Metal-Ion Intercalation Mechanisms in Vanadium Pentoxide and Its New Perspectives

 ,
,  , , , , and
, , , , and 
Abstract
:1. Introduction

2. Synthetic Methods
2.1. Sol–Gel
2.2. Hydrothermal Process
2.3. Spray-Drying
2.4. Electrospinning
2.5. Electrochemical Deposition
2.6. Template
3. Intercalation in Battery Electrode
3.1. Univalent Cations
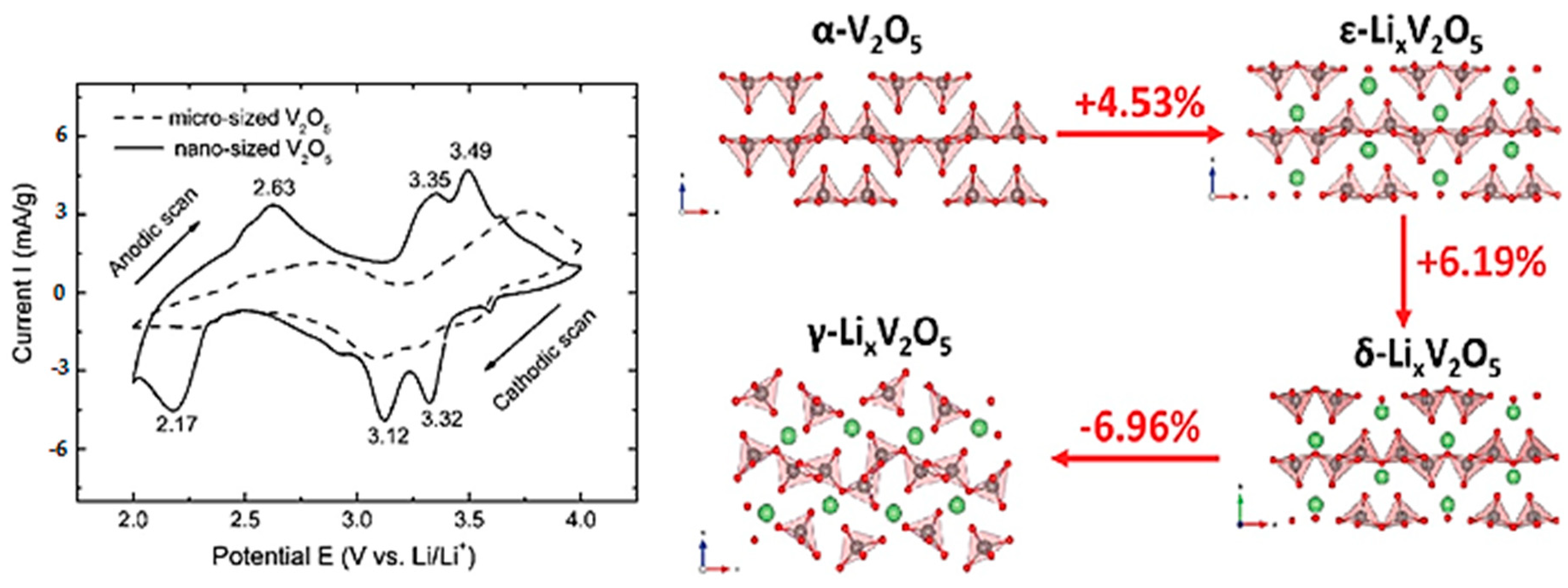
3.1.1. Lithium
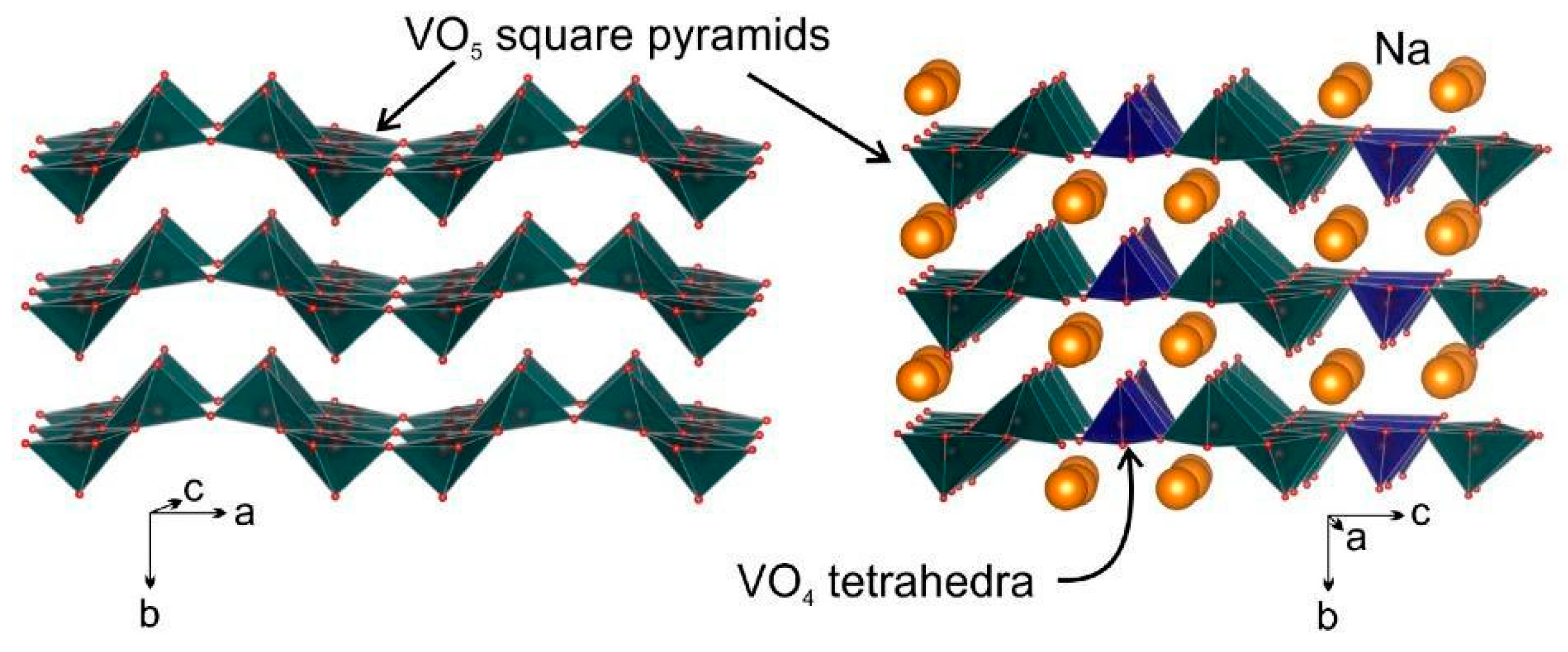
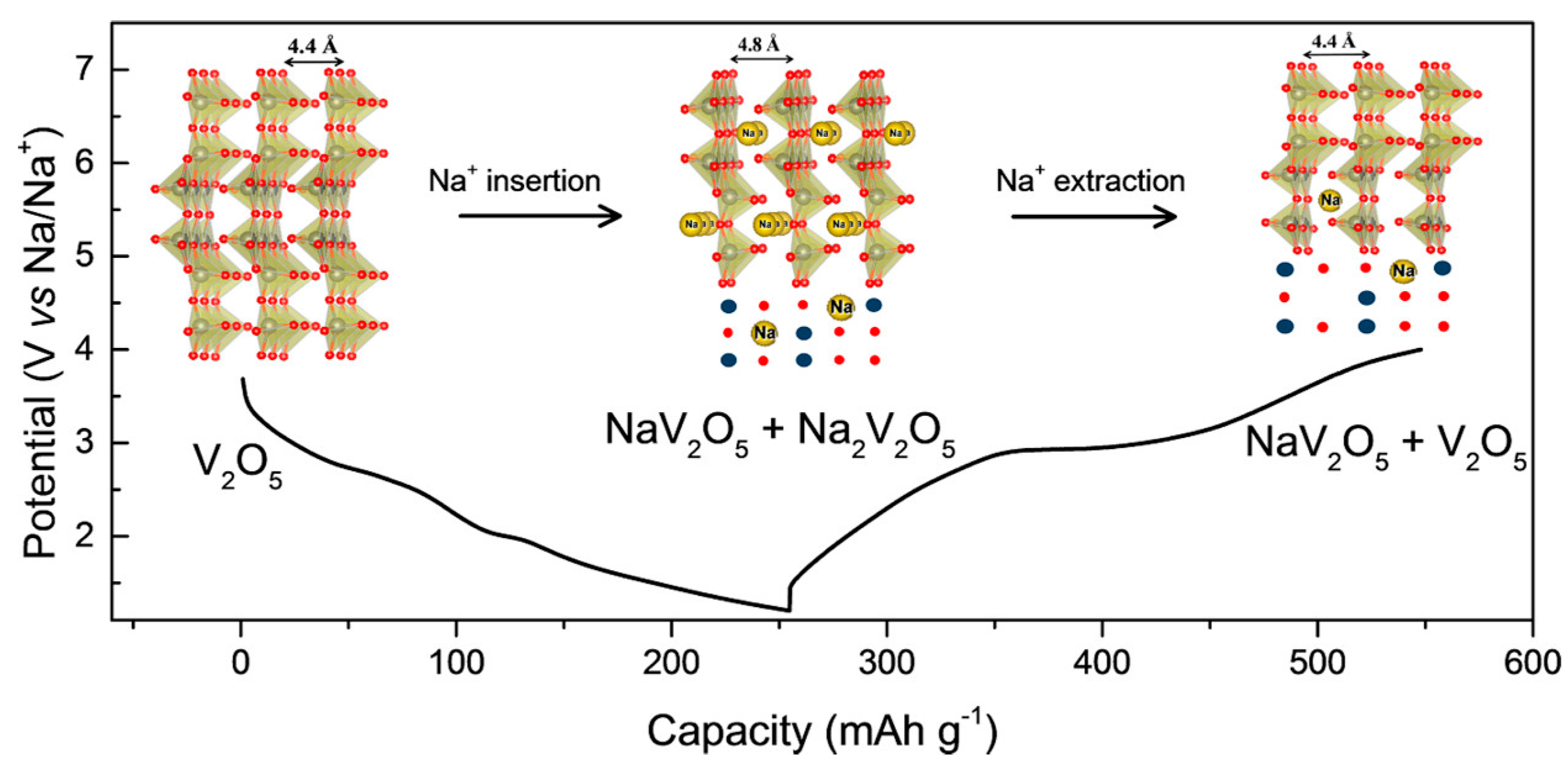
3.1.2. Sodium
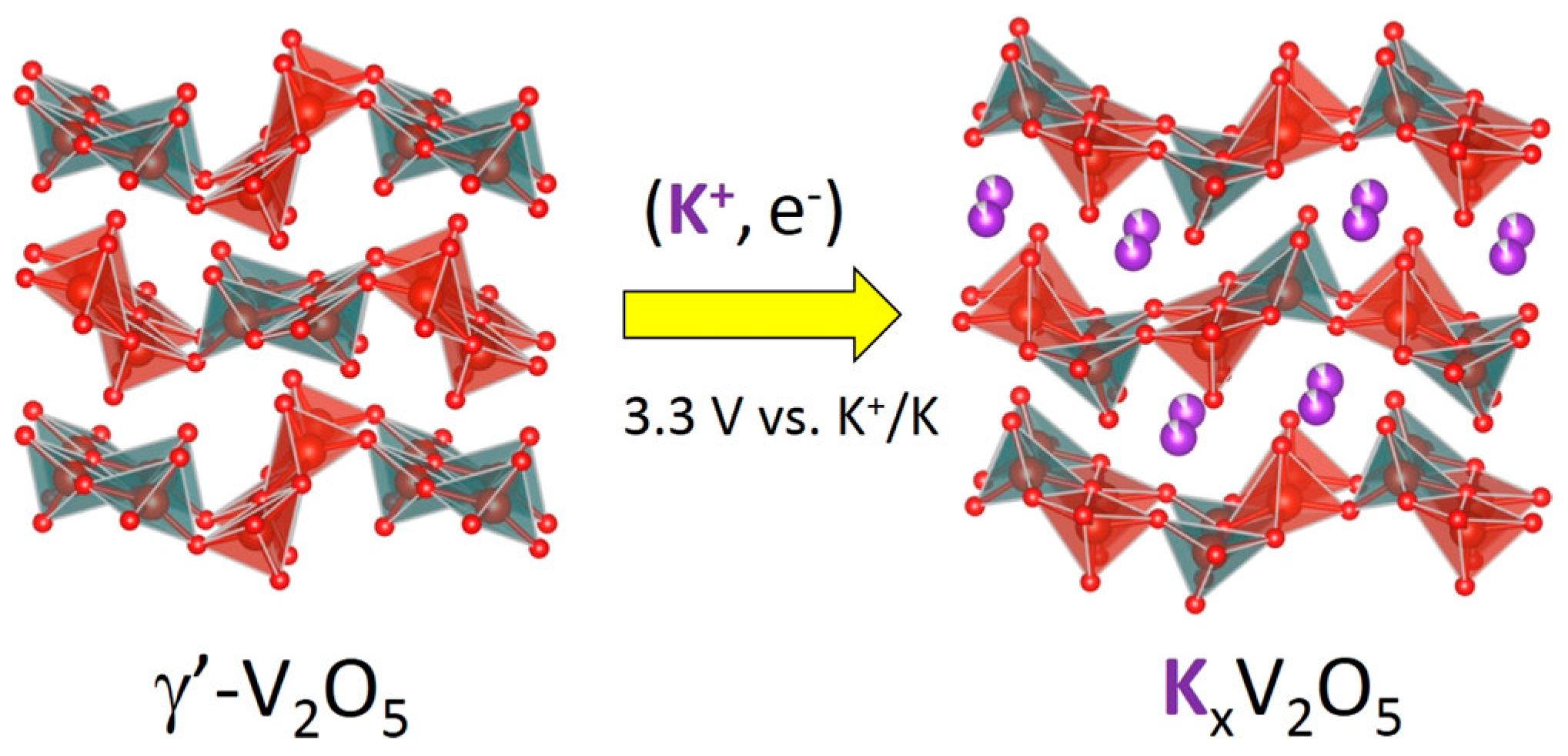
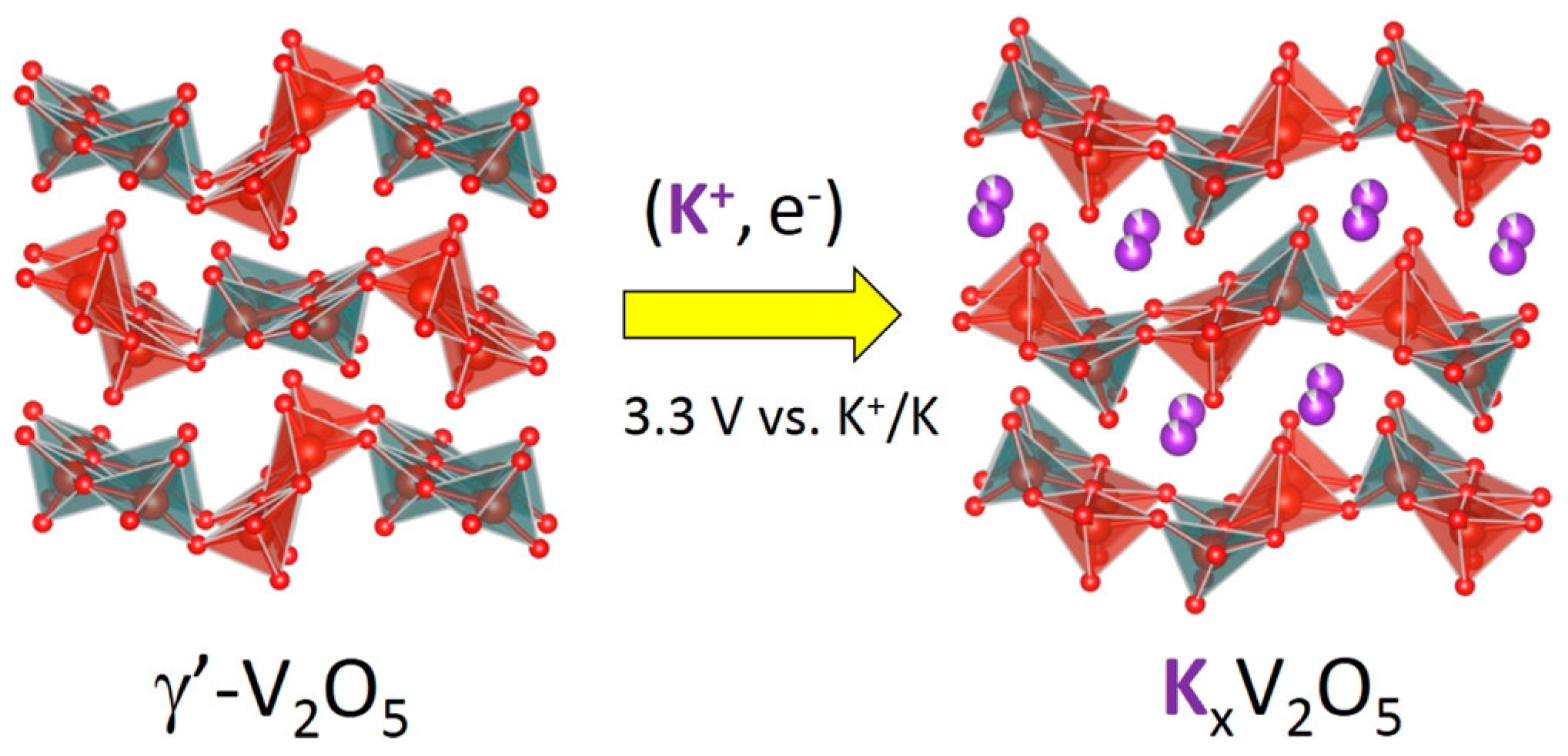
3.1.3. Potassium
3.2. Divalent Cations
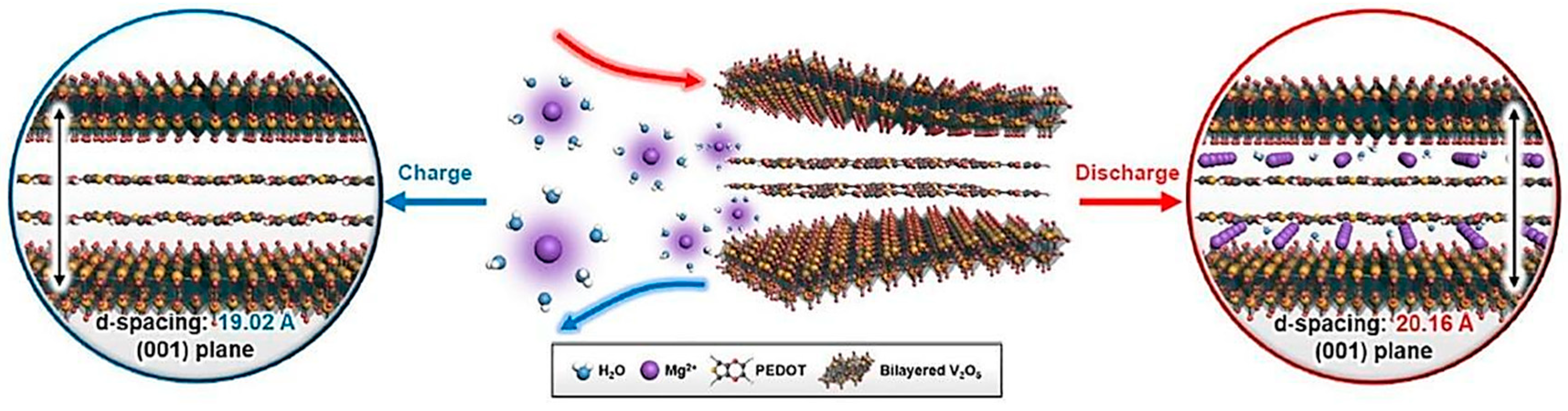
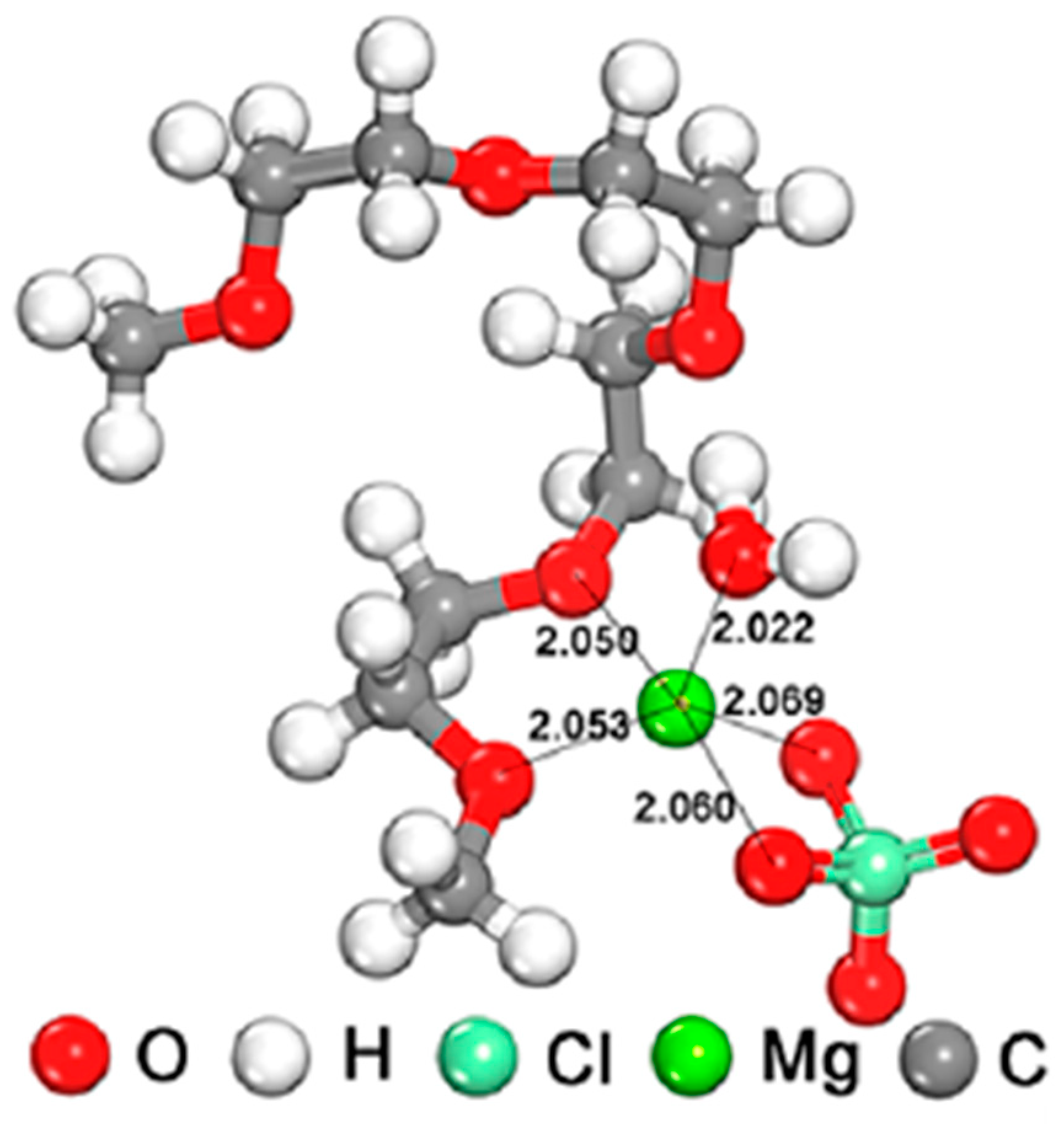
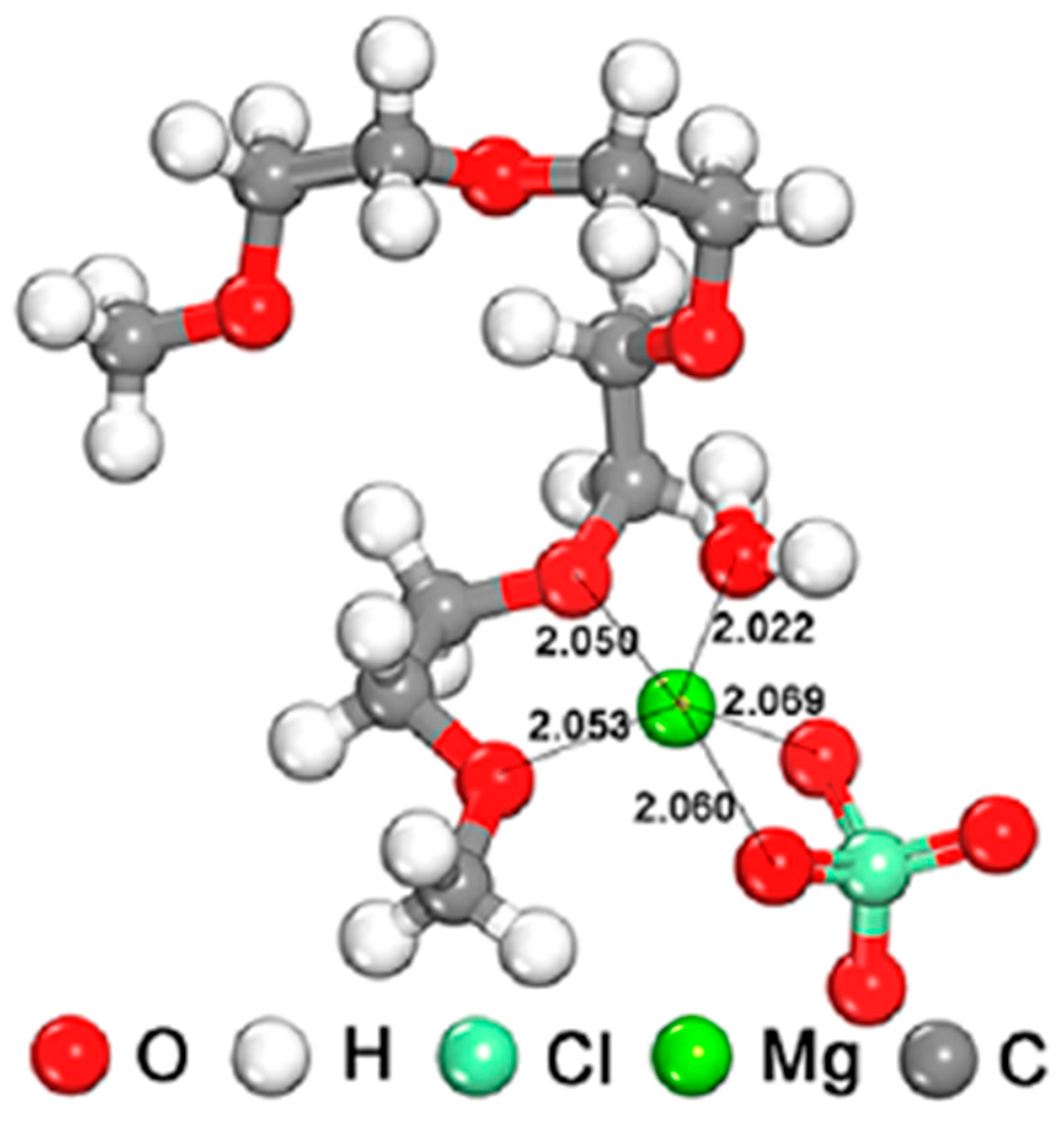
3.2.1. Magnesium
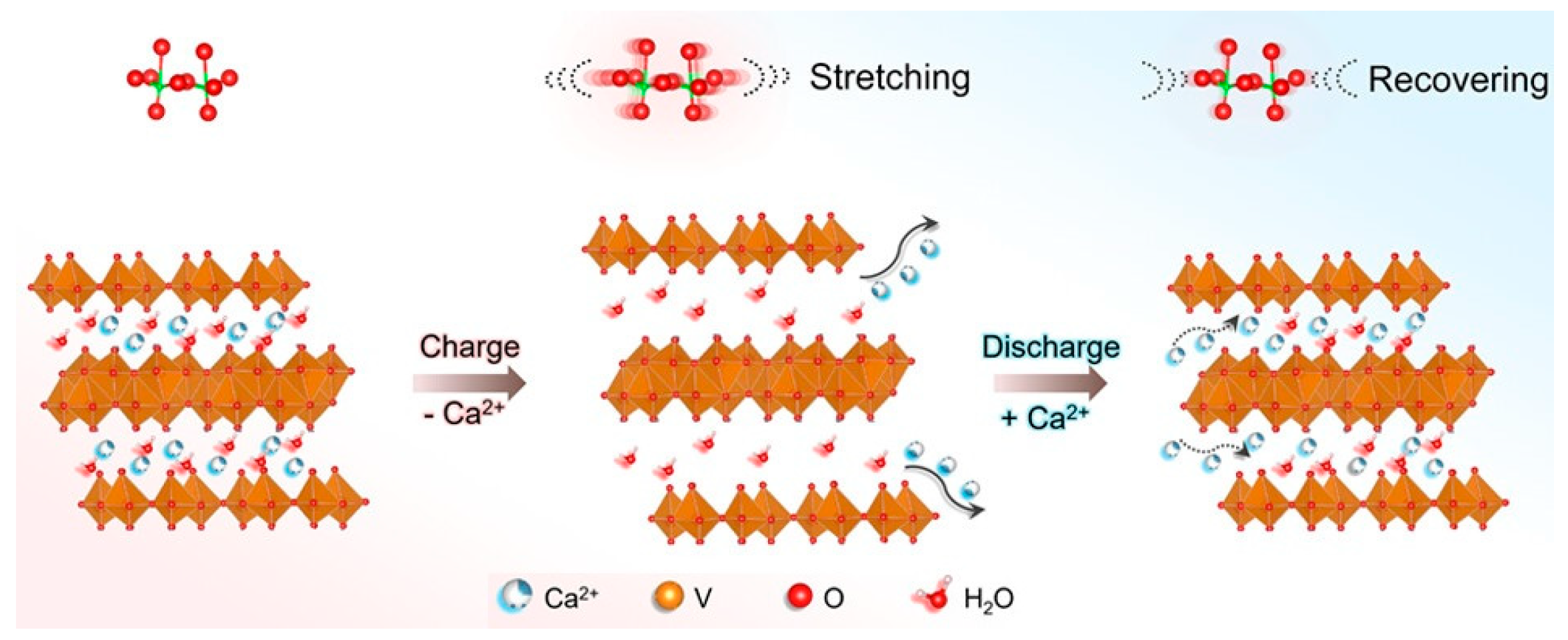
3.2.2. Calcium
3.3. Trivalent Cations
3.4. Dual-Ion Intercalation
4. Recent Advances in the Intercalation Properties of Vanadium Pentoxides
4.1. Cationic Doping
4.2. Electrolytes and Interfaces
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Enjalbert, R.; Galy, J. A refinement of the structure of V2O5. Acta Crystallogr. C 1986, 42, 1467–1469. [Google Scholar] [CrossRef]
- Livage, J. Vanadium pentoxide gels. Chem. Mater. 1991, 3, 578–593. [Google Scholar] [CrossRef]
- Kristoffersen, H.H.; Metiu, H. Reconstruction of low-index α-V2O5 surfaces. J. Phys. Chem. C 2016, 120, 3986–3992. [Google Scholar] [CrossRef]
- González, J.R.; Nacimiento, F.; Cabello, M.; Acántara, R.; Lavela, P.; Tirado, J.L. Reversible intercalation of aluminium into vanadium pentoxide xerogel for aqueous rechargeable batteries. RSC Adv. 2016, 6, 62157–62164. [Google Scholar] [CrossRef]
- Moretti, A.; Maroni, F.; Osada, I.; Nobili, F.; Passerini, S. V2O5 Aerogel as a Versatile Cathode Material for Lithium and Sodium Batteries. ChemElectroChem 2015, 2, 529–537. [Google Scholar] [CrossRef]
- Luo, W.; Gaumet, J.-J.; Mai, L. Nanostructured layered vanadium oxide as cathode for high-performance sodium-ion batteries: A perspective. MRS Commun. 2017, 7, 152–165. [Google Scholar] [CrossRef]
- Vernardou, D.; Drosos, C.; Kafizas, A.; Pemble, M.E.; Koudoumas, E. Towards high performance chemical vapour deposition V2O5 cathodes for batteries employing aqueous media. Molecules 2020, 25, 5558. [Google Scholar] [CrossRef]
- Cestarolli, D.T.; Guerra, E.M. Vanadium pentoxide (V2O5): Their obtaining methods and wide applications. In Transition Metal Compounds, 1st ed.; Haider, S., Haider, A., Eds.; IntechOpen: London, UK, 2021; pp. 1–11. [Google Scholar]
- Liu, H.; Wu, Y.P.; Rahm, E.; Holze, R.; Wu, H.Q. Cathode materials for lithium ion batteries prepared by sol-gel methods. J. Solid State Electrochem. 2004, 8, 450–466. [Google Scholar] [CrossRef]
- Fu, L.J.; Liu, H.; Li, C.; Wu, Y.P.; Rahm, E.; Holze, R.; Wu, H.Q. Electrode materials for lithium secondary batteries prepared by sol-gel. Prog. Mater. Sci. 2005, 50, 881–928. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Xiong, Z.; Hu, Y.; Song, W.; Huang, Q.; Cheng, X.; Chen, L.-Q.; Sun, C.; Gu, H. V2O5 Nanowire Composite Paper as a High-Performance Lithium-Ion Battery Cathode. ACS Omega 2017, 2, 793–799. [Google Scholar] [CrossRef]
- Mohan, V.M.; Hu, B.; Qiu, W.; Chen, W. Synthesis, structural, and electrochemical performance of V2O5 nanotubes as cathode material for lithium battery. J. Appl. Electrochem. 2009, 39, 2001–2006. [Google Scholar] [CrossRef]
- Lin, S.; Shao, B.; Taniguchi, I. One-step synthesis of dense and spherical nanostructured V2O5 particles for cathode of lithium batteries and their electrochemical properties. Mater. Res. Bull. 2014, 49, 291–296. [Google Scholar] [CrossRef]
- Ng, S.H.; Patey, T.J.; Büchel, R.; Krumeich, F.; Wang, J.Z.; Liu, H.K.; Pratsinis, S.E.; Novak, P. Flame spray-pyrolyzed vanadium oxide nanoparticles for lithium battery cathodes. Phys. Chem. Chem. Phys. 2009, 11, 3748–3755. [Google Scholar] [CrossRef]
- Roex, E.; Boschini, F.; Delaval, V.; Schrijnemakers, A.; Cloots, R.; Mahmoud, A. Spray-dried V2O5 as cathode material for high-performance aqueous zinc-ion batteries. J. Electroanal. Chem. 2023, 929, 117133. [Google Scholar] [CrossRef]
- Peng, X.; Zhang, S.; Zou, Z.; Ling, W.; Geng, J.; Zhong, S.; Liang, F. A min-review: Electrospun vanadium-based materials for lithium-ion batteries. J. Electron. Mater. 2023, 52, 4413–4433. [Google Scholar] [CrossRef]
- Armer, C.F.; Yeoh, J.S.; Li, X.; Lowe, A. Electrospun vanadium-based oxides as electrode materials. J. Power Sources 2018, 395, 414–429. [Google Scholar] [CrossRef]
- Viswanathamurthi, P.; Narayan, B.; Hak Yong, K.; Douk Rae, L. Vanadium pentoxide nanofibers by electrospinning. Scr. Mater. 2003, 49, 577–581. [Google Scholar]
- Tolosa, A.; Fleischmann, S.; Grobelsek, I.; Presser, V. Electrospun hybrid vanadium oxide/carbon fiber mats for lithium and sodium-ion battery electrodes. ACS Appl. Energy Mater. 2018, 1, 3790–3801. [Google Scholar] [CrossRef]
- Zou, Z.; Zhang, S.; Li, S. A review of the preparation and performance improvement of V6O13 as a cathode material for lithium-ion batteries. Mater. Technol. 2020, 35, 300–315. [Google Scholar] [CrossRef]
- Rasoulis, M.; Vernardou, D. Electrodeposition of vanadium oxides at room temperature as cathodes in lithium-ion batteries. Coatings 2017, 7, 100. [Google Scholar] [CrossRef]
- Vernardou, D.; Spanakis, E.; Katsarakis, N.; Koudoumas, E. Electrodeposition of V2O5 using ammonium metavanadate at room temperature. Adv. Mater. Lett. 2014, 5, 569–572. [Google Scholar] [CrossRef]
- Tepavcevic, S.; Xiong, H.; Stamenkovic, V.R.; Zuo, X.; Balasubramanian, M.; Prakapenka, V.B.; Johnson, C.S.; Rajh, T. Nanostructured bilayered vanadium oxide electrodes for rechargeable sodium-ion batteries. ACS Nano 2012, 6, 530–538. [Google Scholar] [CrossRef]
- Cheng, F.; Tao, Z.; Liang, J.; Chen, J. Template-directed materials for rechargeable lithium-ion batteries. Chem. Mater. 2008, 20, 667–681. [Google Scholar] [CrossRef]
- Spahr, M.E.; Bitterli, P.; Nesper, R.; Haas, O.; Novák, P. Vanadium oxide nanotubes a new nanostructured redox-active material for the electrochemical insertion of lithium. J. Electrochem. Soc. 1999, 146, 2780–2783. [Google Scholar] [CrossRef]
- Youn, C.; Ko, W.; Cho, A.; Lee, J.; Young, S.; Seo, Y.-Y.; Lee, J.; Lee, B.-S.; Kim, J.; Choi, T. One-dimensional nanostructured vanadium oxides with single-crystalline structure synthesized by cellulose nanocrystal-template-assisted hydrothermal method for li-ion battery cathodes. Cellulose 2023, 30, 7177–7191. [Google Scholar] [CrossRef]
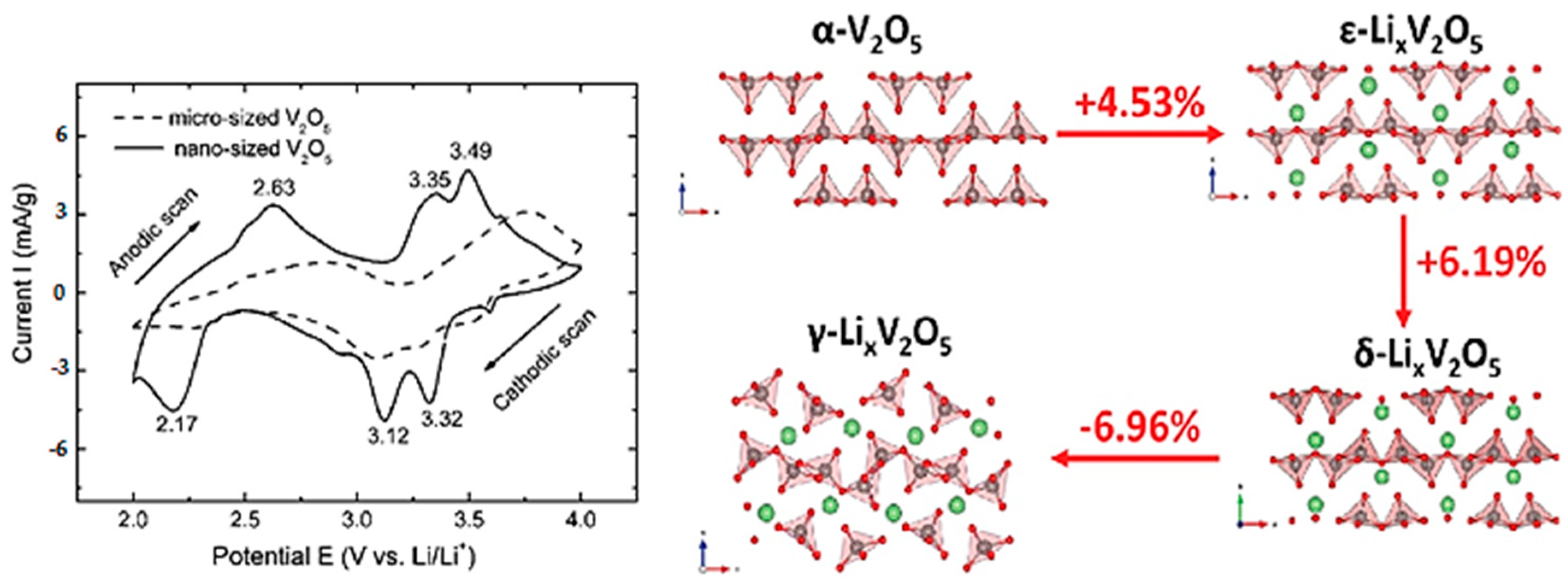
- Delmas, C.; Cognac-Auradou, H.; Cocciantelli, J.M.; Ménétrier, M.; Doumerc, J.P. The LixV2O5 system: An overview of the structure modifications induced by the lithium intercalation. Solid State Ion. 1994, 69, 257–264. [Google Scholar] [CrossRef]
- Zhang, Y.; Luo, Y.; Fincher, C.; Banerjee, S.; Pharr, M. Chemo-mechanical degradation in V2O5 thin film cathodes of Li-ion batteries during electrochemical cycling. J. Mater. Chem. A 2019, 7, 23922–23930. [Google Scholar] [CrossRef]
- Pan, A.; Zhang, J.-G.; Nie, Z.; Cao, G.; Arey, B.W.; Li, G.; Liang, S.-Q.; Liu, J. Facile synthesized nanorod structured vanadium pentoxide for high-rate lithium batteries. J. Mater. Chem. 2010, 20, 9193–9199. [Google Scholar] [CrossRef]
- Chernova, N.A.; Roppolo, M.; Dillon, A.C.; Whittingham, M.S. Layered vanadium and molybdenum oxides: Batteries and electrochromics. J. Mater. Chem. 2009, 19, 2526–2552. [Google Scholar] [CrossRef]
- Yao, J.; Li, Y.; Massé, R.C.; Uchaker, E.; Cao, G. Revitalized interest in vanadium pentoxide as cathode material for lithium-ion batteries and beyond. Energy Storage Mater. 2018, 11, 205–259. [Google Scholar] [CrossRef]
- Pecquenard, B.; Gourier, D.; Baffier, N. EPR identification of LixV2O5 phases generated by chemical and electrochemical lithium intercalation in V2O5. Solid State Ion. 1995, 78, 287–303. [Google Scholar] [CrossRef]
- Cocciantelli, J.M.; Doumerc, J.P.; Pouchard, M.; Broussely, M.; Labat, J. Crystal chemistry of electrochemically inserted LixV2O5. J. Power Sources 1991, 34, 103–111. [Google Scholar] [CrossRef]
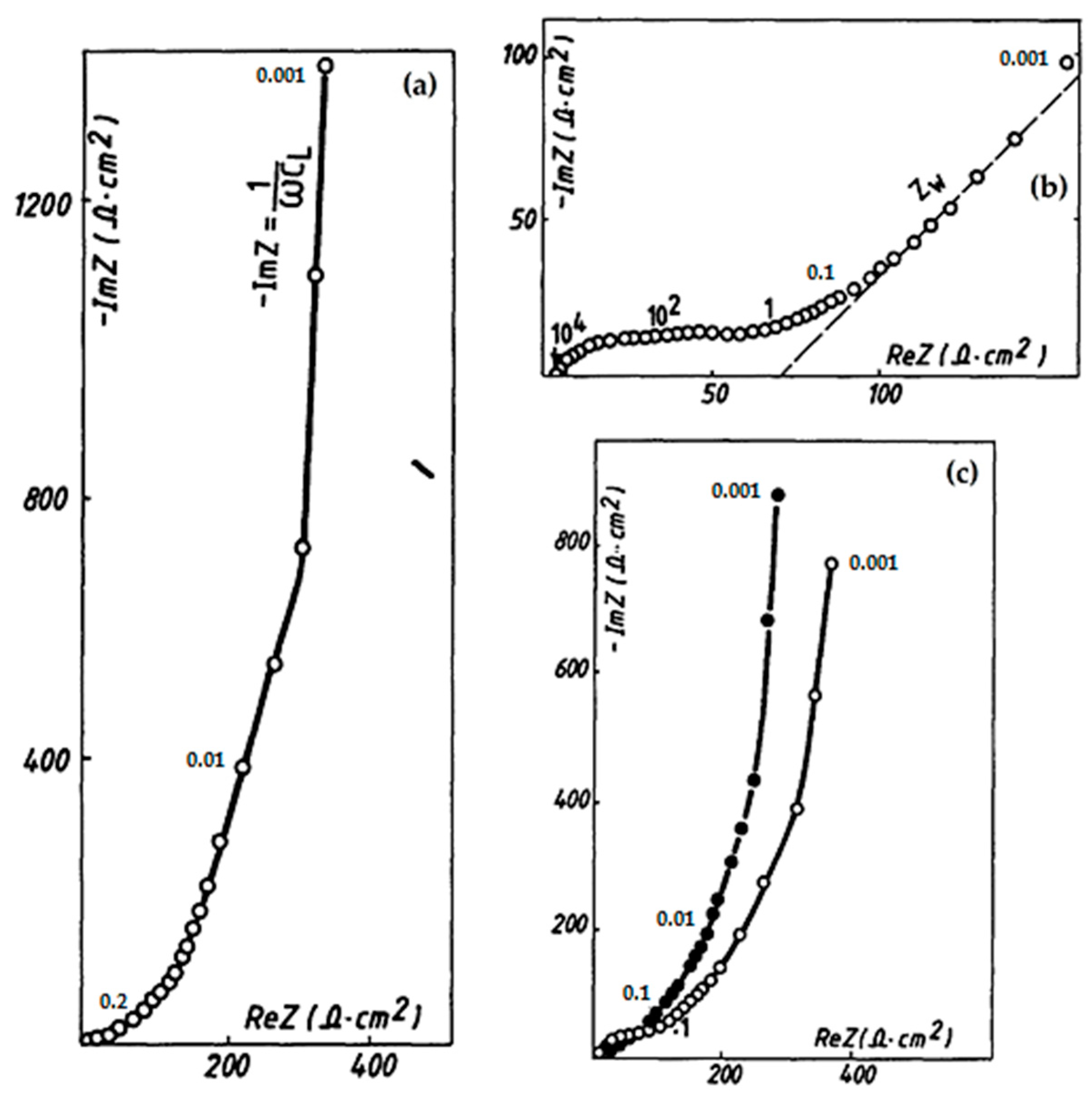
- Farcy, J.; Messina, R.; Perichon, J. Kinetic study of the lithium electroinsertion in V2O5 by impedance spectroscopy. J. Electrochem. Soc. 1990, 137, 1337–1341. [Google Scholar] [CrossRef]
- Keddam, M.; Rakotomavo, C.; Takenouti, H. Impedance of a porous electrode with an axial gradiaent of concentration. J. Appl. Electrochem. 1984, 14, 437–448. [Google Scholar] [CrossRef]
- Hub, S.; Tranchant, A.; Messina, R. X-ray investigations on electroformed LixV2O5 bronzes. Electrochim. Acta 1988, 33, 997–1002. [Google Scholar] [CrossRef]
- Światowska-Mrpwiecka, J.; Maurice, V.; Zanna, S.; Klein, L.; Marcus, P. XPS study of Li ion intercalation in V2O5 thin films prepared by thermal oxidation of vanadium metal. Electrochim. Acta 2007, 52, 5644–5653. [Google Scholar] [CrossRef]
- Parant, J.-P.; Olazcuaga, R.; Devalette, M.; Fouassier, C.; Hagenmuller, P. Sur quelques nouvelles phases de formule NaxMnO2 (x ≤ 1). J. Solid State Chem. 1971, 3, 1–11. [Google Scholar] [CrossRef]
- Kubota, K.; Dahbi, M.; Hosaka, T.; Kumakura, S.; Komaba, S. Towards K-ion and Na-ion batteries as “beyond Li-ion”. Chem. Rec. 2018, 4, 459–479. [Google Scholar] [CrossRef]
- Hwang, J.-Y.; Myung, S.-T.; Sun, Y.-K. Sodium-ion batteries: Present and future. Chem. Soc. Rev. 2017, 46, 3529–3614. [Google Scholar] [CrossRef]
- Byström, A.; Wilhelmi, K.-a.; Brotzen, O. Vanadium pentoxide—A compound with five –coordinated vanadium atoms. Acta Chem. Scand. 1950, 4, 1119–1130. [Google Scholar] [CrossRef]
- Haberkorn, R.; Bauer, J.; Kickelbick, G. Chemical sodiation of V2O5 by Na2S. Z. Anorg. Allg. Chem. 2014, 640, 3197–3202. [Google Scholar] [CrossRef]
- Kirsanova, M.A.; Akmaev, A.S.; Gorbunov, M.V.; Mkhailova, D.; Abakumov, A.M. Sodium-vanadium bronze Na9V14O35: An electrode material for Na-ion batteries. Molecules 2022, 27, 86. [Google Scholar] [CrossRef]
- Pouchard, M.; Casalot, A.; Galy, J.; Hagenmuller, P. Vanadium bronzes with NaxV2O5 formula. Bull. Société Chim. Fr. 1967, 11, 4343–4348. [Google Scholar]
- Kanke, Y.; Takayama-Muromachi, E.; Kato, K.; Matsui, Y. Phase equilibrium study of the system NaV2O5–V2O3–V2O5 at 923 K. J. Solid State Chem. 1990, 89, 130–137. [Google Scholar] [CrossRef]
- Savariault, J.M.; Parize, J.L.; Tkatchenko, D.B.; Galy, V. τ-NaxV2O5 (x = 0.64): A vanadium bronze with an original intergrowth structure. J. Solid State Chem. 1996, 122, 1–6. [Google Scholar] [CrossRef]
- Renard, M.S.; Emery, N.; Roginskii, E.M.; Baddour-Hadjeana, R.; Pereira-Ramos, J.-P. Crystal structure determination of a new sodium vanadium bronze electrochemically formed. J. Solid State Chem. 2017, 254, 62–68. [Google Scholar] [CrossRef]
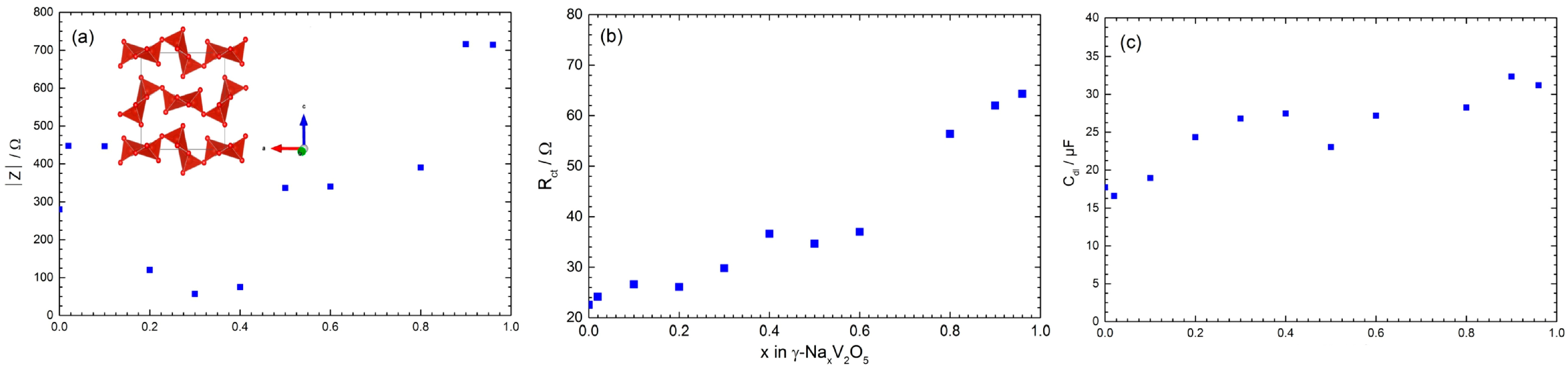
- Emery, N.; Baddour-Hadjean, R.; Batyrbekuly, D.; Laïk, B.; Bakenov, Z.; Pereira-Ramos, J.-P. γ-Na0.96V2O5: A new competitive cathode material for sodium ion battery synthesized by a soft chemistry route. Chem. Mater. 2018, 30, 5305–5314. [Google Scholar] [CrossRef]
- West, K.; Zachou-Christiansen, B.; Jacobsen, T.; Skaarup, S. Sodium insertion in vanadium oxides. Solid State Ion. 1988, 28–30, 1128–1131. [Google Scholar] [CrossRef]
- Millet, P.; Henry, J.-Y.; Galy, J. The vanadium oxide bronze η-NaxV2O5 (x = 1.286). Acta Crystallogr. 1999, C55, 276–279. [Google Scholar] [CrossRef]
- Safrany Renard, M.; Baddour-Hadjean, R.; Pereira-Ramos, J.P. Kinetic insight into the electrochemical sodium insertion-extraction mechanism of the puckered γ’-V2O5 polymorph. Electrochem. Acta 2019, 322, 134670. [Google Scholar] [CrossRef]
- Huo, D.; Laïk, B.; Bonnet, P.; Guérin, K.; Baddour-Hadjean, R.; Pereira-Ramos, J.-P. Electrochemical kinetics of Li insertion in nanosized high performance V2O5 obtained via fluorine chemistry. Electrochim. Acta 2017, 253, 472–478. [Google Scholar] [CrossRef]
- Pereira-Ramos, J.P.; Soudan, P.; Baddour-Hadjean, R.; Bach, S. A kinetic study of lithium transport in the sol-gel Cr0.11V2O5.16 mixed oxide. Electrochim. Acta 2011, 56, 1381–1386. [Google Scholar] [CrossRef]
- Ali, G.; Lee, J.-H.; Oh, S.H.; Cho, B.W.; Nam, K.-W.; Chung, K.Y. Investigation of the Na intercalation mechanism into nanosized V2O5/C composite cathode material for Na-ion batteries. ACS Appl. Mater. Interfaces 2016, 8, 6032–6039. [Google Scholar] [CrossRef] [PubMed]
- Muller-Bouvet, D.; Baddour-Hadjean, R.; Tanabe, M.; Huynh, L.T.N.; Le, M.L.P.; Pereira-Ramos, J.P. Electrochemically formed α’-NaV2O5: A new sodium intercalation compound. Electrochim. Acta 2015, 176, 586–593. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, G.; Wu, X.; Li, L.; Yue, J.; Guan, Y.; Hou, J.; Shi, F.; Liang, J. Research progress on avanadium oxides for potassium-ion batteries. J. Semicond. 2023, 44, 041701. [Google Scholar] [CrossRef]
- Xu, K.; Hu, S.; Wu, C.; Lin, C.; Lu, X.; Peng, L.; Yang, J.; Xie, Y. Highly entangled K0.5V2O5 superlong nanobelt membranes for flexible nonvolatile memory devices. J. Mater. Chem. 2012, 22, 18214–18220. [Google Scholar] [CrossRef]
- Patridge, C.J.; Wu, T.L.; Jaye, C.; Ravel, B.; Takeuchi, E.S.; Fischer, D.A.; Sambandamurthy, G.; Banerjee, S. Synthesis, spectroscopic characterization, and observation of massive metal insulator transitions in nanowires of a nonstoichiometric vanadium oxide bronze. Nano Lett. 2010, 10, 2448–2453. [Google Scholar]
- Zhu, Y.-H.; Zhang, Q.; Yang, X.; Zhao, E.-Y.; Sun, T.; Zhang, X.-B.; Wang, S.; Yu, X.-Q.; Yan, J.-M.; Jiang, Q. Reconstructed orthorhombic V2O5 polyhedra for fast ion diffusion in K-ion batteries. Chem 2019, 5, 168–179. [Google Scholar] [CrossRef]
- Koch, D.; Kulish, V.V.; Manzhos, S. A first-principles study of potassium insertion in crystalline vanadium oxide phases as possible potassium-ion battery cathode materials. MRS Commun. 2017, 7, 819–825. [Google Scholar] [CrossRef]
- Parija, A.; Prendergast, D.; Banerjee, S. Evaluation of multivalent cation insertion in single- and double-layered polymorphs of V2O5. ACS Appl. Mater. Interfaces 2017, 9, 23756–23765. [Google Scholar] [CrossRef]
- Bhatia, A.; Pereira-Ramos, J.-P.; Emery, N.; Baddour-Hadjean, R. γ’-V2O5 polymorph as a promising host structure for potassium storage: An electrochemical and structural study. Chem. Mater. 2021, 33, 5276–5289. [Google Scholar] [CrossRef]
- Pereira-Ramos, J.-P.; Messina, R.; Perichon, J. Electrochemical formation of vanadium pentoxide bronzes MxV2O5 in molten dimethylsulfone. J. Electrochem. Soc. 1988, 135, 3050–3057. [Google Scholar] [CrossRef]
- Parija, A.; Liang, Y.; Andrews, J.L.; De Jesus, L.R.; Prendergast, D.; Banerjee, S. Topochemically de-intercalated phases of V2O5 as cathode materials or multivalent intercalation batteries: A first principle evaluation. Chem. Mater. 2016, 28, 5611–5620. [Google Scholar] [CrossRef]
- Fu, Q.; Sarapulova, A.; Zhu, L.; Melinte, G.; Missyul, A.; Welter, E.; Luo, X.; Knapp, M.; Ehrenberg, H.; Dsoke, S. In operando study of orthorhombic V2O5 as positive electrode materials for K-ion batteries. J. Energy Chem. 2021, 62, 627–636. [Google Scholar] [CrossRef]
- Clites, M.; Hart, J.L.; Taheri, M.L.; Pomerantseva, E. Chemically preintercalated bilayered KxV2O5•nH2O nanobelts as a high-performing cathode material for K-ion batteries. ACS Energy Lett. 2018, 3, 562–567. [Google Scholar] [CrossRef]
- Petkov, V.; Trikalitis, P.N.; Bozin, E.S.; Billinge, S.J.L.; Vogt, T.; Kanatzidis, M.G. Structure of V2O5·nH2O xerogel solved by the atomic pair distribution function technique. J. Am. Chem. Soc. 2002, 124, 10157–10162. [Google Scholar] [CrossRef] [PubMed]
- Clites, M.; Pomerantseva, E. Bilayered vanadium oxides by chemical pre-intercalation of alkali and alkali-earth ions as battery electrodes. Energy Storage Mater. 2018, 11, 30–37. [Google Scholar] [CrossRef]
- Novák, P.; Imhof, R.; Haas, O. Magnesium insertion electrodes for rechargeable nonaqueous batteries-a competitive alternative to lithium? Electrochim. Acta 1999, 45, 351–367. [Google Scholar] [CrossRef]
- Sai Gautman, G.; Canepa, P.; Abdellahi, A.; Urban, A.; Malik, R.; Ceder, G. The intercalation phase diagram of Mg in V2O5 from first-principles. Chem. Mater. 2015, 27, 3733–3742. [Google Scholar] [CrossRef]
- Verrelli, R.; Black, A.P.; Pattanathummasid, C.; Tchitchekova, D.S.; Ponrouch, A.; Oró-Solé, J.; Frontera, C.; Bardé, F.; Rozier, P.; Palacín, M.R. On the strange case of divalent ions intercalation in V2O5. J. Power Sources 2018, 407, 162–172. [Google Scholar] [CrossRef]
- Yoo, H.D.; Jokisaari, J.R.; Yu, Y.-S.; Kwon, B.J.; Hu, L.; Kim, S.; Han, S.-D.; Lopez, M.; Lapidus, S.H.; Nolis, G.N.; et al. Intercalation of magnesium into a layered vanadium oxide with high capacity. ACS Energy Lett. 2019, 4, 1528–1534. [Google Scholar] [CrossRef]
- Joe, Y.S.; Kang, M.S.; Jang, G.; Lee, S.J.; Nakhanivei, P.; Baek, S.H.; Kim, Y.K.; Jeong, G.; Kim, H.-S.; Park, H.S. Intercalation of bilayered V2O5 by electronically coupled PEDOT for greatly improved kinetic performance of magnesium ion battery. Chem. Eng. J. 2023, 460, 141706. [Google Scholar] [CrossRef]
- Onoda, M.; Nishiguchi, N. Crystal structure and spin gap state of CaV2O5. J. Solid State Chem. 1996, 127, 359–362. [Google Scholar] [CrossRef]
- Jeon, B.; Kwak, H.H.; Hong, S.-T. Bilayered Ca0.28V2O5·H2O: High-capacity cathode material for rechargeable Ca-ion batteries and its charge storage mechanism. Chem. Mater. 2022, 34, 1491–1498. [Google Scholar] [CrossRef]
- Qin, X.; Zhao, X.; Zhang, G.; Wei, Z.; Li, L.; Wang, X.; Zhi, C.; Li, H.; Han, C.; Li, B. Highly reversible intercalation of calcium ions in layered vanadium compounds enabled by acetonitrile-water hybrid electrolyte. ACS Nano 2023, 17, 12040–12051. [Google Scholar] [CrossRef]
- Lin, M.-C.; Gong, M.; Lu, B.; Wu, Y.; Wang, D.-Y.; Guan, M.; Angell, M.; Chen, C.; Yang, J.; Hwang, B.-J.; et al. An ultrafast rechargeable aluminium-ion battery. Nature 2015, 520, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Cahe, M.S.; Heo, J.W.; Hyoung, J.; Hong, S.-T. Double-sheet vanadium oxide as a cathode material for calcium-ion batteries. ChemNanoMat 2020, 6, 1049–1053. [Google Scholar]
- Yao, T.; Oka, Y.; Yamamoto, N. Layered structures of vanadium pentoxide gels. Mater. Res. Bull. 1992, 27, 669–675. [Google Scholar] [CrossRef]
- Le, D.B.; Passerini, S.; Coustier, F.; Guo, J.; Soderstrom, T.; Owens, B.B.; Smyrl, W.H. Intercalation of polyvalent cations into V2O5 aerogels. Chem. Mater. 1998, 10, 682–684. [Google Scholar] [CrossRef]
- Diem, A.M.; Fenk, B.; Bill, J.; Burghard, Z. Binder-free V2O5 cathode for high energy density rechargeable aluminium-ion batteries. Nanomaterials 2020, 10, 247. [Google Scholar] [CrossRef]
- De, P.; Halder, J.; Priya, S.; Kumar Srivastava, A.; Chandra, A. Two-Dimensional V2O5 Nanosheets as an advanced cathode material for realizing low-cost aqueous aluminum-ion batteries. ACS Appl. Energy Mater. 2023, 6, 753–762. [Google Scholar] [CrossRef]
- Wang, H.; Bai, Y.; Chen, S.; Luo, X.; Wu, C.; Wu, F.; Lu, J.; Amine, K. Binder-free V2O5 for greener rechargeable aluminium battery. ACS Appl. Mater. Interfaces 2015, 7, 80–84. [Google Scholar] [CrossRef]
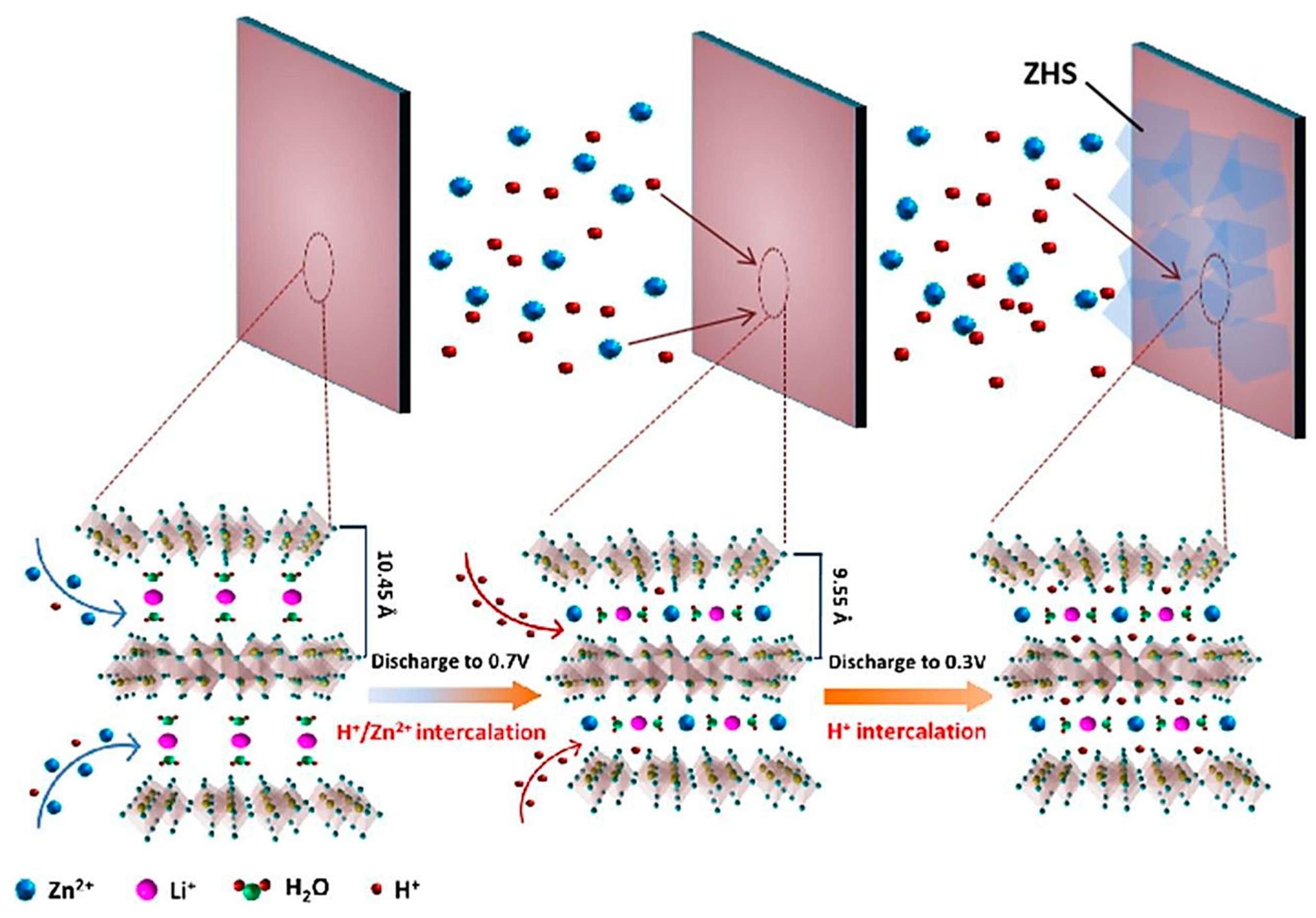
- Feng, Z.; Zhang, Y.; Sun, J.; Liu, Y.; Jiang, H.; Cui, M.; Hu, T.; Meng, C. Dual ions enable vanadium oxide hydration with superior Zn2+ storage for aqueous zinc-ion batteries. Chem. Eng. J. 2022, 433, 133795. [Google Scholar] [CrossRef]
- Tong, Y.; Li, X.; Su, S.; Li, J.; Fang, J.; Liang, B.; Hou, J.; Luo, M. Hydrated lithium ions inercalated V2O5 with dual-ion synergistic insertion mechanism for high-performance aqueous zin-ion batteries. J. Colloid Interface Sci. 2022, 606, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Arroyo y de Dompablo, M.E.; Gallardo-Amores, J.M.; Amador, U.; Morán, E. Are high pressure materials suitable for electrochemical applications? HP-V2O5 as a novel electrode material for Li batteries. Electrochem. Commun. 2007, 9, 1305–1310. [Google Scholar] [CrossRef]
- Córdoba, R.; Kuhn, A.; Pérez-Flores, J.C.; Morán, E.; Gallardo-Amores, J.M.; García-Alvarado, F. Sodium insertion in high pressure β-V2O5: A new high capacity cathode material for sodium ion batteries. J. Power Sources 2019, 422, 42–48. [Google Scholar] [CrossRef]
- Baddour-Hadjean, R.; Renard, M.S.; Pereira-Ramos, J.-P. Enhanced electrochemical properties of ball-milled γ′-V2O5 as cathode material for Na-ion batteries: A structural and kinetic investigation. J. Power Sources 2021, 482, 229017. [Google Scholar] [CrossRef]
- Su, F.; Xing, F.; Wang, X.; Liu, F.; Zhang, L.; Wu, Z.-S. Enabling rapid pseudocapacitive multi-electron reactions by heterostructure engineering of vanadium oxide for high-energy and high-power lithium storage. Energy Environ. Sci. 2023, 16, 222–230. [Google Scholar] [CrossRef]
- McNulty, R.C.; Penston, K.; Amin, S.S.; Stal, S.; Lee, J.Y.; Samperi, M.; Pérez-García, L.; Cameron, J.M.; Johnson, L.R.; Amabilino, D.B.; et al. Self-assembled surfactant-polyoxovanadate soft materials as tuneable vanadium oxide cathode precursors for lithium-ion batteries. Angew. Chem. Int. Ed. 2023, 62, e202216066. [Google Scholar] [CrossRef]
- Jovanović, A.; Dobrota, A.S.; Rafailović, L.D.; Mentus, S.V.; Pašti, I.A.; Johansson, B.; Skorodumova, N.V. Structural and electronic properties of V2O5 and their tuning by doping with 3d elements-modelling using the DFT+U method and dispersion corrction. Phys. Chem. Chem. Phys. 2018, 20, 13934–13943. [Google Scholar] [CrossRef]
- Moretti, A.; Giuli, G.; Trapananti, A.; Passerini, S. Electrochemical and structural investigation of transition metal doped V2O5 sono-aerogel cathodes for lithium metal batteries. Solid State Ion. 2018, 319, 46–52. [Google Scholar] [CrossRef]
- Zhong, W.; Huang, J.; Liang, S.; Liu, J.; Li, Y.; Cai, G.; Jiang, Y.; Liu, K. New prelithiated V2O5 superstructure for lithium-ion batteries with long cycle life and high power. ACS Energy Lett. 2020, 5, 31–38. [Google Scholar] [CrossRef]
- Hu, B.; Li, L.; Xiong, X.; Liu, L.; Huang, C.; Yu, D.; Chen, C. High-performance of copper-doped vanadium pentoxide porous thin films cathode for lithium-ion batteries. J. Solid State Electrochem. 2019, 23, 1315–1324. [Google Scholar] [CrossRef]
- Liu, C.; Yao, J.; Zou, Z.; Li, Y.; Cao, G. Boosting the cycling stability of hydrated vanadium pentoxide by Y3+ pillaring for sodium-ion batteries. Mater. Today Energy 2019, 11, 218–227. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, F.; Gao, W.; Zhan, Z. A high-performance Cu-doped vanadium pentoxide thin-film cathode for lithium-ion batteries. Int. J. Ion. 2021, 27, 2335–2344. [Google Scholar] [CrossRef]
- Wei, Y.; Ryu, C.W.; Kim, K.B. Improvement in electrochemical performance of V2O5 by Cu doping. J. Power Sources 2007, 165, 386–392. [Google Scholar] [CrossRef]
- Gogoi, R.K.; Neog, A.B.; Konch, T.J.; Sarmah, N.; Raidongia, K. A two-dimensional ion-pump of a vanadium pentoxide nanofluidic membrane. J. Mater. Chem. A 2019, 17, 10552–10560. [Google Scholar] [CrossRef]
- Yao, J.H.; Yin, Z.L.; Zou, Z.G.; Li, Y.W. Y-doped V2O5 with enhanced lithium storage performance. RSC Adv. 2017, 7, 32327–32335. [Google Scholar] [CrossRef]
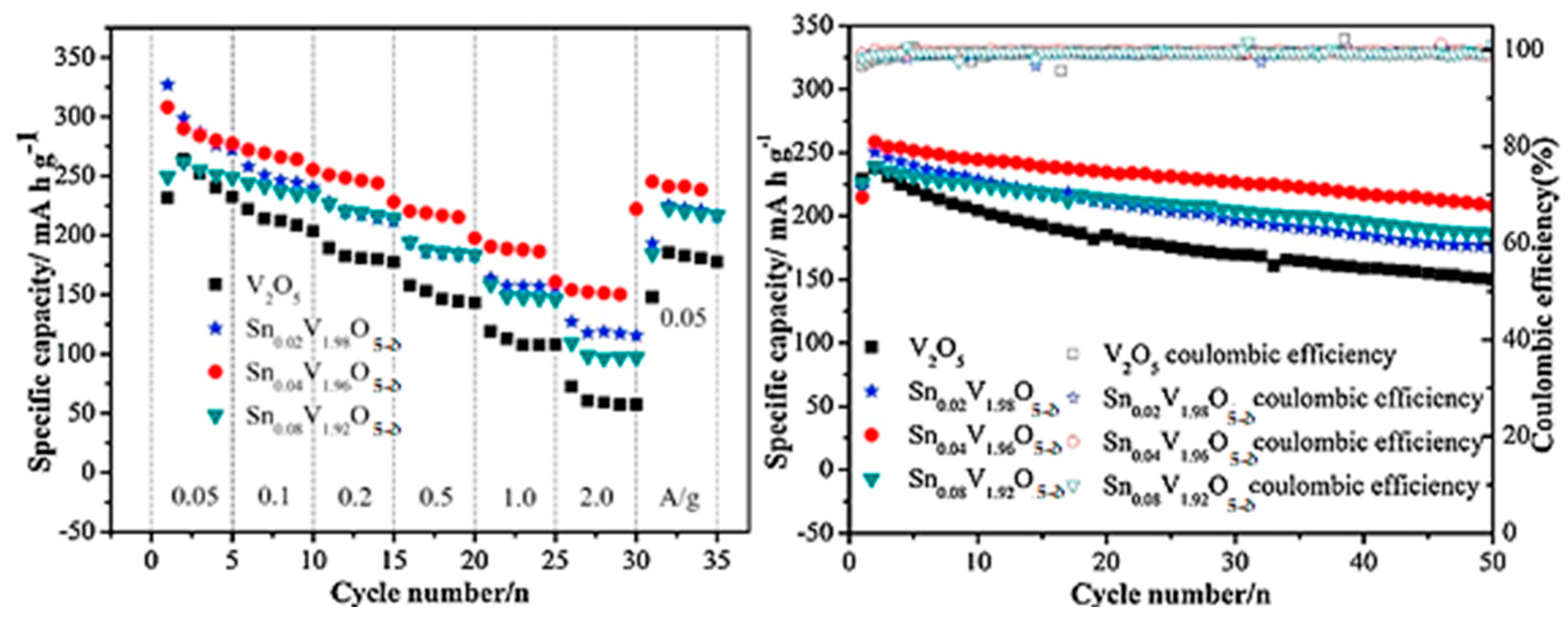
- Li, Z.; Zhang, C.; Liu, C.; Fu, H.; Nan, X.; Wang, K.; Li, X.; Ma, W.; Lu, X.; Cao, G. Enhanced electrochemical properties of Sn-doped V2O5 as a cathode material for lithium ion batteries. Electrochim. Acta 2016, 222, 1831–1838. [Google Scholar] [CrossRef]
- Ngamwongwan, L.; Fongkaew, I.; Phonsuksawang, P.; Siritanon, T.; Hirunsit, P.; Jungthawan, S.; Suhtirakun, S. On the origin of ion intercalation and conductivity enhancement in Sn-doped V2O5 cathodes of Li-ion batteries: A computational study. J. Phys. Chem. C 2023, 127, 11526–11535. [Google Scholar] [CrossRef]
- Gao, H.; Jiao, L.; Yang, J.; Qi, Z.; Wang, Y.; Yuan, H. High rate capability of Co-doped LiFePO4/C. Electrochim. Acta 2013, 97, 143–149. [Google Scholar] [CrossRef]
- Wang, D.Y.; Li, H.; Shi, S.Q.; Huang, X.J.; Chen, L.Q. Improving the rate performance of LiFePO4 by Fe-site doping. Electrochim. Acta 2005, 50, 2955–2958. [Google Scholar] [CrossRef]
- Armstrong, A.R.; Robertson, A.D.; Gitzendanner, R.; Bruce, P.G. The layered intercalation compounds Li(Mn1-yCoy)Oz: Positive electrode materials for lithium–ion batteries. J. Solid State Chem. 1999, 145, 549–556. [Google Scholar] [CrossRef]
- Ji, Y.; Fang, D.; Wang, C.; Zhou, Z.; Luo, Z.; Huang, J.; Yi, J. Cobalt-doped V2O5 nanowire arrays on Ti foil for enhanced lithium-ion storage. J. Alloys Compd. 2018, 742, 567–576. [Google Scholar] [CrossRef]
- Kou, L.Q.; Chen, F.J.; Tao, F.; Dong, Y.; Chen, L. High rate capability and cycle performance of Ce-doped LiMnPO4/C via an efficient solvothermal synthesis in water/diethylene glycol system. Electrochim. Acta 2015, 173, 721–727. [Google Scholar] [CrossRef]
- Li, X.L.; Pan, L.S.; Wang, Y.Y.; Xu, C.S. High efficiency immobilization of sulfur on Ce-doped carbon aerogel for high performance lithium-sulfur batteries. Electrochim. Acta 2016, 190, 548–555. [Google Scholar] [CrossRef]
- Chen, P.; Zheng, G.; Guo, G.; Wang, Z.; Tang, J.; Li, S.; Wen, Z.; Ji, S.; Sun, J. Ce-doped V2O5 microspheres with improved electrochemical performance for high-power rechargeable lithium ion batteries. J. Alloys Compd. 2019, 784, 574–583. [Google Scholar] [CrossRef]
- Zhao, N.; Li, Y.; Zhi, X.; Wang, L.; Zhao, X.; Wang, Y.; Liang, G. Effect of Ce3+ doping on the properties of LiFePO4 cathode material. J. Rare Earths 2016, 34, 174–180. [Google Scholar] [CrossRef]
- Qiu, H.; Yue, H.; Zhang, T.; Ju, Y.; Zhang, Y.; Guo, Z.; Wang, C.; Chen, G.; Wei, Y.; Zhang, D. Enhanced electrochemical performance of Li2FeSiO4/C positive electrodes for lithium-ion batteries via yttrium doping. Electrochim. Acta 2016, 188, 636–644. [Google Scholar] [CrossRef]
- Amatucci, G.G.; Badway, F.; Singhal, A.; Beaudoin, B.; Skandan, G.; Bowmer, T.; Plitz, I.; Pereira, N.; Chapman, T.; Jaworski, R. Investigation of yttrium and polyvalent ion intercalation into nanocrystalline vanadium oxide. J. Electrochem. Soc. 2001, 148, A940–A950. [Google Scholar] [CrossRef]
- Hao, X.; Zheng, L.; Hu, S.; Wu, Y.; Zhang, G.; Li, B.; Yang, M.; Han, C. Stabilizing Ca-Ion Batteries with a 7000-cycle Lifespan and Superior Rate Capability by a Superlattice-like Vanadium Heterostructure. Mater. Today Energy 2023, 101456. [Google Scholar] [CrossRef]
- Cambaz, M.A.; Vinayan, B.P.; Pervez, S.A.; Johnsen, R.E.; Geßwein, H.; Guda, A.A.; Rusalev, Y.V.; Kinyanjui, M.K.; Kaiser, U.; Fichtner, M. Suppressing dissolution of vanadium from cation-disordered Li2−xVO2F via a concentrated electrolyte approach. Chem. Mater. 2019, 31, 7941–7950. [Google Scholar] [CrossRef]
- Untarabut, P.; Singsen, S.; Ngamwongwan, L.; Fongkaew, I.; Junkaew, A.; Suthirakun, S. Unraveling the role of hydrogen insertion in enhancing the electrochemical performance of the V2O5 cathode for Mg-ion batteries: A first-principle study. ACS Appl. Energy Mater. 2023, 6, 8666–8676. [Google Scholar] [CrossRef]
- Song, X.; Li, X.; Shan, H.; Wang, H.; Wang, J.; Li, W.; Xu, K.; Zhang, K.; Maleki Kheimeh Sari, H.; Lei, L.; et al. V-O-C bonding of heterointerface boosting kinetics of free-standing Na5V12O32 cathode for ultralong lifespan sodium-ion batteries. Adv. Funct. Mater. 2023, 2303211. [Google Scholar] [CrossRef]
- Liu, F.; Xu, H.; He, Y.; Bian, H.; Li, D.; Wang, A.; Sun, D. Synergistic effect of composite V2O5/Ca0.17V2O5 film electrodes as high-performance cathodes of SIBs. Energy Fuels 2023, 37, 11355–11366. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cathode | Specific Capacity/mAh g−1 | Capacity Retention/% | References |
|---|---|---|---|
| Ni0.1V2O5 vs. Li/Li+ | 275 at 560 mA g−1 | 69.9 after 140 cycles | [92] |
| Mn0.1V2O5 vs. Li/Li+ | 225 at 560 mA g−1 | 76.4 after 140 cycles | [92] |
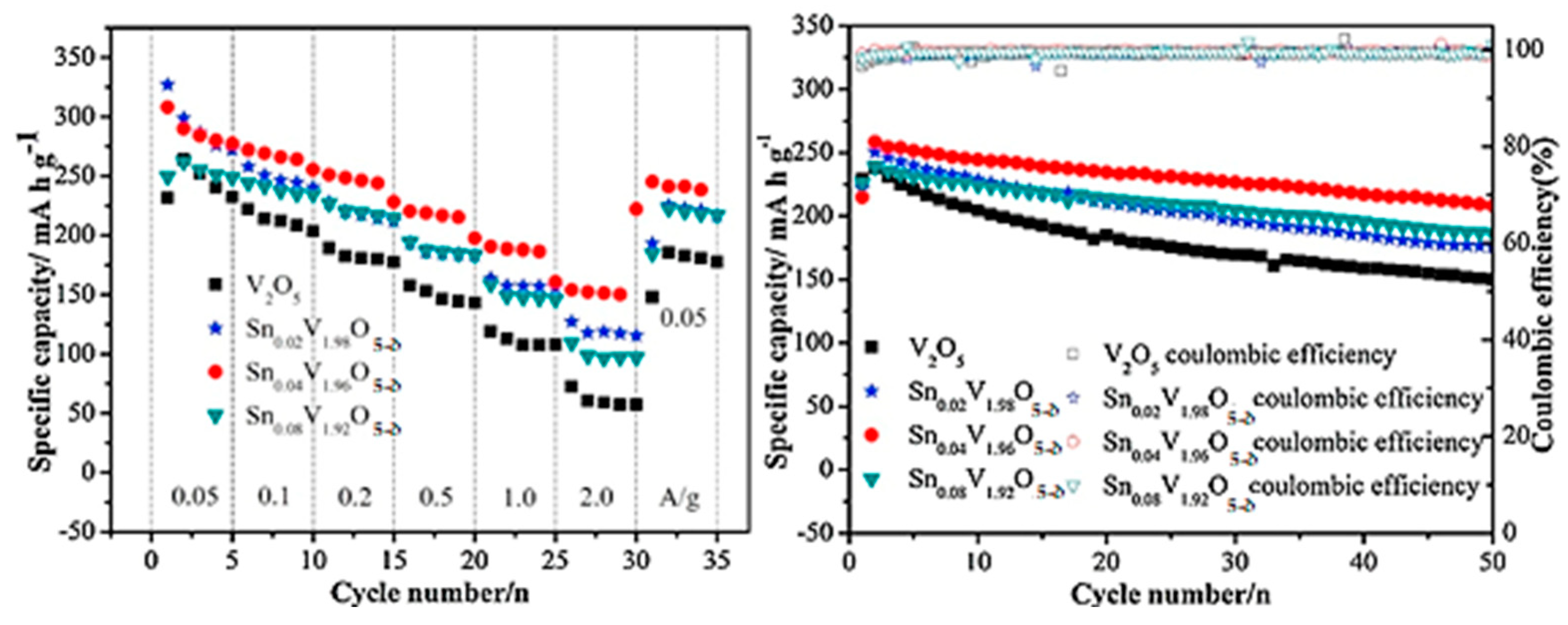
| Sn0.04V1.96O5−δ vs. Li/Li+ | 292 at 200 mA g−1 | 0.36 per cycle | [96] |
| Co-doped V2O5 vs. Li/Li+ | 394.98 at 200 mA g−1 | 94 after 100 cycles | [105] |
| Ce0.1V2O5 vs. Li/Li+ | 260 at 294 mA g−1 | 88.93 after 200 cycles | [106] |
| Y0.02V2O5 vs. Na/Na+ | 119 at 100 mA g−1 | 0.12 per cycle between 10 and 100 cycles | [95] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alcántara, R.; Lavela, P.; Edström, K.; Fichtner, M.; Le, T.K.; Floraki, C.; Aivaliotis, D.; Vernardou, D. Metal-Ion Intercalation Mechanisms in Vanadium Pentoxide and Its New Perspectives. Nanomaterials 2023, 13, 3149. https://doi.org/10.3390/nano13243149
Alcántara R, Lavela P, Edström K, Fichtner M, Le TK, Floraki C, Aivaliotis D, Vernardou D. Metal-Ion Intercalation Mechanisms in Vanadium Pentoxide and Its New Perspectives. Nanomaterials. 2023; 13(24):3149. https://doi.org/10.3390/nano13243149
Chicago/Turabian StyleAlcántara, Ricardo, Pedro Lavela, Kristina Edström, Maximilian Fichtner, Top Khac Le, Christina Floraki, Dimitris Aivaliotis, and Dimitra Vernardou. 2023. "Metal-Ion Intercalation Mechanisms in Vanadium Pentoxide and Its New Perspectives" Nanomaterials 13, no. 24: 3149. https://doi.org/10.3390/nano13243149
APA StyleAlcántara, R., Lavela, P., Edström, K., Fichtner, M., Le, T. K., Floraki, C., Aivaliotis, D., & Vernardou, D. (2023). Metal-Ion Intercalation Mechanisms in Vanadium Pentoxide and Its New Perspectives. Nanomaterials, 13(24), 3149. https://doi.org/10.3390/nano13243149