

Effects of Structure and Composition of Adsorbents on Competitive Adsorption of Gaseous Emissions: Experiment and Modeling

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
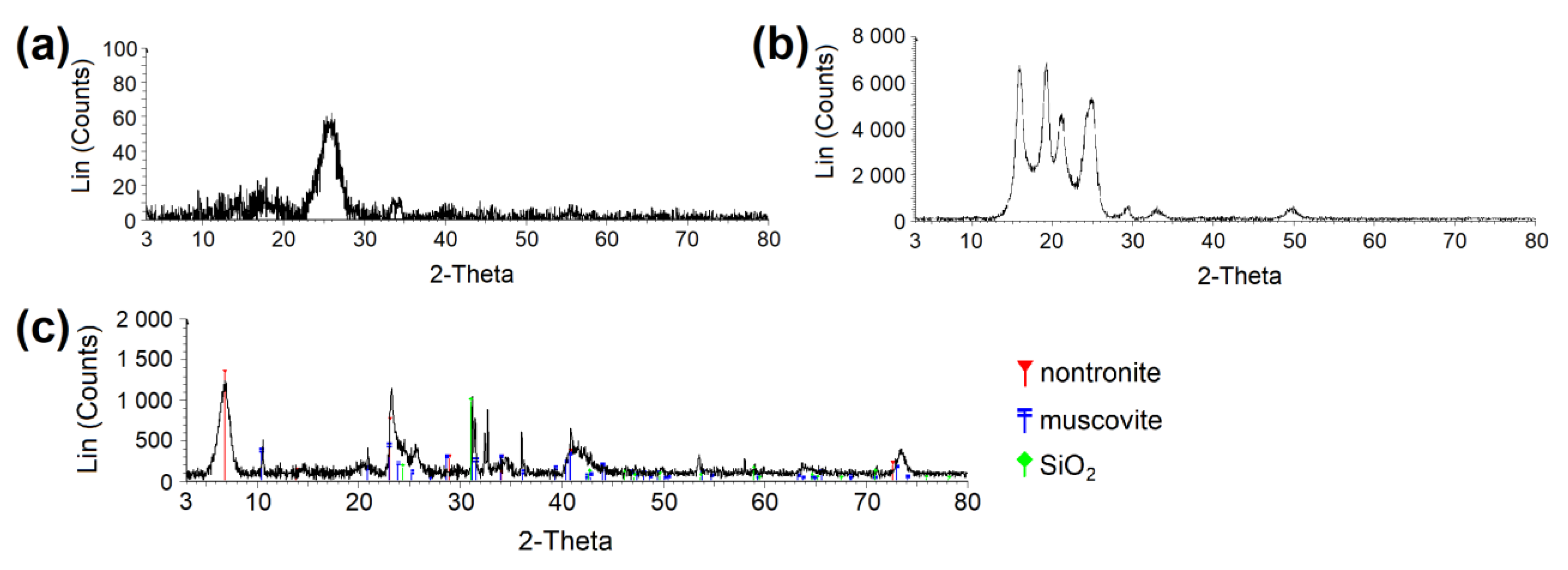
2.2. Characterization of Samples
2.3. Sample Preparation
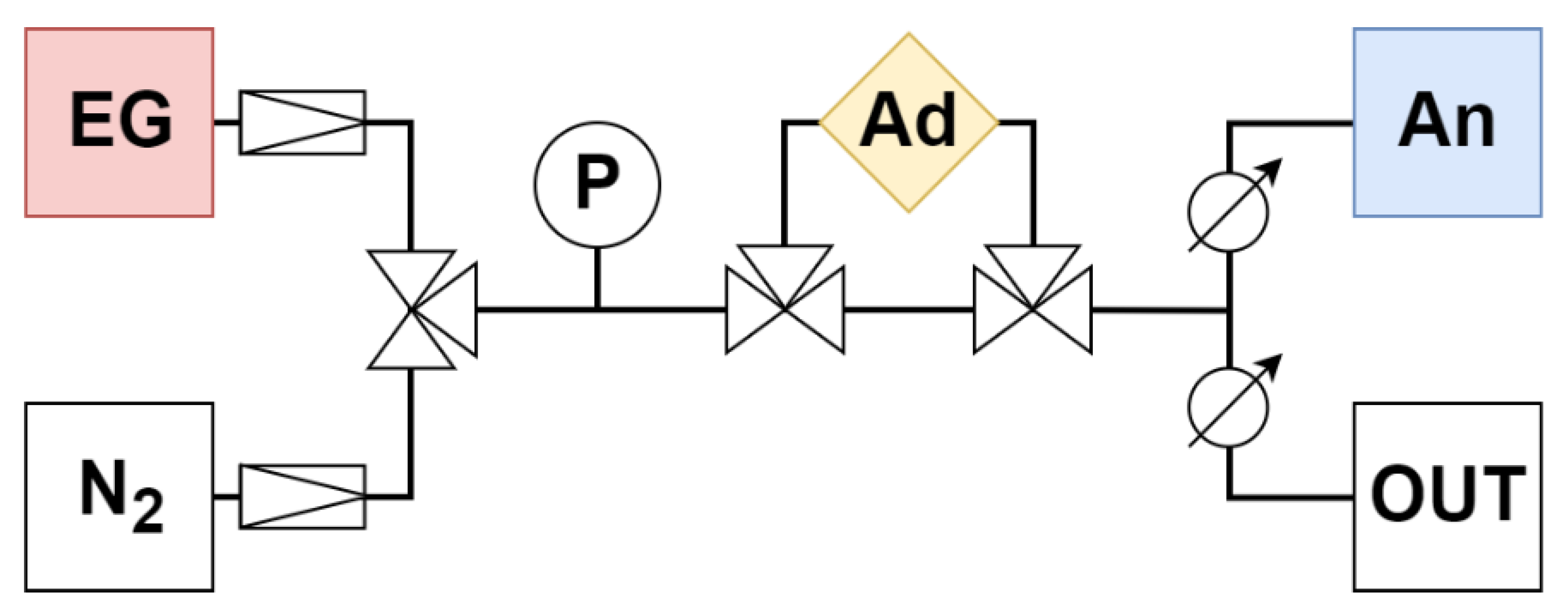
2.4. Adsorption System
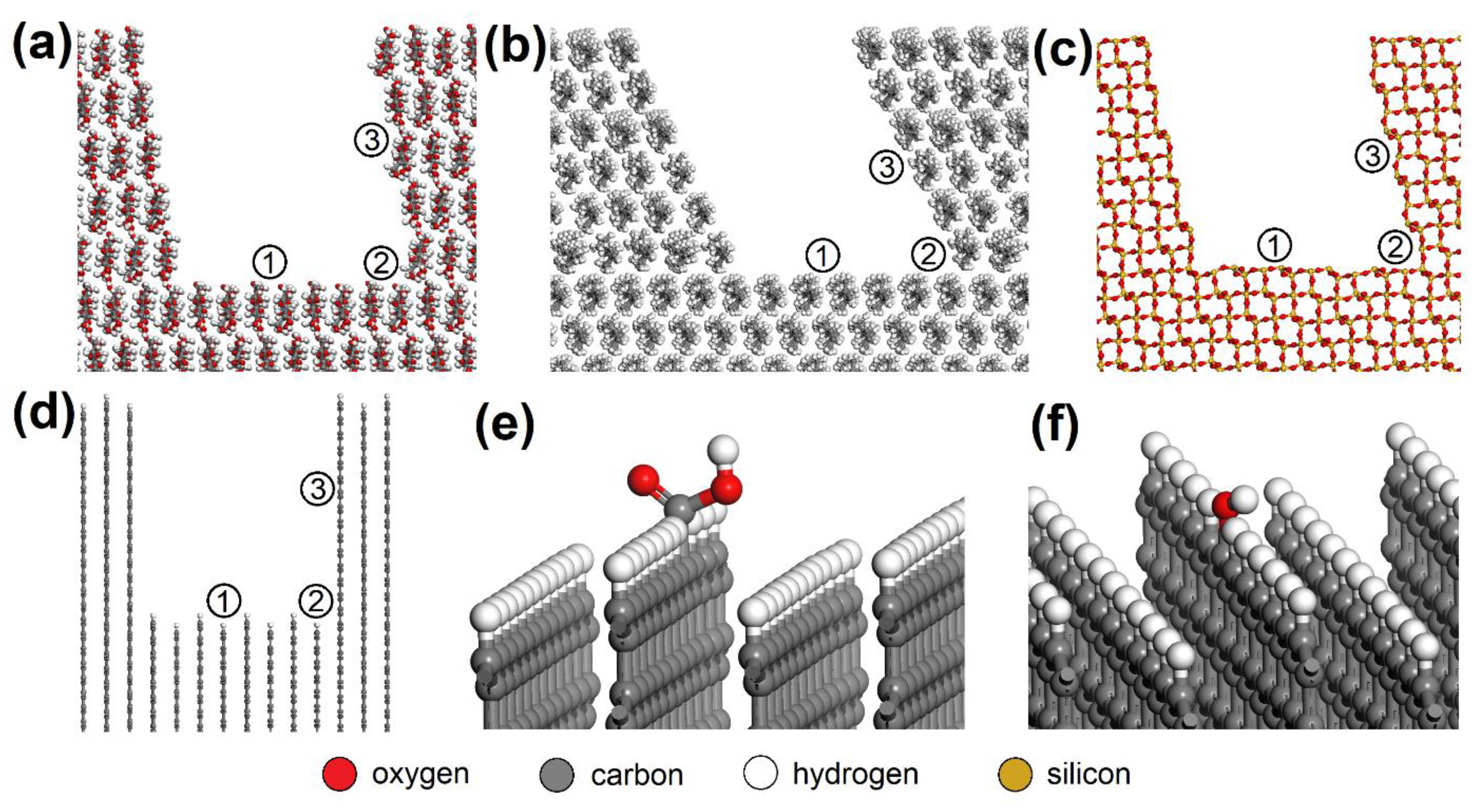
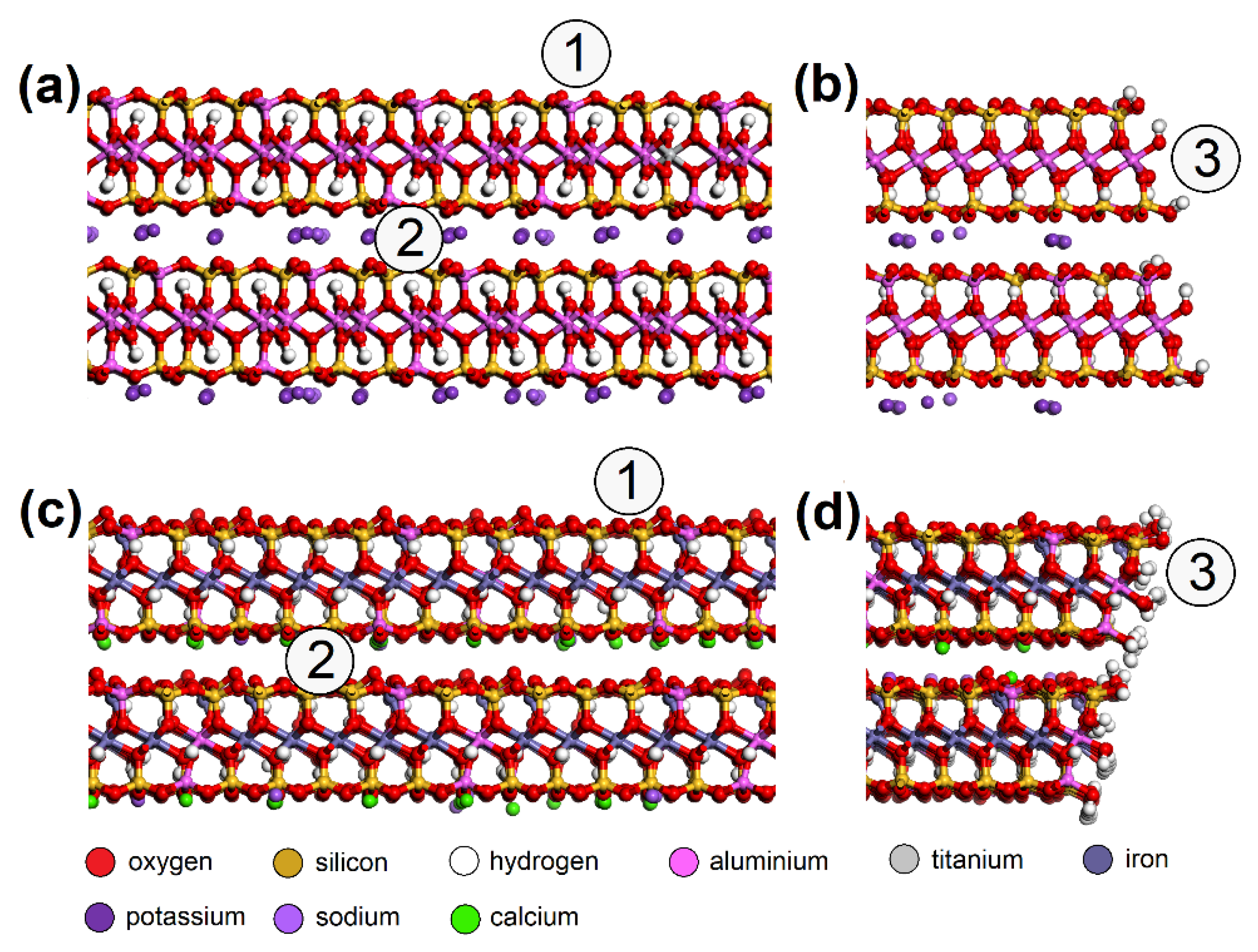
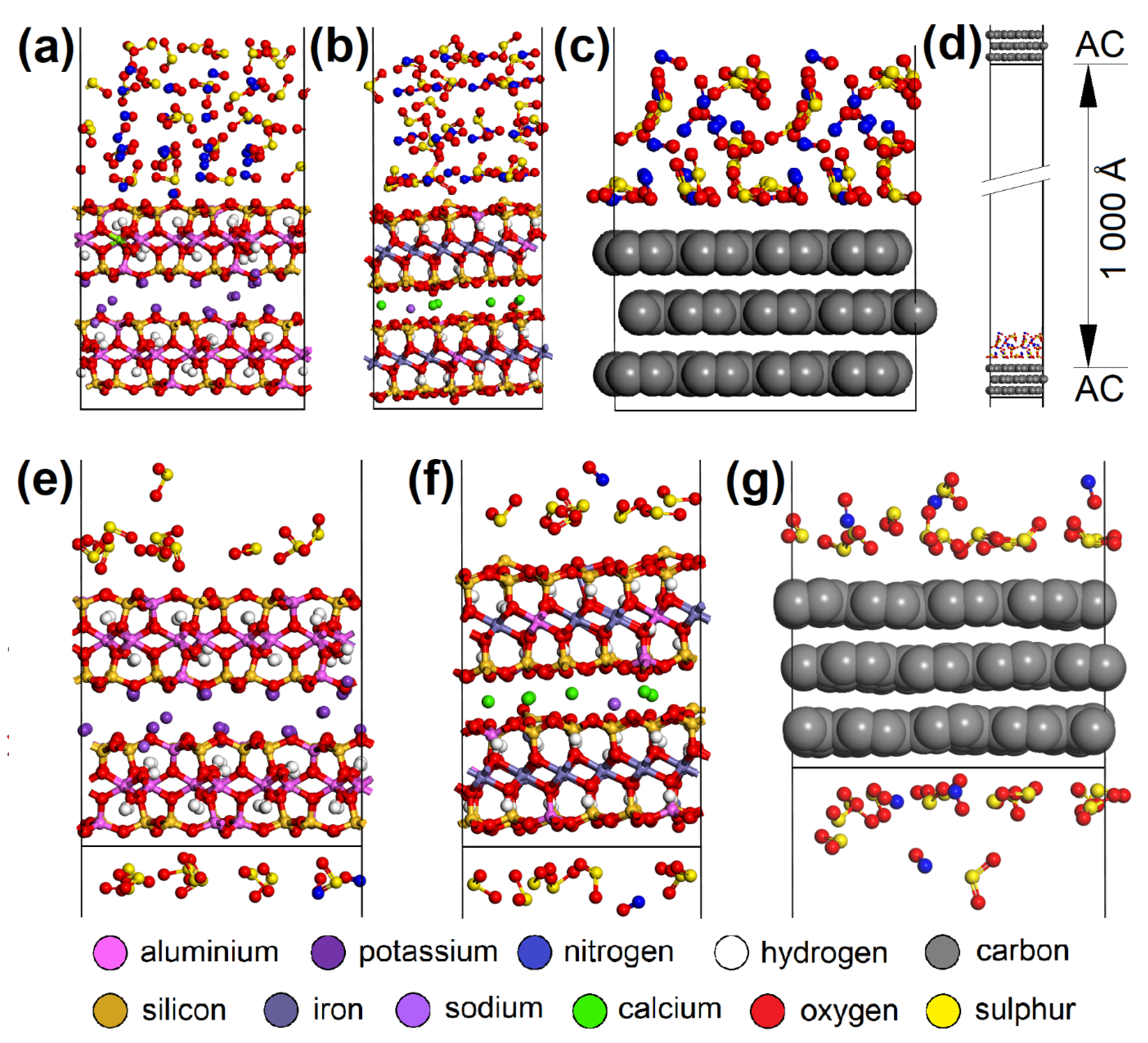
2.5. Atomistic Models and Modeling Strategy
3. Results and Discussion
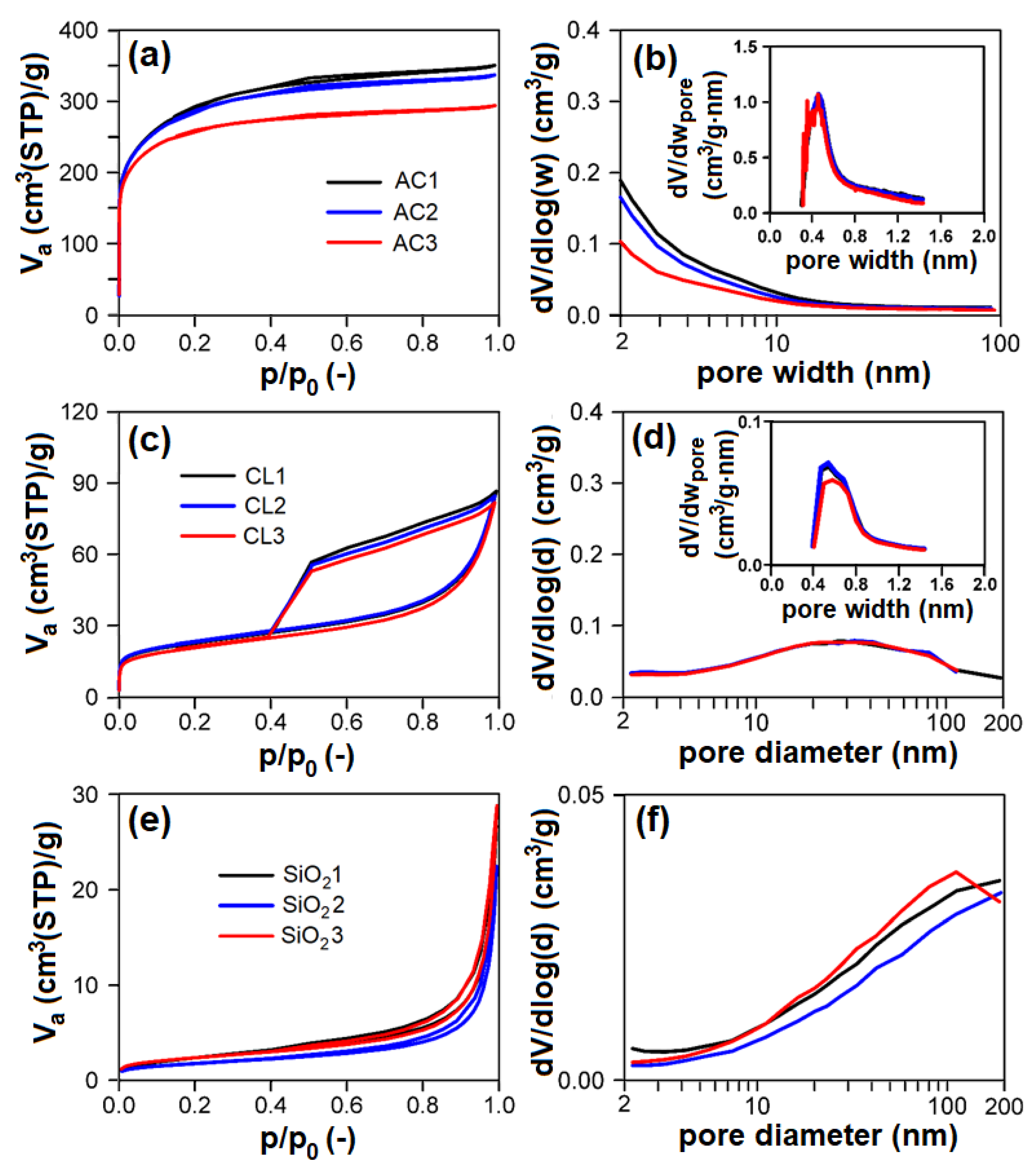
3.1. Physisorption
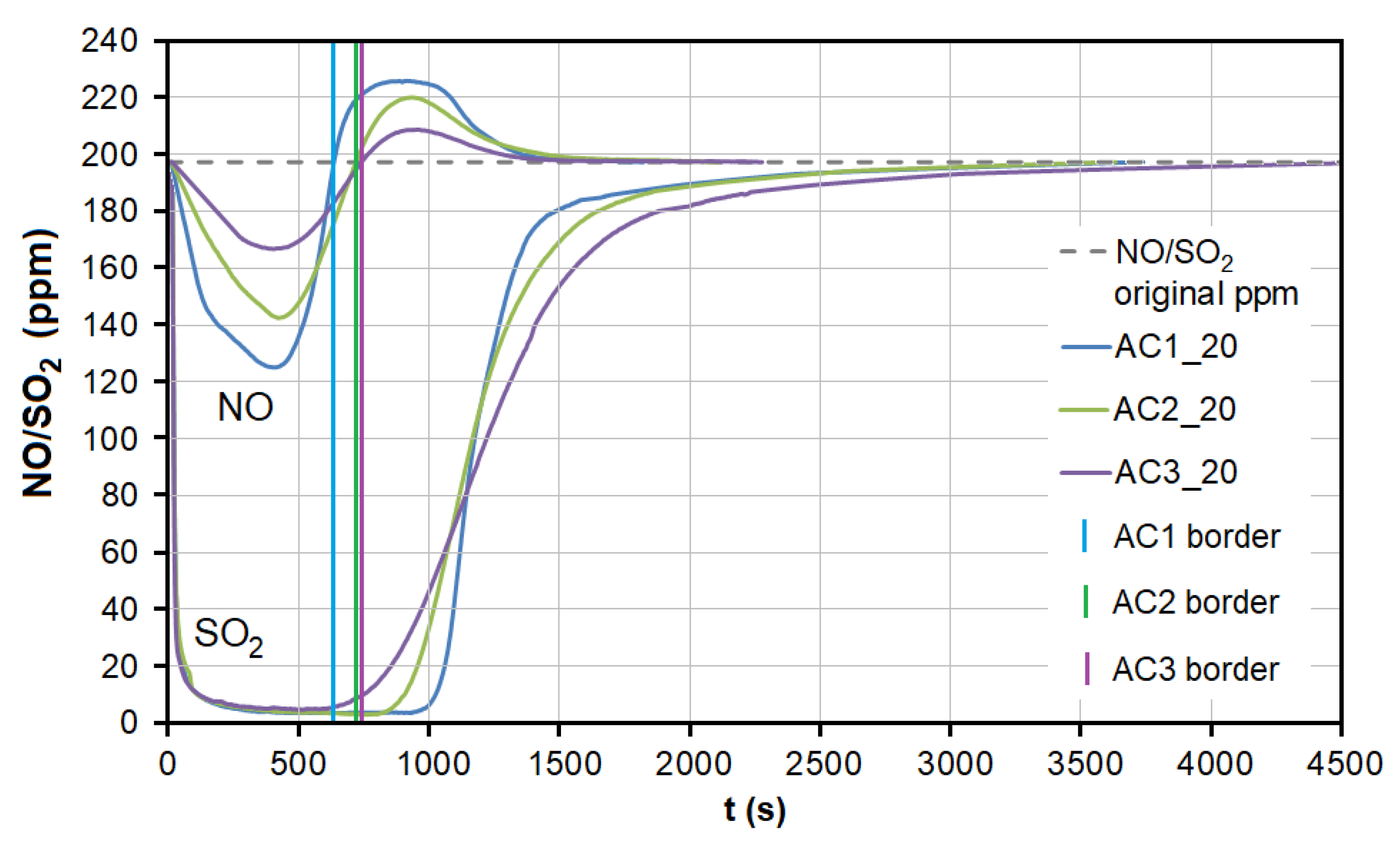
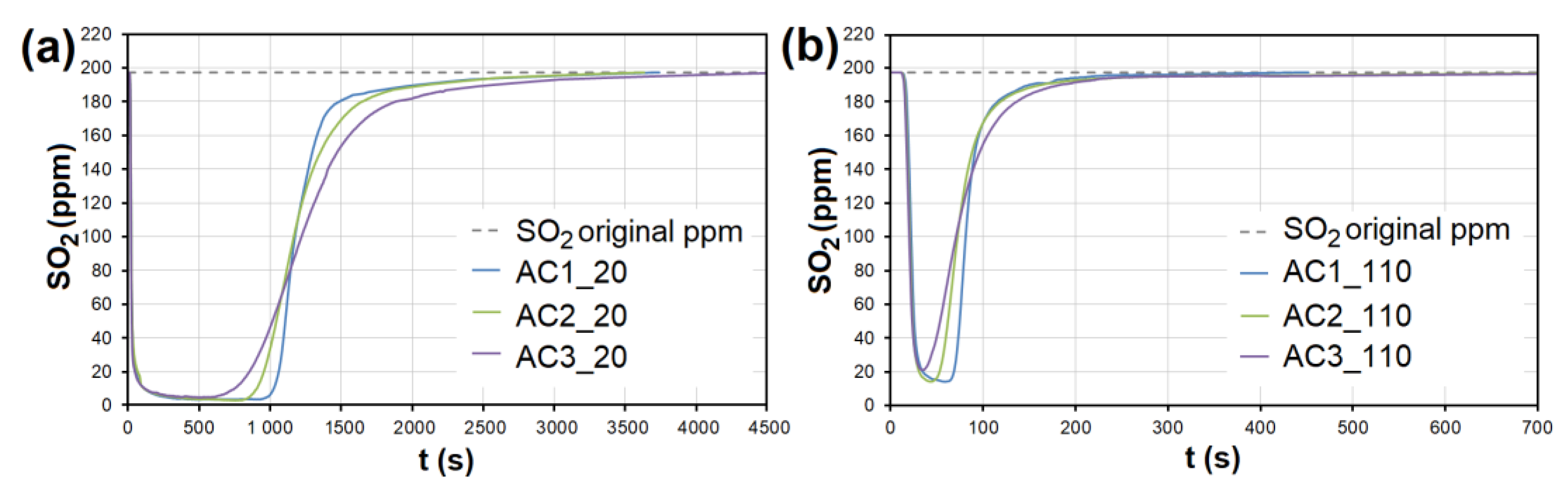
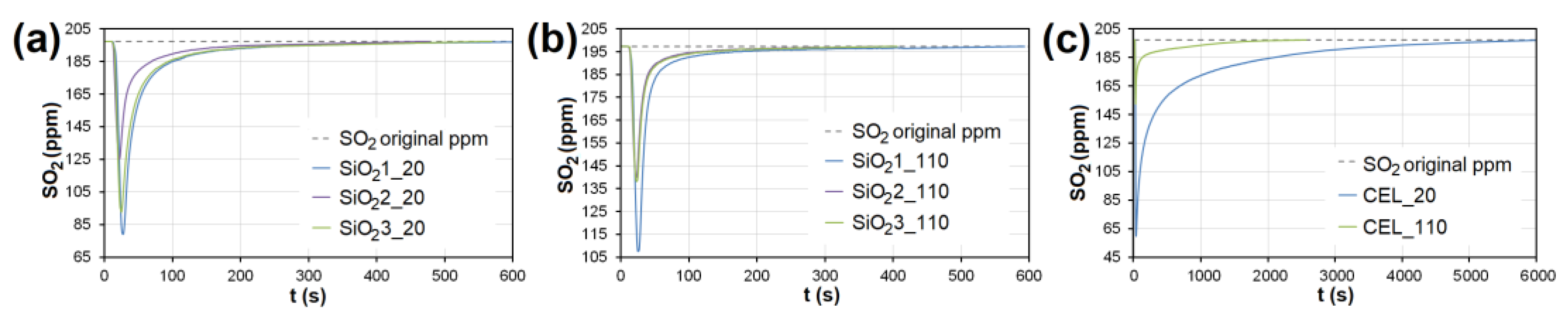
3.2. Emission Gas Adsorption
3.3. Molecular Modeling
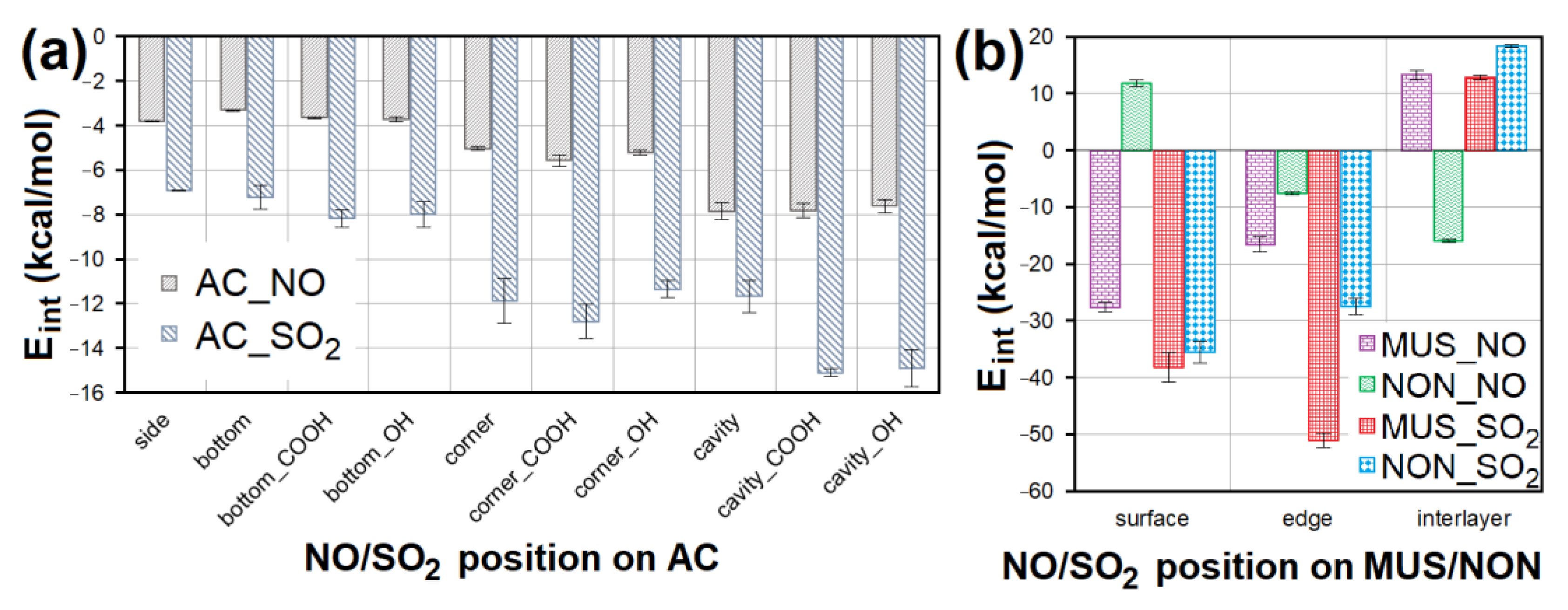
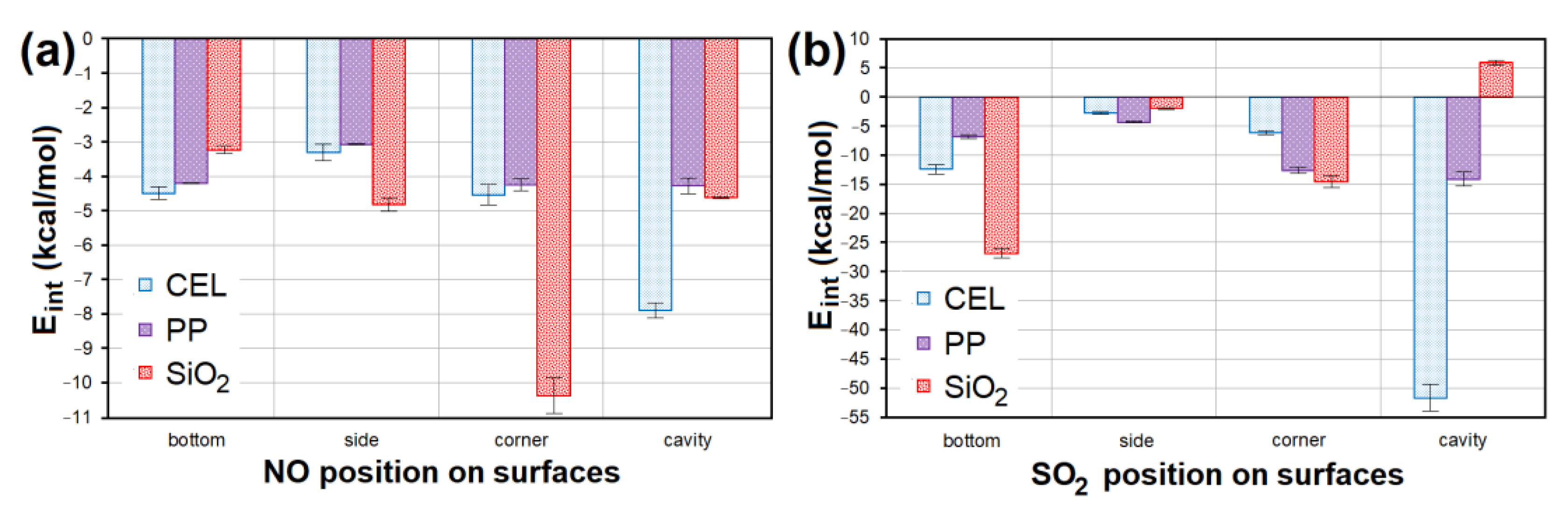
3.4. Adsorption Factors
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Brusseau, M.L.; Pepper, I.L.; Gerba, C.P. Environmental and Pollution Science, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Henzel, C.K. Investigating Manufacturing Pollution, 1st ed.; Child’s World: Parker, CO, USA, 2022. [Google Scholar]
- Tillman, D.A. Coal-Fired Electricity and Emissions Control: Efficiency and Effectiveness, 1st ed.; Butterworth-Heinemann: Oxford, UK, 2018. [Google Scholar]
- Matz, C.J.; Egyed, M.; Hocking, R.; Seenundun, S.; Charman, N.; Edmonds, N. Human health effects of traffic-related air pollution (TRAP): A scoping review protocol. Syst. Rev. 2019, 8, 223. [Google Scholar] [CrossRef]
- Boot-Handford, M.E.; Abanades, J.C.; Anthony, E.J.; Blunt, M.J.; Brandani, S.; Mac Dowell, N.; Fernández, J.R.; Ferrari, M.C.; Gross, R.; Hallett, J.P.; et al. Carbon capture and storage update. Energy Environ. Sci. 2014, 7, 130–189. [Google Scholar] [CrossRef]
- Werther, J.; Saenger, M.; Hartge, E.U.; Ogada, T.; Siagi, Z. Combustion of agricultural residues. Prog. Energy Combust. Sci. 2000, 26, 1–27. [Google Scholar] [CrossRef]
- Obey, G.; Adelaide, M.; Ramaraj, R. Biochar derived from non-customized matamba fruit shell as an adsorbent for wastewater treatment. J. Bioresour. Bioprod. 2022, 7, 109–115. [Google Scholar] [CrossRef]
- Zheng, Q.; Li, Z.; Watanabe, M. Production of solid fuels by hydrothermal treatment of wastes of biomass, plastic, and biomass/plastic mixtures: A review. J. Bioresour. Bioprod. 2022, 7, 221–244. [Google Scholar] [CrossRef]
- Bai, J.; Yu, C.; Li, L.; Wu, P.; Luo, Z.; Ni, M. Experimental study on the NO and N2O formation characteristics during biomass combustion. Energy Fuels 2013, 27, 515–522. [Google Scholar] [CrossRef]
- Jones, J.M.; Lea-Langton, A.R.; Ma, L.; Pourkashanian, M.; Williams, A. The Combustion of Solid Biomass. In Pollutants Generated by the Combustion of Solid Biomass Fuels, 1st ed.; Springer: London, UK, 2014; pp. 25–43. [Google Scholar]
- Yu, C.H.; Huang, C.H.; Tan, C.S. A review of CO2 capture by absorption and adsorption. Aerosol Air Qual. Res. 2012, 12, 745–769. [Google Scholar] [CrossRef]
- Sircar, S. Basic research needs for design of adsorptive gas separation processes. Ind. Eng. Chem. Res. 2006, 45, 5435–5448. [Google Scholar] [CrossRef]
- Zhu, B.; Liu, Q.; Zhou, Q.; Yang, J.; Ding, J.; Wen, J. Absorption of carbon dioxide from flue gas using blended amine solutions. Chem. Eng. Technol. 2014, 37, 635–642. [Google Scholar] [CrossRef]
- Choi, S.; Drese, J.H.; Jones, C.W. Adsorbent materials for carbon dioxide capture from large anthropogenic point sources. ChemSusChem 2009, 2, 796–854. [Google Scholar] [CrossRef]
- Samanta, A.; Zhao, A.; Shimizu, G.K.; Sarkar, P.; Gupta, R. Post-combustion CO2 capture using solid sorbents: A review. Ind. Eng. Chem. Res. 2012, 51, 1438–1463. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, Y.; Liu, C.; Zhou, L. Comparative studies of CO2 and CH4 sorption on activated carbon in presence of water. Colloids Surfaces A Physicochem. Eng. Asp. 2008, 322, 14–18. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, K.; Clennell, M.B.; Dewhurst, D.N.; Pan, Z.; Pervukhina, M.; Han, T. Molecular simulation studies of hydrocarbon and carbon dioxide adsorption on coal. Pet. Sci. 2015, 12, 692–704. [Google Scholar] [CrossRef]
- Bezerra, D.P.; Oliveira, R.S.; Vieira, R.S.; Cavalcante, C.L.; Azevedo, D. Adsorption of CO2 on nitrogen-enriched activated carbon and zeolite 13X. Adsorption 2011, 17, 235–246. [Google Scholar] [CrossRef]
- Li, Z.S.; Cai, N.S.; Huang, Y.Y.; Han, H.J. Synthesis, experimental studies, and analysis of a new calcium-based carbon dioxide absorbent. Energy Fuels 2005, 19, 1447–1452. [Google Scholar] [CrossRef]
- Franchi, R.S.; Harlick, P.J.; Sayari, A. Applications of pore-expanded mesoporous silica. 2. Development of a high-capacity, water-tolerant adsorbent for CO2. Ind. Eng. Chem. Res. 2005, 44, 8007–8013. [Google Scholar] [CrossRef]
- Ding, Y.; Alpay, E. Equilibria and kinetics of CO2 adsorption on hydrotalcite adsorbent. Chem. Eng. Sci. 2000, 55, 3461–3474. [Google Scholar] [CrossRef]
- Srivatsa, S.C.; Bhattacharya, S. Amine-based CO2 capture sorbents: A potential CO2 hydrogenation catalyst. J. CO2 Util. 2018, 26, 397–407. [Google Scholar] [CrossRef]
- Li, J.R.; Kuppler, R.J.; Zhou, H.C. Selective gas adsorption and separation in metal–organic frameworks. Chem. Soc. Rev. 2009, 38, 1477–1504. [Google Scholar] [CrossRef]
- Wang, H.; Bai, J.Q.; Yin, Y.; Wang, S.F. Experimental and numerical study of SO2 removal from a CO2/SO2 gas mixture in a Cu-BTC metal organic framework. J. Mol. Graph. Model. 2020, 96, 107533. [Google Scholar] [CrossRef]
- Ozensoy, E.; Goodman, D.W. Vibrational spectroscopic studies on CO adsorption, NO adsorption CO+ NO reaction on Pd model catalysts. Phys. Chem. Chem. Phys. 2004, 6, 3765–3778. [Google Scholar] [CrossRef]
- Joshi, A.M.; Tucker, M.H.; Delgass, W.N.; Thomson, K.T. CO adsorption on pure and binary-alloy gold clusters: A quantum chemical study. J. Chem. Phys. 2006, 125, 194707. [Google Scholar] [CrossRef] [PubMed]
- Abdulrasheed, A.A.; Jalil, A.A.; Triwahyono, S.; Zaini, M.A.A.; Gambo, Y.; Ibrahim, M. Surface modification of activated carbon for adsorption of SO2 and NOX: A review of existing and emerging technologies. Renew. Sustain. Energ. Rev. 2018, 94, 1067–1085. [Google Scholar] [CrossRef]
- Niu, J.; Miao, J.; Zhang, H.; Guo, Y.; Li, L.; Cheng, F. Focusing on the impact of inherent minerals in coal on activated carbon production and its performance: The role of trace sodium on SO2 and/or NO removal. Energy 2023, 263, 125638. [Google Scholar] [CrossRef]
- Rodriguez, J.A. The chemical properties of bimetallic surfaces: Importance of ensemble and electronic effects in the adsorption of sulfur and SO2. Prog. Surf. Sci. 2006, 81, 141–189. [Google Scholar] [CrossRef]
- Yi, H.; Deng, H.; Tang, X.; Yu, Q.; Zhou, X.; Liu, H. Adsorption equilibrium and kinetics for SO2, NO, CO2 on zeolites FAU and LTA. J. Hazard. Mater. 2012, 203, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xuan, C.; Zhang, X.; Sun, R.; Cheng, X.; Wang, Z.; Ma, C. NOx Adsorption Mechanism of Coal-Based Activated Carbon Modified with Trace Potassium: In Situ DRIFTS and DFT Study. Energy Fuels 2022, 36, 7633–7650. [Google Scholar] [CrossRef]
- Zhao, R.; Liu, G.; Wei, G.; Gao, J.; Lu, H. Analysis of SO2 Physisorption by Edge-Functionalized Nanoporous Carbons Using Grand Canonical Monte Carlo Methods and Density Functional Theory: Implications for SO2 Removal. ACS Omega 2021, 6, 33735–33746. [Google Scholar] [CrossRef]
- Zeng, W.; Tan, S.J.; Liu, M.; Zhang, D.; Liu, L.; Do, D.D. New Insights into the Capture of Low-level Gaseous Pollutants in Indoor Environment by Carbonaceous Materials: Effects of Functional Groups, Pore Size, and Presence of Moist. Sep. Purif. Technol. 2022, 298, 121652. [Google Scholar] [CrossRef]
- Wang, J.; Yang, M.; Deng, D.; Qiu, S. The adsorption of NO, NH3, N2 on carbon surface: A density functional theory study. J. Mol. Model. 2017, 23, 262. [Google Scholar] [CrossRef]
- Najser, T.; Gaze, B.; Knutel, B.; Verner, A.; Najser, J.; Mikeska, M.; Chojnacki, J.; Němček, O. Analysis of the Effect of Catalytic Additives in the Agricultural Waste Combustion Process. Materials 2022, 15, 3526. [Google Scholar] [CrossRef]
- Towler, G.; Sinnott, R. Process Flowsheet Development. In Chemical Engineering Design: Principles, Practice and Economic of Plant and Process Design, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 33–102. [Google Scholar]
- Das, T.K. Industrial Pollution Sources, Its Characterization, Estimation, and Treatment. In Industrial Environmental Management: Engineering, Science, and Policy, 1st ed.; Wiley: Hoboken, NJ, USA, 2020; pp. 71–113. [Google Scholar]
- BIOVIA. Materials Studio 7.0; Dassault Systèmes: San Diego, CA, USA, 2013. [Google Scholar]
- Chen, P.; Nishiyama, Y.; Putaux, J.L.; Mazeau, K. Diversity of potential hydrogen bonds in cellulose I revealed by molecular dynamics simulation. Cellulose 2014, 21, 897–908. [Google Scholar] [CrossRef]
- Verenich, S.; Paul, S.; Pourdeyhimi, B. Surface and bulk properties of glycidyl methacrylate modified polypropylene: Experimental and molecular modeling studies. J. Appl. Polym. Sci. 2008, 108, 2983–2987. [Google Scholar] [CrossRef]
- Wyckoff, R.W.G. Crystal Structures, 2nd ed.; Wiley: New York, NY, USA, 1963. [Google Scholar]
- Weiss, Z.; Kužvart, M. Clay Minerals: Their Nanostructure and Utilization, 1st ed.; Karolinum: Prague, Czech Republic, 2005. [Google Scholar]
- Bednárek, J.; Matějová, L.; Jankovská, Z.; Vaštyl, M.; Sokolová, B.; Peikertová, P.; Šiler, P.; Verner, A.; Tokarský, J.; Koutník, I.; et al. The Influence of Structural Properties on the Adsorption Capacities of Microwave-assisted Biochars for Metazachlor Removal from Aqueous Solutions. J. Environ. Chem. Eng. 2022, 10, 108003. [Google Scholar] [CrossRef]
- Sun, H. COMPASS: An ab initio force-field optimized for condensed-phase applications—Overview with details on alkane and benzene compounds. J. Phys. Chem. 1998, 102, 7338–7364. [Google Scholar] [CrossRef]
- Bazooyar, F.; Momany, F.A.; Bolton, K. Validating empirical force fields for molecular-level simulation of cellulose dissolution. Comput. Theor. Chem. 2012, 984, 119–127. [Google Scholar] [CrossRef]
- Deckers, F.; Rasim, K.; Schröder, C. Molecular dynamics simulation of polypropylene: Diffusion and sorption of H2O, H2O2, H2, O2 and determination of the glass transition temperature. J. Polym. Res. 2022, 29, 463. [Google Scholar] [CrossRef]
- Vijayakumar, S.D.; Ridzuan, N. Molecular interaction study on Gemini surfactant and nanoparticles in wax inhibition of Malaysian crude oil. Asia-Pac. J. Chem. Eng. 2021, 16, e2700. [Google Scholar] [CrossRef]
- Rai, B.; Sathish, P.; Tanwar, J.; Moon, K.S.; Fuerstenau, D.W. A molecular dynamics study of the interaction of oleate and dodecylammonium chloride surfactants with complex aluminosilicate minerals. J. Colloid Interface Sci. 2011, 362, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
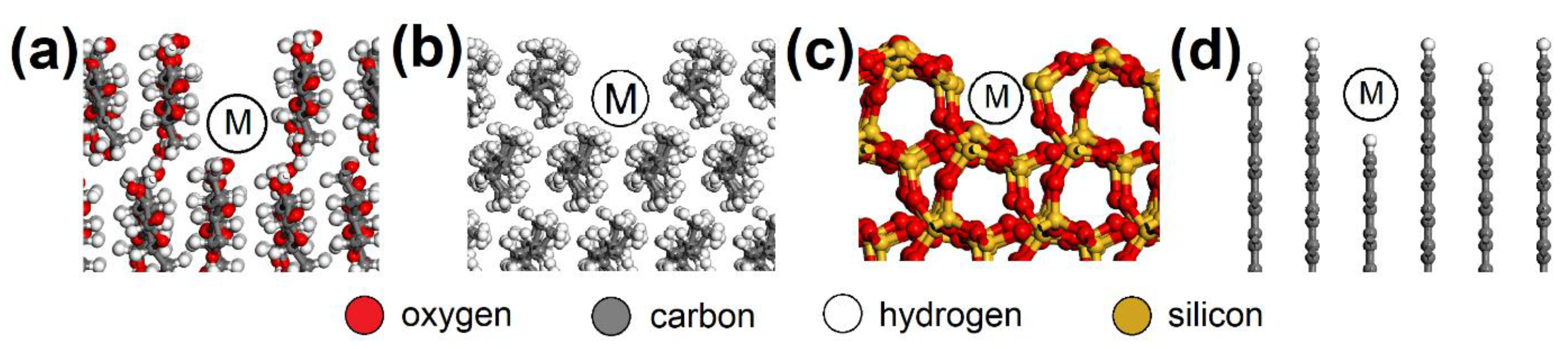{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adsorbing Material | Particle Density (g/cm3) | Humidity (%) | Adsorbing Material Form | Granulometric Fraction Labels | ||
|---|---|---|---|---|---|---|
| 0.16–0.315 mm | 0.315–0.63 mm | 0.63–1.00 mm | ||||
| AC | 1.354 | 3.318 | powder | AC1 | AC2 | AC3 |
| CL | 2.214 | 11.992 | powder | CL1 | CL2 | CL3 |
| SiO2 | 1.754 | 0.285 | powder | SiO21 | SiO22 | SiO23 |
| CEL | 1.225 | 15.339 | pellets | - | - | - |
| PP | 1.007 | 1.204 | nonwoven | - | - | - |
| Adsorbent | SBET (m2/g) | Smeso (m2/g) | Vmicro (mm3liq/g) | Vnet (mm3liq/g) |
|---|---|---|---|---|
| AC1 | 999 | 209 | 373 | 542 |
| AC2 | 975 | 180 | 376 | 521 |
| AC3 | 868 | 114 | 359 | 455 |
| CL1 | 76 | 49 | 14 | 134 |
| CL2 | 78 | 50 | 14 | 131 |
| CL3 | 70 | 47 | 12 | 127 |
| SiO21 | 8.9 | 8.9 * | n. d. | 41 |
| SiO22 | 6.4 | 6.4 * | n. d. | 35 |
| SiO23 | 8.5 | 8.5 * | n. d. | 45 |
| CEL | 1.6 ± 0.1 | n. d. | n. d. | n. d. |
| PP | 0.4 ± 0.1 | n. d. | n. d. | 1.02 |
| Adsorbent | Mass | Adsorption Capacity | |||||
|---|---|---|---|---|---|---|---|
| Wet Sample (g) | Dried Sample (g) | NO | SO2 | CO | |||
| 20 °C (μg/g) | 110 °C (μg/g) | 20 °C (μg/g) | 110 °C (μg/g) | 110 °C (μg/g) | |||
| AC1 | 1.5277 | 1.4770 | 120.83 | 2.27 | 3549.61 | 190.00 | 0.47 |
| AC2 | 1.5370 | 1.4860 | 102.23 | 1.85 | 3482.41 | 182.27 | 0.17 |
| AC3 | 1.5404 | 1.4893 | 75.65 | 2.01 | 3724.97 | 186.58 | 0.22 |
| CL1 | 2.8859 | 2.5398 | 20.28 | 6.20 | 2771.37 | 1182.46 | 5.69 |
| CL2 | 2.7871 | 2.4529 | 8.16 | 4.53 | 2281.26 | 1110.76 | 4.86 |
| CL3 | 2.7690 | 2.4369 | 4.98 | 3.43 | 2242.24 | 888.29 | 3.80 |
| SiO21 | 1.6116 | 1.6070 | 2.11 | - | 71.68 | 28.01 | - |
| SiO22 | 1.6741 | 1.6693 | 0.33 | - | 68.42 | 5.95 | - |
| SiO23 | 1.5494 | 1.5450 | 0.10 | - | 59.72 | 4.69 | - |
| CEL | 1.6520 | 1.3986 | 5.09 | 1.13 | 1141.61 | 154.89 | 1.48 |
| PP | 0.7907 | 0.7812 | - | - | - | - | - |
| Adsorbing Material | Adsorption Capacity (μg/g) | Lowest Eint (kcal/mol) | SBET (m2/g) | Smeso (m2/g) | Vmicro (mm3liq/g) | Vnet (mm3liq/g) | ||
|---|---|---|---|---|---|---|---|---|
| NO | SO2 | NO | SO2 | |||||
| AC | 120.83 | 3549.61 | −8.31 | −15.36 | 999 * | 209 * | 373 * | 542 * |
| CL | 20.28 | 2771.37 | −28.79 * | −52.36 * | 76 * | 49 * | 14 * | 134 * |
| SiO2 | 2.11 | 71.68 | −10.94 | −27.87 | 8.9 * | 8.9 * | - | 41 * |
| CEL | 5.09 | 1141.61 | −8.14 * | −55.49 * | 1.6 ± 0.1 | - | - | - |
| PP | - | - | −4.60 | −16.23 | 0.4 ± 0.1 * | - | - | 1.02 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verner, A.; Tokarský, J.; Najser, T.; Matějová, L.; Kutláková, K.M.; Kielar, J.; Peer, V. Effects of Structure and Composition of Adsorbents on Competitive Adsorption of Gaseous Emissions: Experiment and Modeling. Nanomaterials 2023, 13, 724. https://doi.org/10.3390/nano13040724
Verner A, Tokarský J, Najser T, Matějová L, Kutláková KM, Kielar J, Peer V. Effects of Structure and Composition of Adsorbents on Competitive Adsorption of Gaseous Emissions: Experiment and Modeling. Nanomaterials. 2023; 13(4):724. https://doi.org/10.3390/nano13040724
Chicago/Turabian StyleVerner, Adam, Jonáš Tokarský, Tomáš Najser, Lenka Matějová, Kateřina Mamulová Kutláková, Jan Kielar, and Václav Peer. 2023. "Effects of Structure and Composition of Adsorbents on Competitive Adsorption of Gaseous Emissions: Experiment and Modeling" Nanomaterials 13, no. 4: 724. https://doi.org/10.3390/nano13040724
APA StyleVerner, A., Tokarský, J., Najser, T., Matějová, L., Kutláková, K. M., Kielar, J., & Peer, V. (2023). Effects of Structure and Composition of Adsorbents on Competitive Adsorption of Gaseous Emissions: Experiment and Modeling. Nanomaterials, 13(4), 724. https://doi.org/10.3390/nano13040724