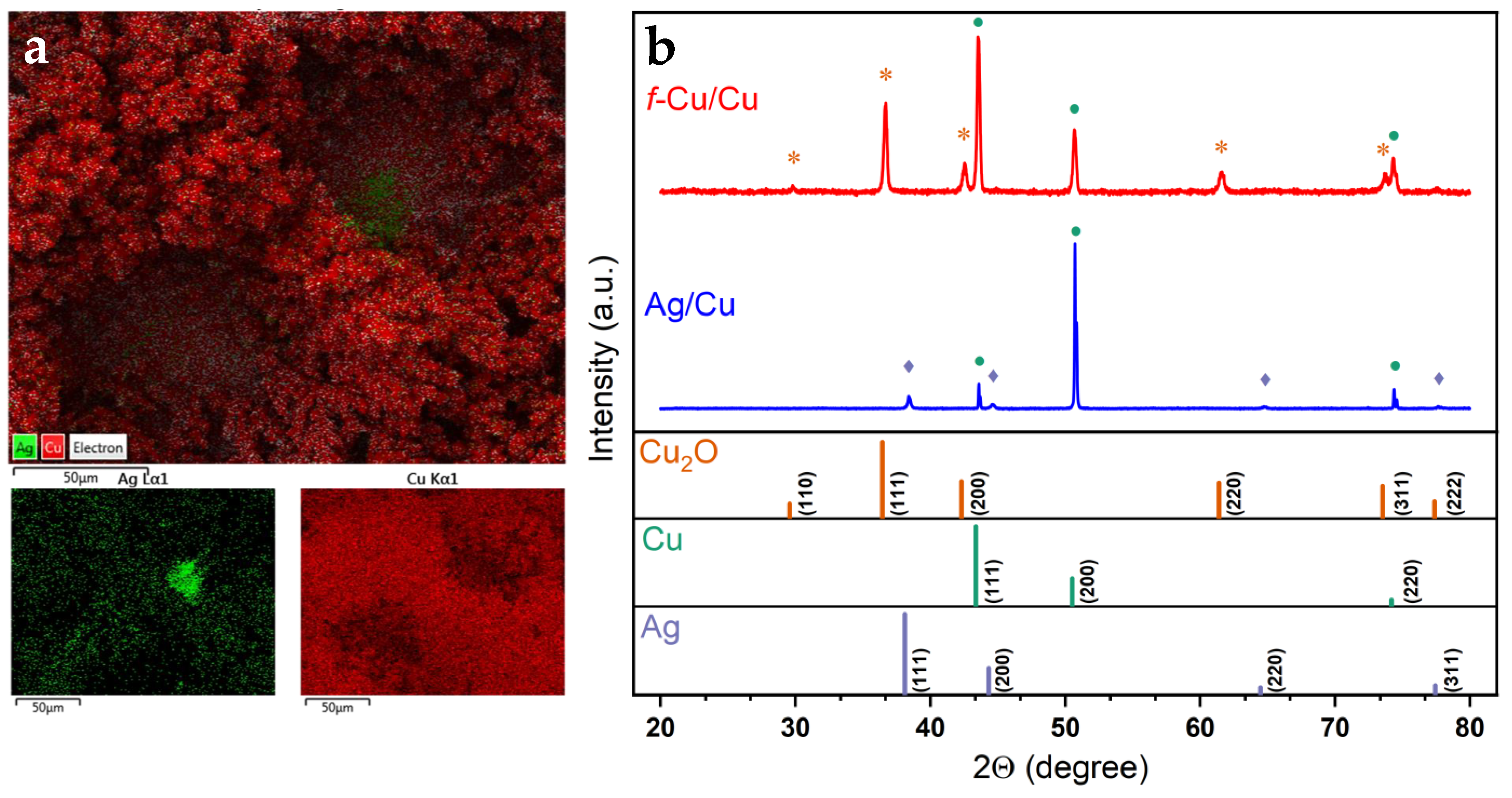
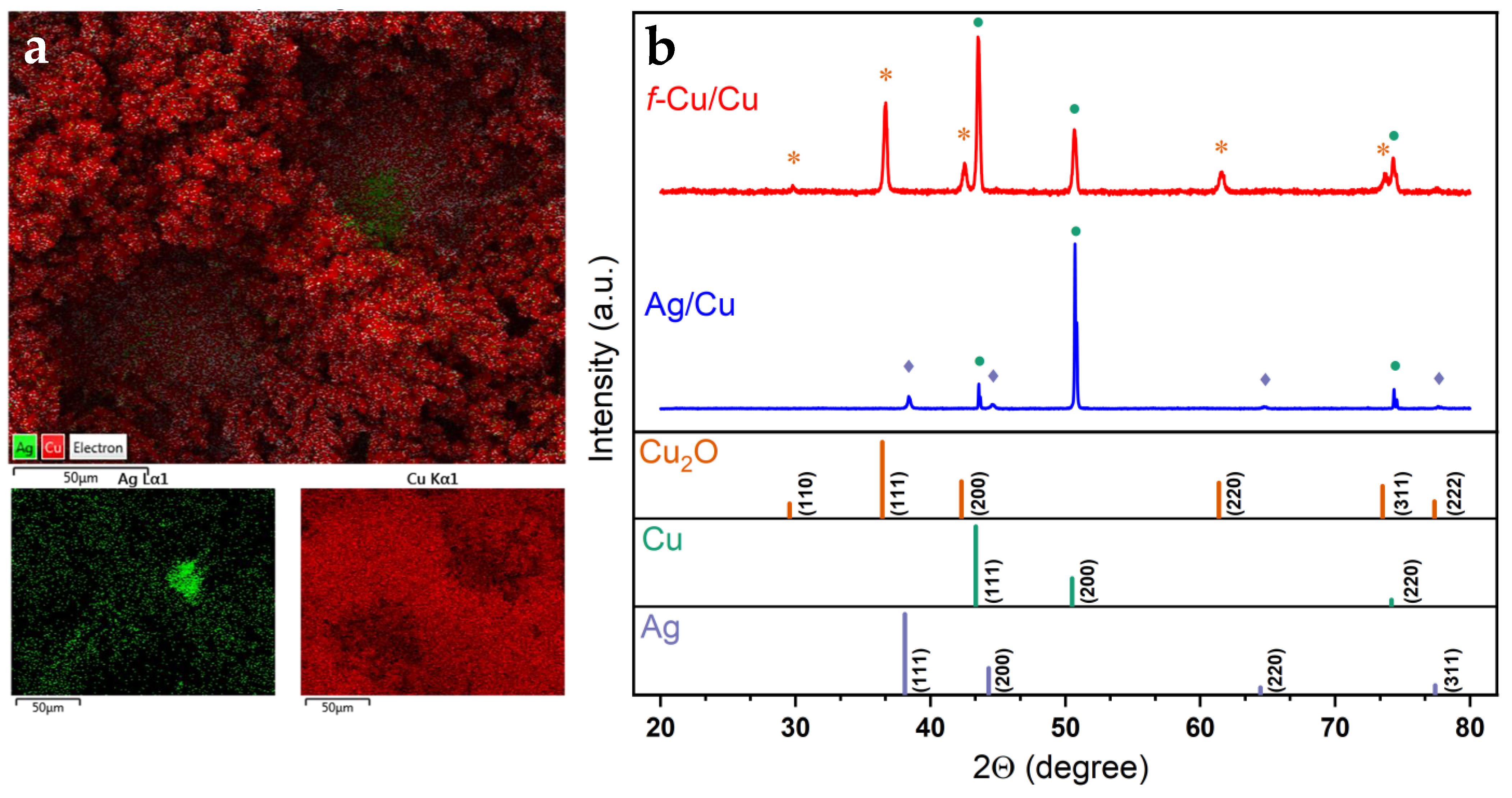
3.4. Surface Characterization
3.5. ECSA and DLC
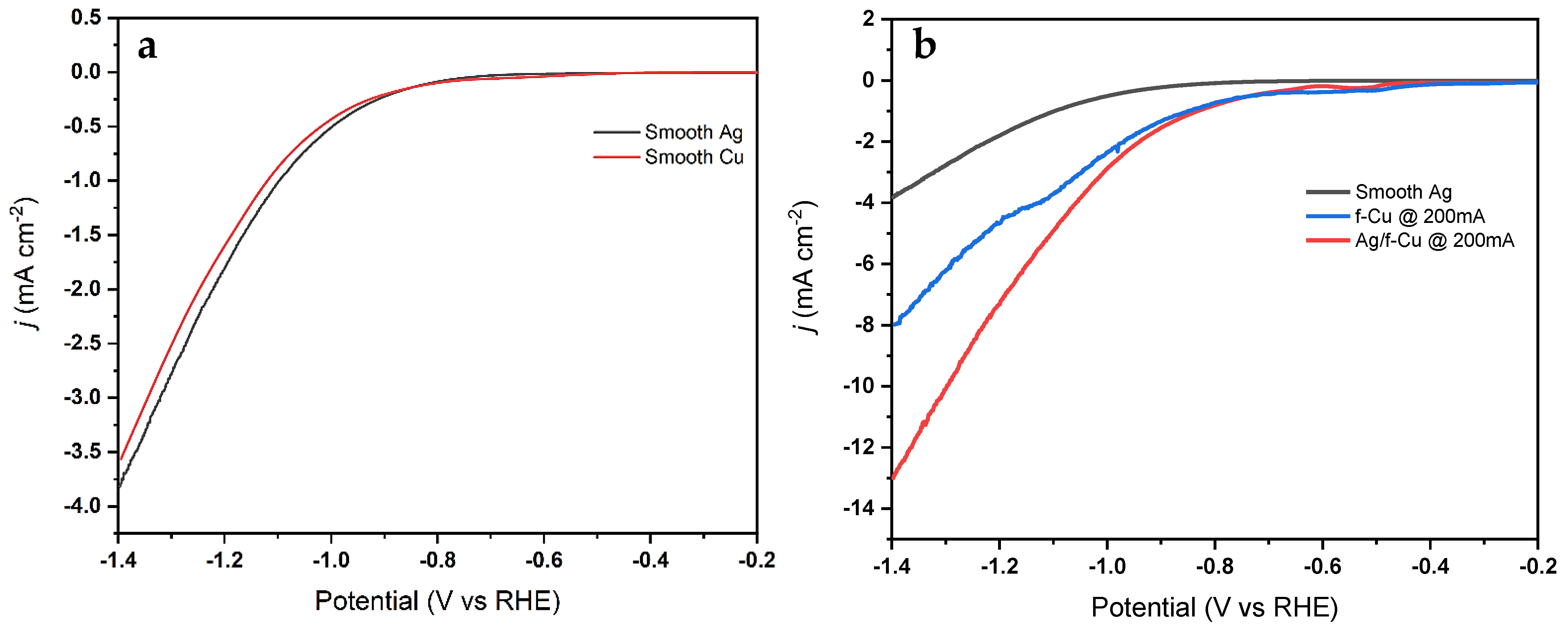
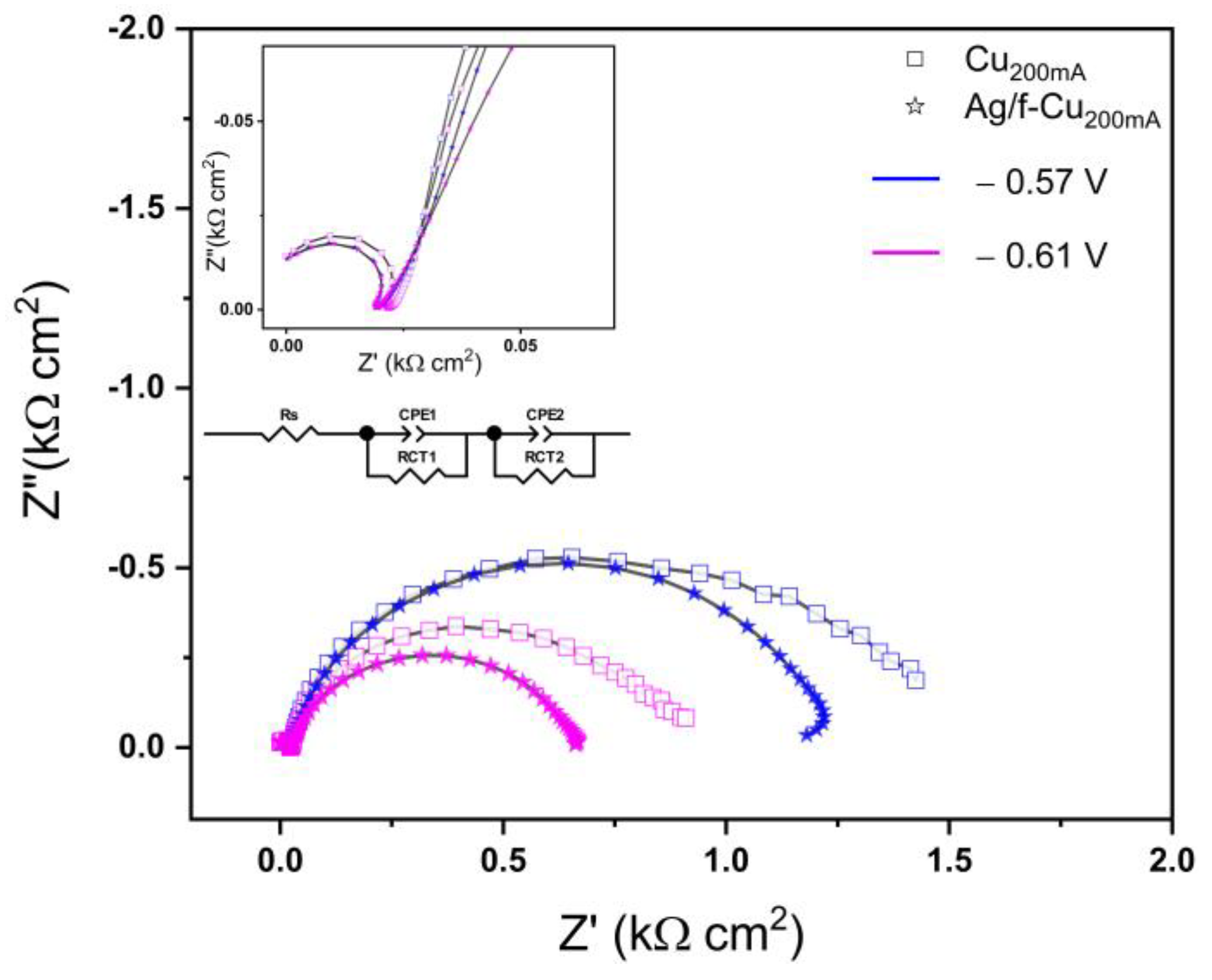
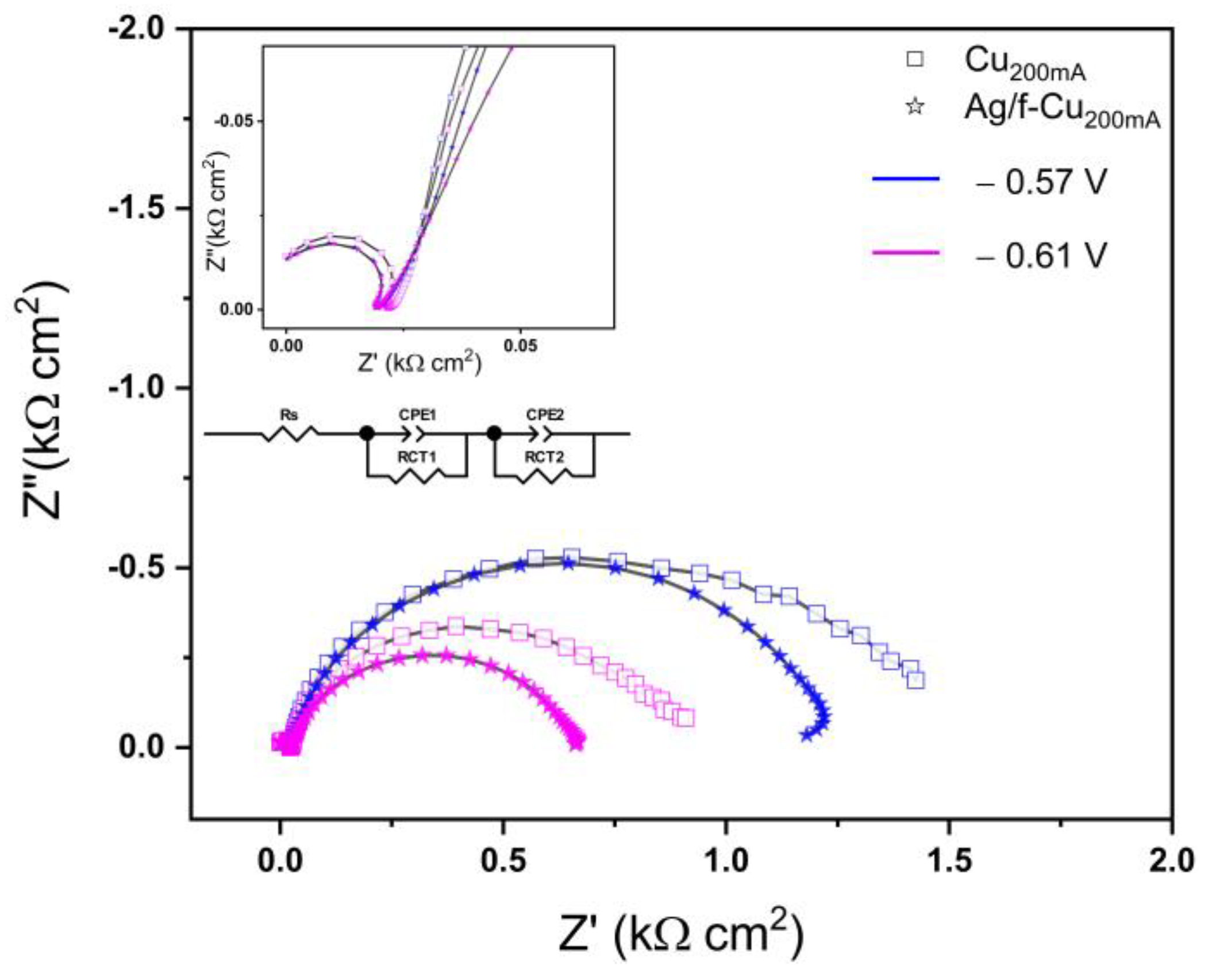
3.6. Electrochemical Reduction Performance Analysis
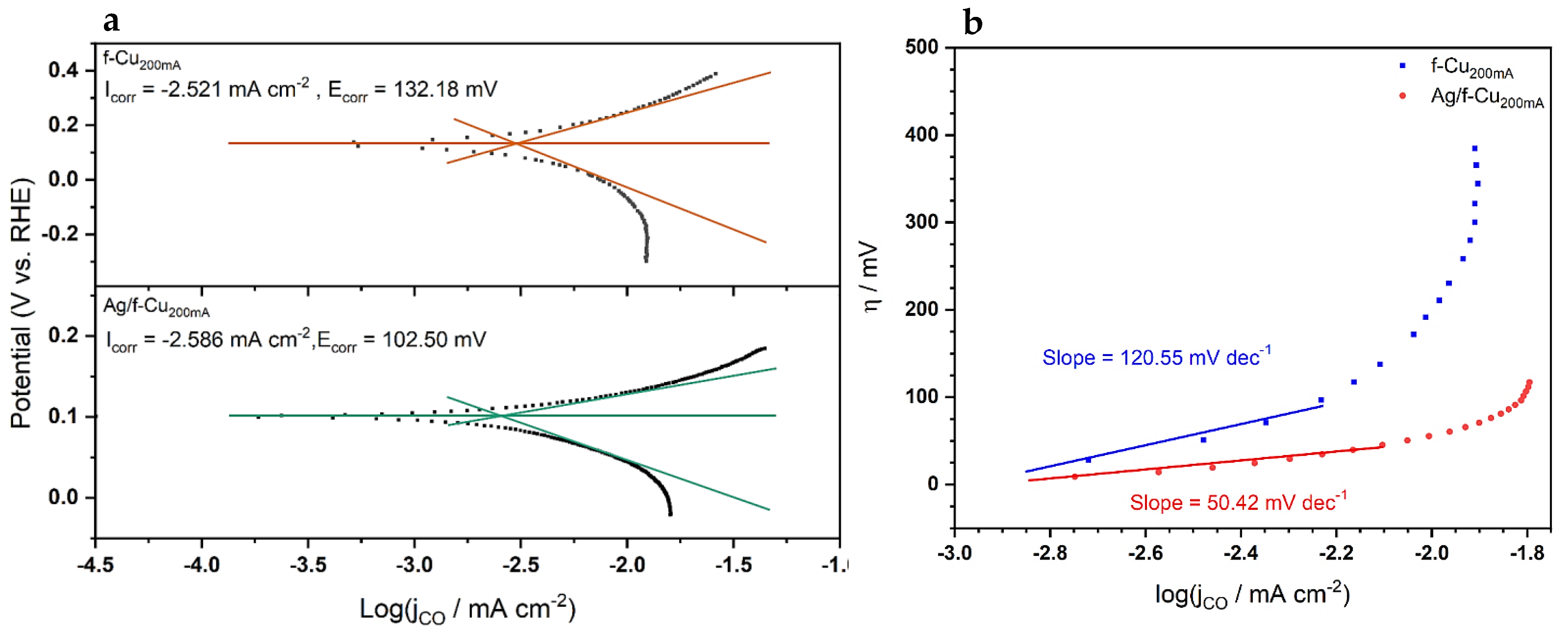
3.6.1. Oxide Passivation Layer
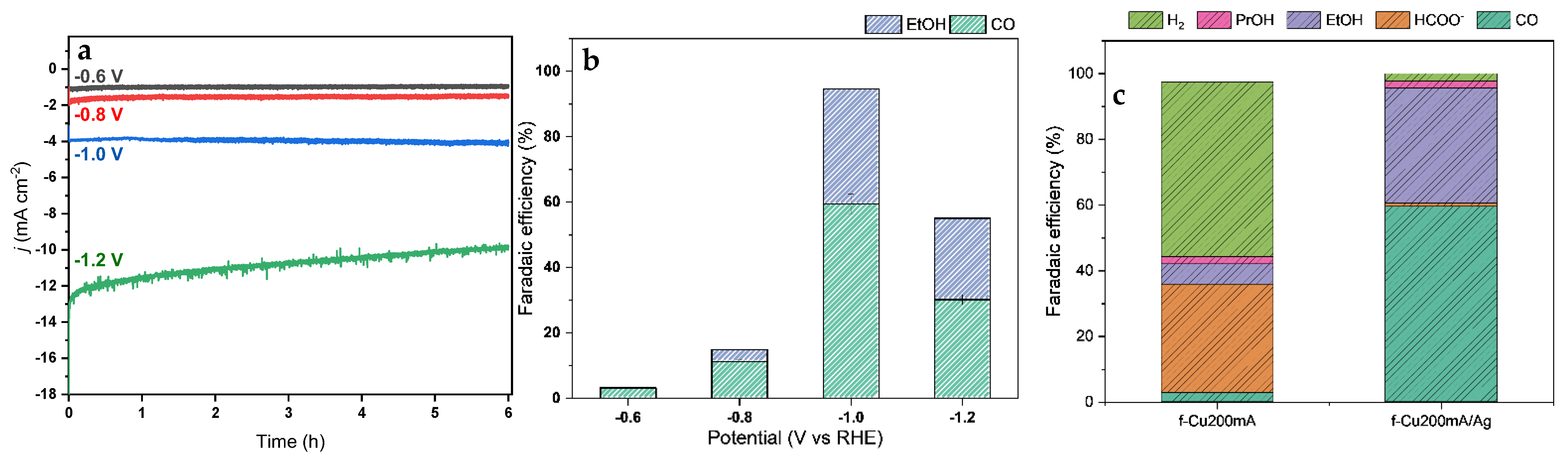
3.6.2. CO2RR Performance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, Y.; Cao, S.; Hou, J.; Li, Z.; Zhang, B.; Zhai, P.; Zhang, Y.; Sun, L. Rational Design of Nanocatalysts with Nonmetal Species Modification for Electrochemical CO2 Reduction. Adv. Energy Mater. 2020, 10, 2000588. [Google Scholar] [CrossRef]
- Popović, S.; Smiljanić, M.; Jovanovič, P.; Vavra, J.; Buonsanti, R.; Hodnik, N. Stability and Degradation Mechanisms of Copper-Based Catalysts for Electrochemical CO2 Reduction. Angew. Chem. Int. Ed. 2020, 59, 14736–14746. [Google Scholar] [CrossRef]
- Tufa, R.A.; Chanda, D.; Ma, M.; Aili, D.; Demissie, T.B.; Vaes, J.; Li, Q.; Liu, S.; Pant, D. Towards Highly Efficient Electrochemical CO2 Reduction: Cell Designs, Membranes and Electrocatalysts. Appl. Energy 2020, 277, 115557. [Google Scholar] [CrossRef]
- Dutta, A.; Montiel, I.Z.; Erni, R.; Kiran, K.; Rahaman, M.; Drnec, J.; Broekmann, P. Activation of Bimetallic AgCu Foam Electrocatalysts for Ethanol Formation from CO2 by Selective Cu Oxidation/Reduction. Nano Energy 2020, 68, 104331. [Google Scholar] [CrossRef]
- Nitopi, S.; Bertheussen, E.; Scott, S.B.; Liu, X.; Engstfeld, A.K.; Horch, S.; Seger, B.; Stephens, I.E.; Chan, K.; Hahn, C.; et al. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 2019, 119, 7610–7672. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.-X.; Liu, K.-H.; Zhang, Q.; Yan, J.-M.; Jiang, Q. Efficient CO2 Reduction to HCOOH with High Selectivity and Energy Efficiency over Bi/RGO Catalyst. Small Methods 2020, 4, 1900846. [Google Scholar] [CrossRef]
- Zahid, A.; Shah, A.; Shah, I. Oxide Derived Copper for Electrochemical Reduction of CO2 to C(2+) Products. Nanomaterials 2022, 12, 1380. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, R.; Zhu, X.; Han, N.; Song, B.; Liu, T.; Hu, G.; Li, Y.; Lu, J.; Li, Y. Mesoporous PdAg Nanospheres for Stable Electrochemical CO2 Reduction to Formate. Adv. Mater. 2020, 32, 2000992. [Google Scholar] [CrossRef]
- Zhao, S.; Liu, A.; Li, Y.; Wen, Y.; Gao, X.; Chen, Q. Boosting the Electrocatalytic CO2 Reduction Reaction by Nanostructured Metal Materials via Defects Engineering. Nanomaterials 2022, 12, 2389. [Google Scholar] [CrossRef]
- Hori, Y.; Murata, A.; Takahashi, R. Formation of Hydrocarbons in the Electrochemical Reduction of Carbon Dioxide at a Copper Electrode in Aqueous Solution. J. Chem. Soc. Faraday Trans. 1989, 85, 2309–2326. [Google Scholar] [CrossRef]
- Dewulf, D.W.; Jin, T.; Bard, A.J. Electrochemical and Surface Studies of Carbon Dioxide Reduction to Methane and Ethylene at Copper Electrodes in Aqueous Solutions. J. Electrochem. Soc. 1989, 136, 1686–1691. [Google Scholar] [CrossRef]
- Peterson, A.A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Nørskov, J.K. How Copper Catalyzes the Electroreduction of Carbon Dioxide into Hydrocarbon Fuels. Energy Environ. Sci. 2010, 3, 1311–1315. [Google Scholar] [CrossRef]
- Peng, X.; Karakalos, S.G.; Mustain, W.E. Preferentially Oriented Ag Nanocrystals with Extremely High Activity and Faradaic Efficiency for CO2 Electrochemical Reduction to CO. ACS Appl. Mater. Interfaces 2018, 10, 1734–1742. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Niu, C.; Zhu, Y. Copper–Silver Bimetallic Nanowire Arrays for Electrochemical Reduction of Carbon Dioxide. Nanomaterials 2019, 9, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, M.N.; Wen, J.; Konda, S.K.; Govindhan, M.; Chen, A. Electrochemical and FTIR Spectroscopic Study of CO2 Reduction at a Nanostructured Cu/Reduced Graphene Oxide Thin Film. Electrochem. Commun. 2017, 82, 16–20. [Google Scholar] [CrossRef]
- Ye, W.; Guo, X.; Ma, T. A Review on Electrochemical Synthesized Copper-Based Catalysts for Electrochemical Reduction of CO2 to C2+ Products. Chem. Eng. J. 2021, 414, 128825. [Google Scholar] [CrossRef]
- Woldu, A.R.; Huang, Z.; Zhao, P.; Hu, L.; Astruc, D. Electrochemical CO2 Reduction (CO2RR) to Multi-Carbon Products over Copper-Based Catalysts. Coord. Chem. Rev. 2022, 454, 214340. [Google Scholar] [CrossRef]
- Pinthong, P.; Klongklaew, P.; Praserthdam, P.; Panpranot, J. Effect of the Nanostructured Zn/Cu Electrocatalyst Morphology on the Electrochemical Reduction of CO2 to Value-Added Chemicals. Nanomaterials 2021, 11, 1671. [Google Scholar] [CrossRef]
- Gao, F.-Y.; Bao, R.-C.; Gao, M.-R.; Yu, S.-H. Electrochemical CO2-to-CO Conversion: Electrocatalysts, Electrolytes, and Electrolyzers. J. Mater. Chem. A 2020, 8, 15458–15478. [Google Scholar] [CrossRef]
- Dutta, A.; Rahaman, M.; Hecker, B.; Drnec, J.; Kiran, K.; Montiel, I.Z.; Weber, D.J.; Zanetti, A.; López, A.C.; Martens, I.; et al. CO2 Electrolysis—Complementary Operando XRD, XAS and Raman Spectroscopy Study on the Stability of CuxO Foam Catalysts. J. Catal. 2020, 389, 592–603. [Google Scholar] [CrossRef]
- Gu, Y.; Li, J.; Wang, L.; Xie, M.; Wu, X.; Xie, F.; Ryan, M.P. Template-Free Cu Electrodeposition to Prepare Cu–Micro-Brush Electrodes for Electrochemical CO2 Reduction. ChemistrySelect 2019, 4, 10995–11001. [Google Scholar] [CrossRef]
- Ni, Z.; Liang, H.T.; Yi, Z.; Guo, R.; Liu, C.; Liu, Y.; Sun, H.; Liu, X. Research Progress of Electrochemical CO2 Reduction for Copper-Based Catalysts to Multicarbon Products. Coord. Chem. Rev. 2021, 441, 213983. [Google Scholar] [CrossRef]
- Das, M.; Biswas, A.; Purkait, T.; Boruah, T.; Bhardwaj, S.; Das, S.K.; Dey, R.S. The Versatility of the Dynamic Hydrogen Bubble Template Derived Copper Foam on the Emerging Energy Applications: Progress and Future Prospects. J. Mater. Chem. A 2022, 10, 13589–13624. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Y.; Zhao, Y.; Yu, J.; Liu, Z.; Shi, Y.; Liu, H.; Li, X.; Zhou, W. Laser-fabricated Channeled Cu6Sn5/Sn as Electrocatalyst and Gas Diffusion Electrode for Efficient CO2 Electroreduction to Formate. Appl. Catal. B Environ. 2022, 307, 120991. [Google Scholar] [CrossRef]
- Du, D.; Lan, R.; Humphreys, J.; Sengodan, S.; Xie, K.; Wang, H.; Tao, S. Achieving Both High Selectivity and Current Density for CO2 Reduction to Formate on Nanoporous Tin Foam Electrocatalysts. ChemistrySelect 2016, 1, 1711–1715. [Google Scholar] [CrossRef] [Green Version]
- Hui, S.R.; Shaigan, N.; Neburchilov, V.; Zhang, L.; Malek, K.; Eikerling, M.; Luna, P.D. Three-Dimensional Cathodes for Electrochemical Reduction of CO2: From Macro- to Nano-Engineering. Nanomaterials 2020, 10, 1884. [Google Scholar] [CrossRef]
- Sen, S.; Liu, D.; Palmore, G.T.R. Electrochemical Reduction of CO2 at Copper Nanofoams. ACS Catal. 2014, 4, 3091–3095. [Google Scholar] [CrossRef]
- Kottakkat, T.; Klingan, K.; Jiang, S.; Jovanov, Z.P.; Davies, V.H.; El-Nagar, G.A.; Dau, H.; Roth, C. Electrodeposited AgCu Foam Catalysts for Enhanced Reduction of CO2 to CO. ACS Appl. Mater. Interfaces 2019, 11, 14734–14744. [Google Scholar] [CrossRef]
- Hossain, M.N.; Wen, J.; Chen, A. Unique copper and reduced graphene oxide nanocomposite toward the efficient electrochemical reduction of carbon dioxide. Sci. Rep. 2017, 7, 3184. [Google Scholar] [CrossRef] [Green Version]
- Abner, S.; Chen, A. Design and Mechanistic Study of Advanced Cobalt-Based Nanostructured Catalysts for Electrochemical Carbon Dioxide Reduction. Appl. Catal. B Environ. 2022, 301, 120761. [Google Scholar] [CrossRef]
- Tang, W.; Peterson, A.A.; Varela, A.S.; Jovanov, Z.P.; Bech, L.; Durand, W.J.; Dahl, S.; Nørskov, J.K.; Chorkendorff, I. The Importance of Surface Morphology in Controlling the Selectivity of Polycrystalline Copper for CO2 Electroreduction. Phys. Chem. Chem. Phys. 2012, 14, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Min, S.; Yang, X.; Lu, A.-Y.; Tseng, C.-C.; Hedhili, M.N.; Li, L.-J.; Huang, K.-W. Low Overpotential and High Current CO2 Reduction with Surface Reconstructed Cu Foam Electrodes. Nano Energy 2016, 27, 121–129. [Google Scholar] [CrossRef] [Green Version]
- Lum, Y.; Yue, B.; Lobaccaro, P.; Bell, A.T.; Ager, J.W. Optimizing C–C Coupling on Oxide-Derived Copper Catalysts for Electrochemical CO2 Reduction. J. Phys. Chem. C 2017, 121, 14191–14203. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Cheng, T.; Goddard, W.A., III; Sundararaman, R. Mechanistic Explanation of the pH Dependence and Onset Potentials for Hydrocarbon Products from Electrochemical Reduction of CO on Cu (111). J. Am. Chem. Soc. 2016, 138, 483–486. [Google Scholar] [CrossRef]
- Hossain, M.N.; Choueiri, R.M.; Abner, S.; Chen, L.D.; Chen, A. Electrochemical Reduction of Carbon Dioxide at TiO2/Au Nanocomposites. ACS Appl. Mater. Interfaces 2022, 14, 51889–51899. [Google Scholar] [CrossRef] [PubMed]
- Dunwell, M.; Luc, W.; Yan, Y.; Jiao, F.; Xu, B. Understanding Surface-Mediated Electrochemical Reactions: CO2 Reduction and Beyond. ACS Catal. 2018, 8, 8121–8129. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Applied Bias (V) | RS (Ω/cm2) | RCT1 (Ω/cm2) | CPE-T1 (mF cm−2) | RCT2 (Ω/cm2) | CPE-T2 (mF cm−2) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cu200mA/Ag/Cu | Cu200mA/Cu | Cu200mA/Ag/Cu | Cu200mA/Cu | Cu200mA/Ag/Cu | Cu200mA/Cu | Cu200mA/Ag/Cu | Cu200mA/Cu | Cu200mA/Ag/Cu | Cu200mA/Cu | |
| −0.57 | 18.75 ± 0.21 | 22.63 ± 0.32 | 210.5 ± 21.1 | 506.2 ± 7.9 | 25.81 ± 1.03 | 9.559 ± 0.013 | 1098.3 ± 6.4 | 1031.67 ± 51 | 2.54 ± 0.18 | 9.94 ± 0.49 |
| −0.61 | 18.75 ± 0.21 | 22.36 ± 0.29 | 108.8 ± 6.5 | 387.5 ± 3.1 | 19.24 ± 0.76 | 6.007 ± 0.012 | 557.3 ± 4.1 | 579.8 ± 28 | 2.38 ± 0.15 | 6.01 ± 0.24 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahmati, F.; Sabouhanian, N.; Lipkowski, J.; Chen, A. Synthesis of 3D Porous Cu Nanostructures on Ag Thin Film Using Dynamic Hydrogen Bubble Template for Electrochemical Conversion of CO2 to Ethanol. Nanomaterials 2023, 13, 778. https://doi.org/10.3390/nano13040778
Rahmati F, Sabouhanian N, Lipkowski J, Chen A. Synthesis of 3D Porous Cu Nanostructures on Ag Thin Film Using Dynamic Hydrogen Bubble Template for Electrochemical Conversion of CO2 to Ethanol. Nanomaterials. 2023; 13(4):778. https://doi.org/10.3390/nano13040778
Chicago/Turabian StyleRahmati, Farnood, Negar Sabouhanian, Jacek Lipkowski, and Aicheng Chen. 2023. "Synthesis of 3D Porous Cu Nanostructures on Ag Thin Film Using Dynamic Hydrogen Bubble Template for Electrochemical Conversion of CO2 to Ethanol" Nanomaterials 13, no. 4: 778. https://doi.org/10.3390/nano13040778
APA StyleRahmati, F., Sabouhanian, N., Lipkowski, J., & Chen, A. (2023). Synthesis of 3D Porous Cu Nanostructures on Ag Thin Film Using Dynamic Hydrogen Bubble Template for Electrochemical Conversion of CO2 to Ethanol. Nanomaterials, 13(4), 778. https://doi.org/10.3390/nano13040778