
Acid-Modulated Peptide Synthesis for Application on Oxide Biosensor Interfaces

 , , , ,
, , , , 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
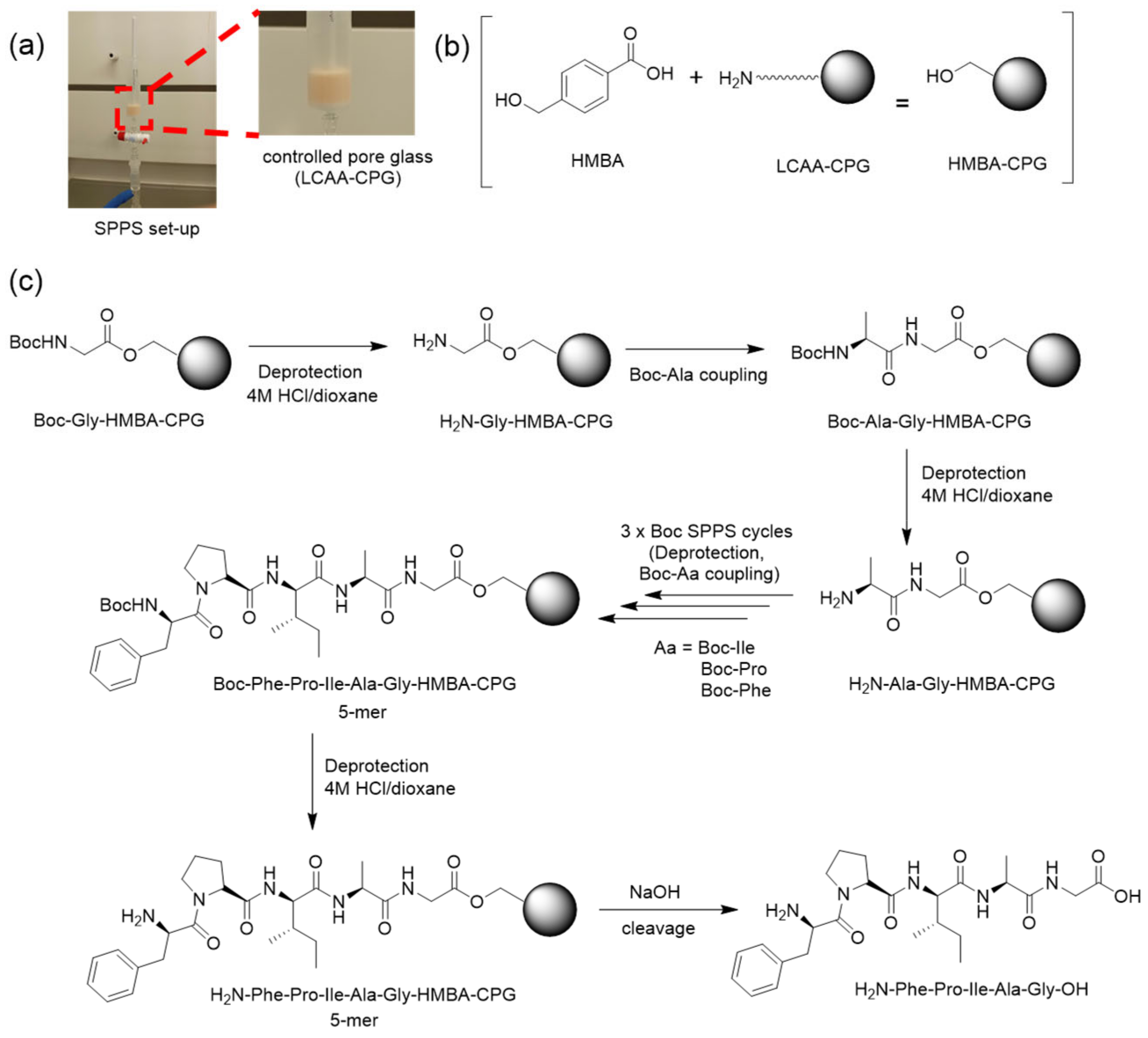
2.2. Peptide Synthesis on CPG
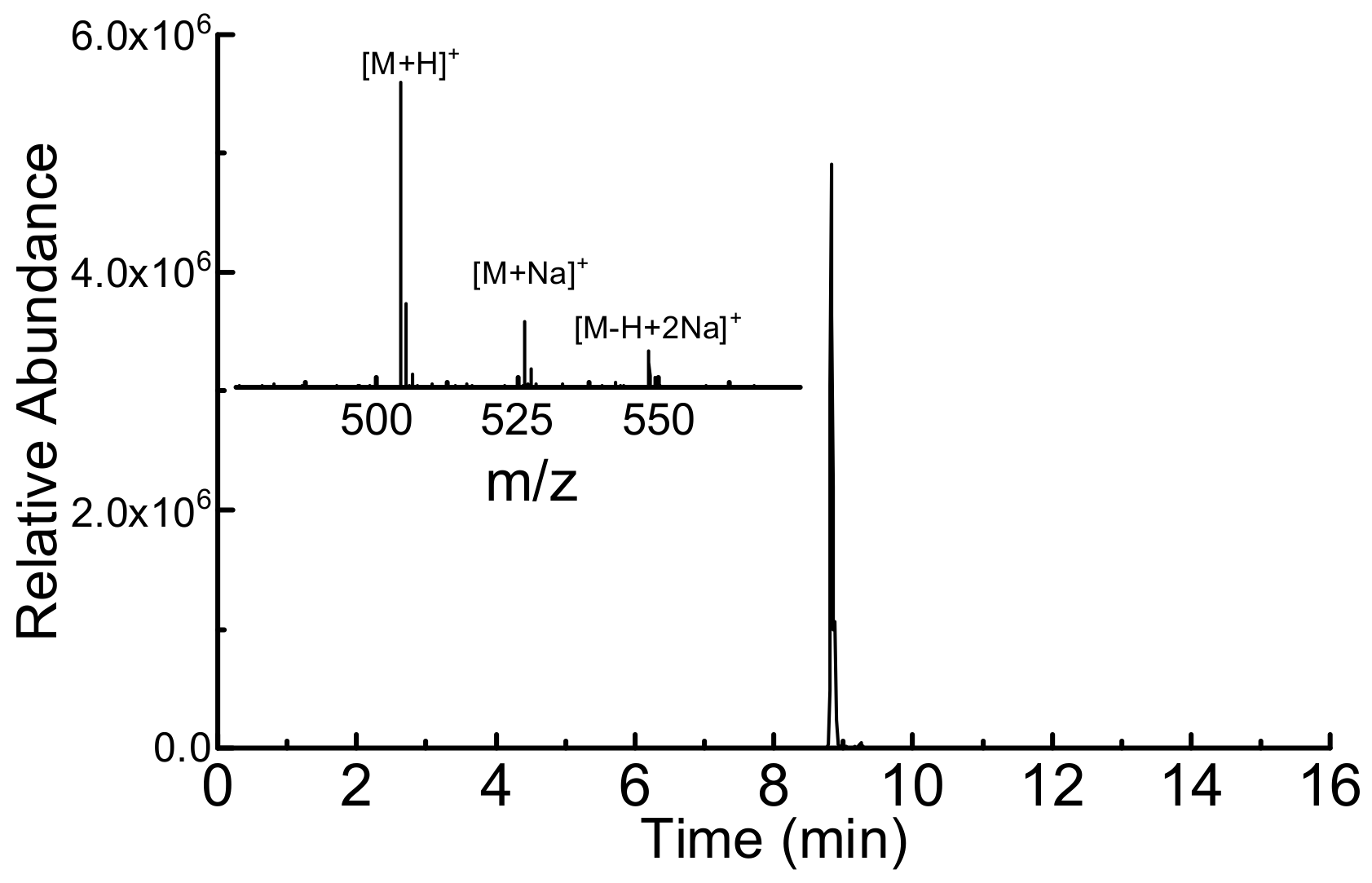
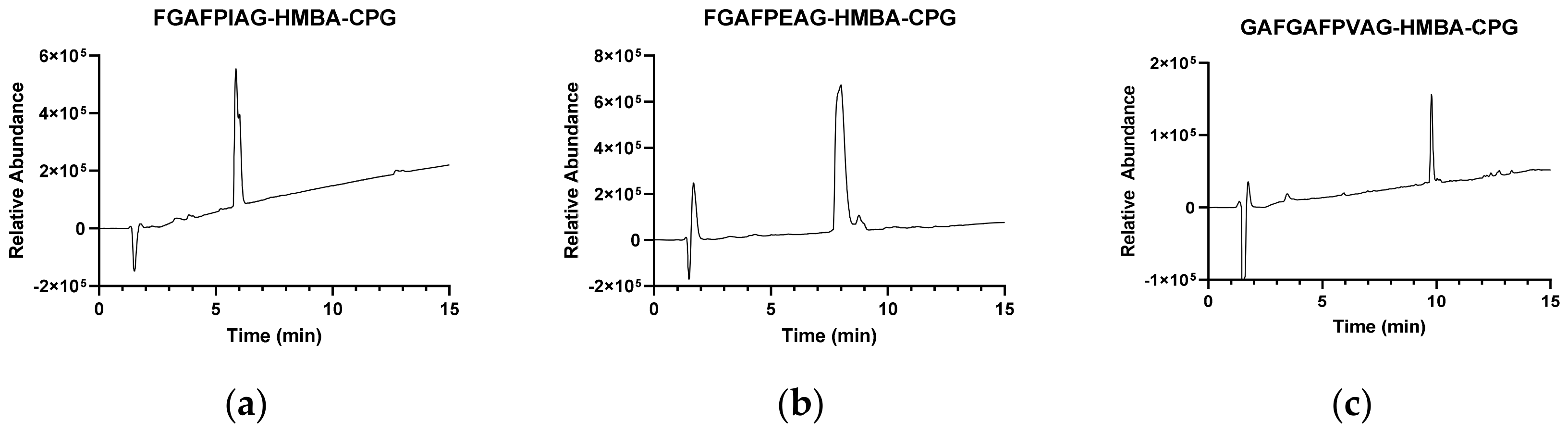
2.3. HPLC-MS Analysis
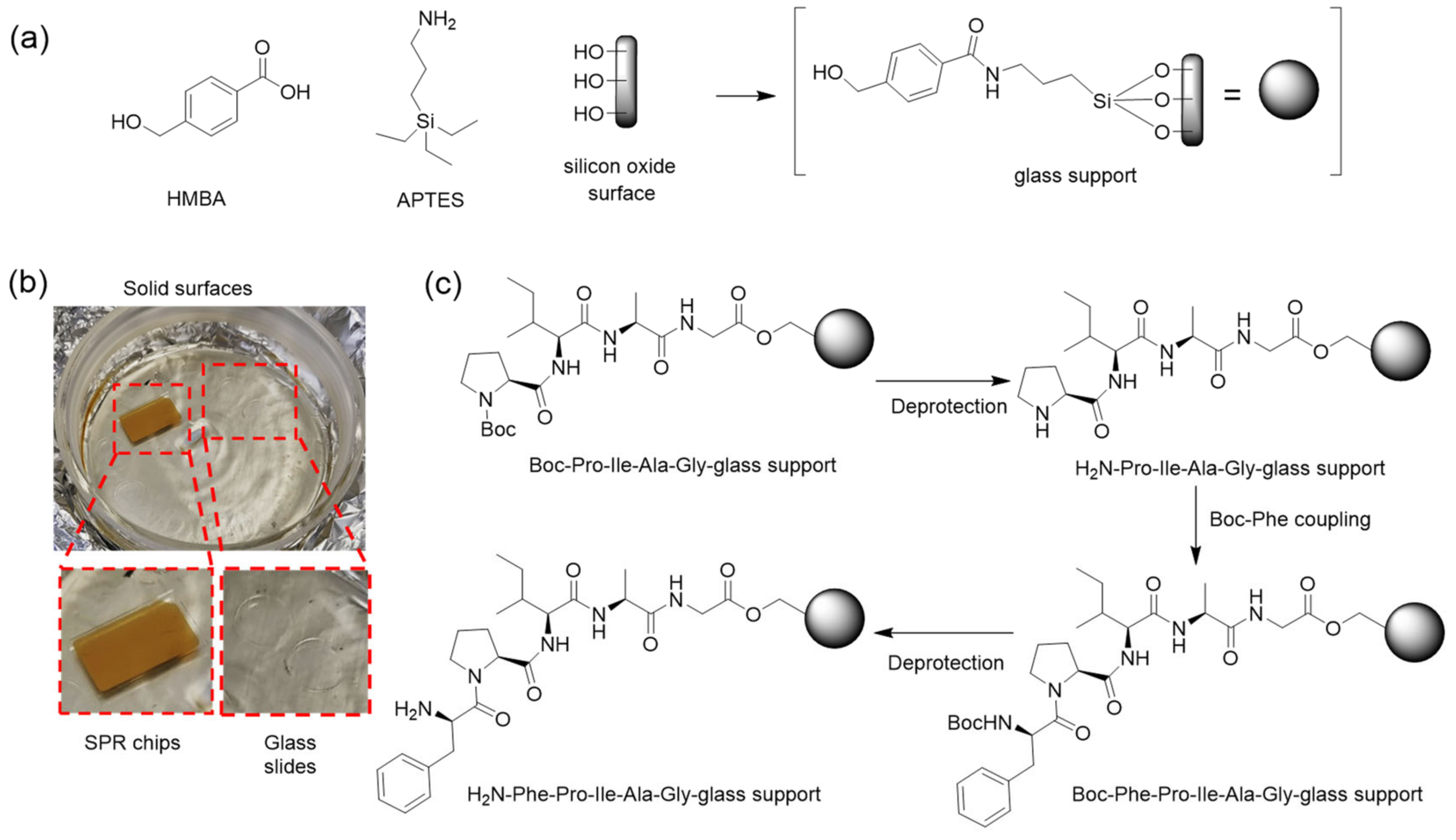
2.4. Adapting SPPS to Glass Surfaces
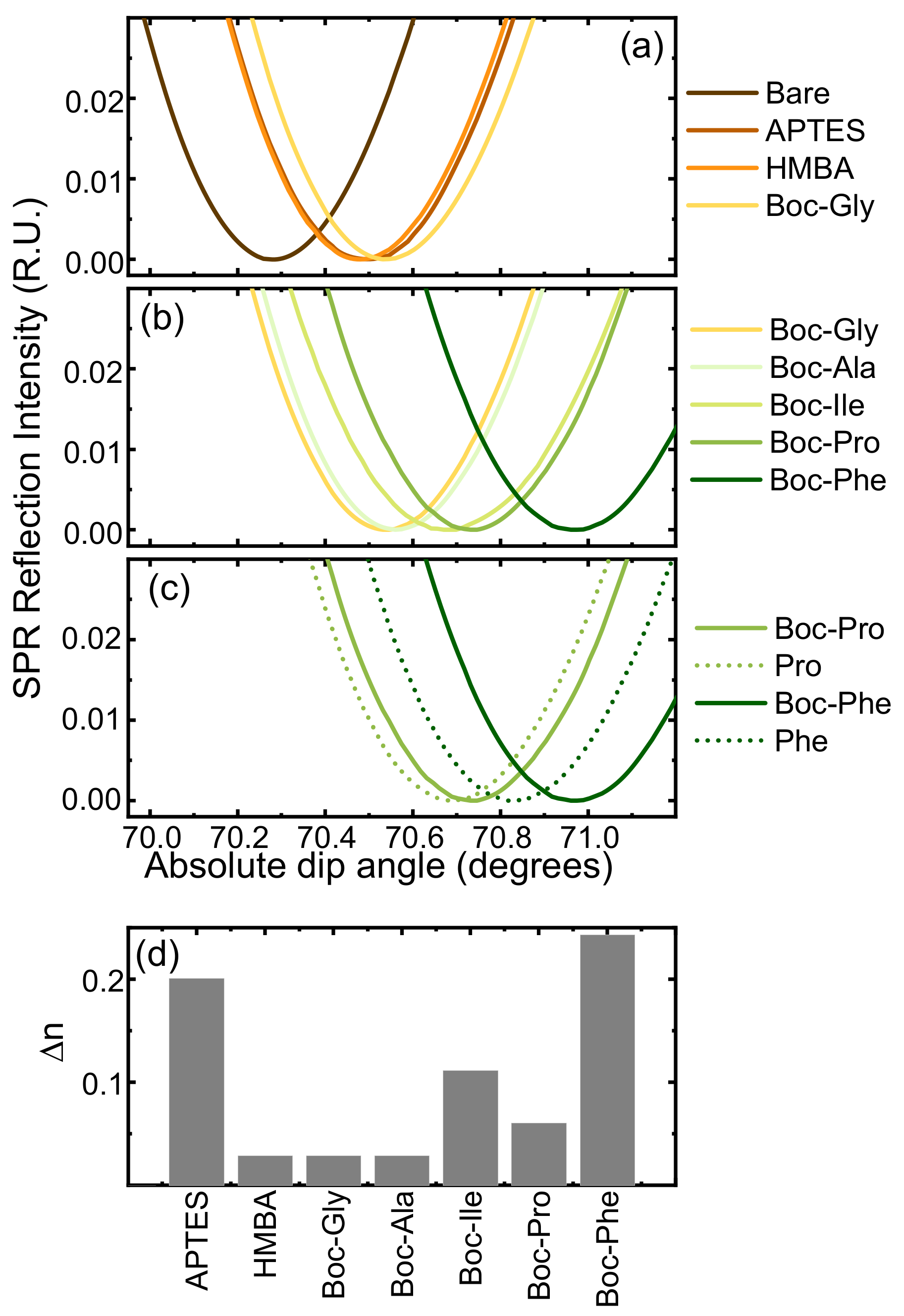
2.5. Surface Plasmon Resonance Analysis
3. Results and Discussion
3.1. Validation of CPG as SPPS Support
3.2. Boc-Based (Acid-Controlled) SPPS on CPG
3.3. Model Boc Syntheses of FPXAG on CPG
3.3.1. Bifunctional Amino Acids at X Position
3.3.2. Trifunctional Amino Acids at X Positions
3.4. DYK Epitope Synthesis for Immunodetection
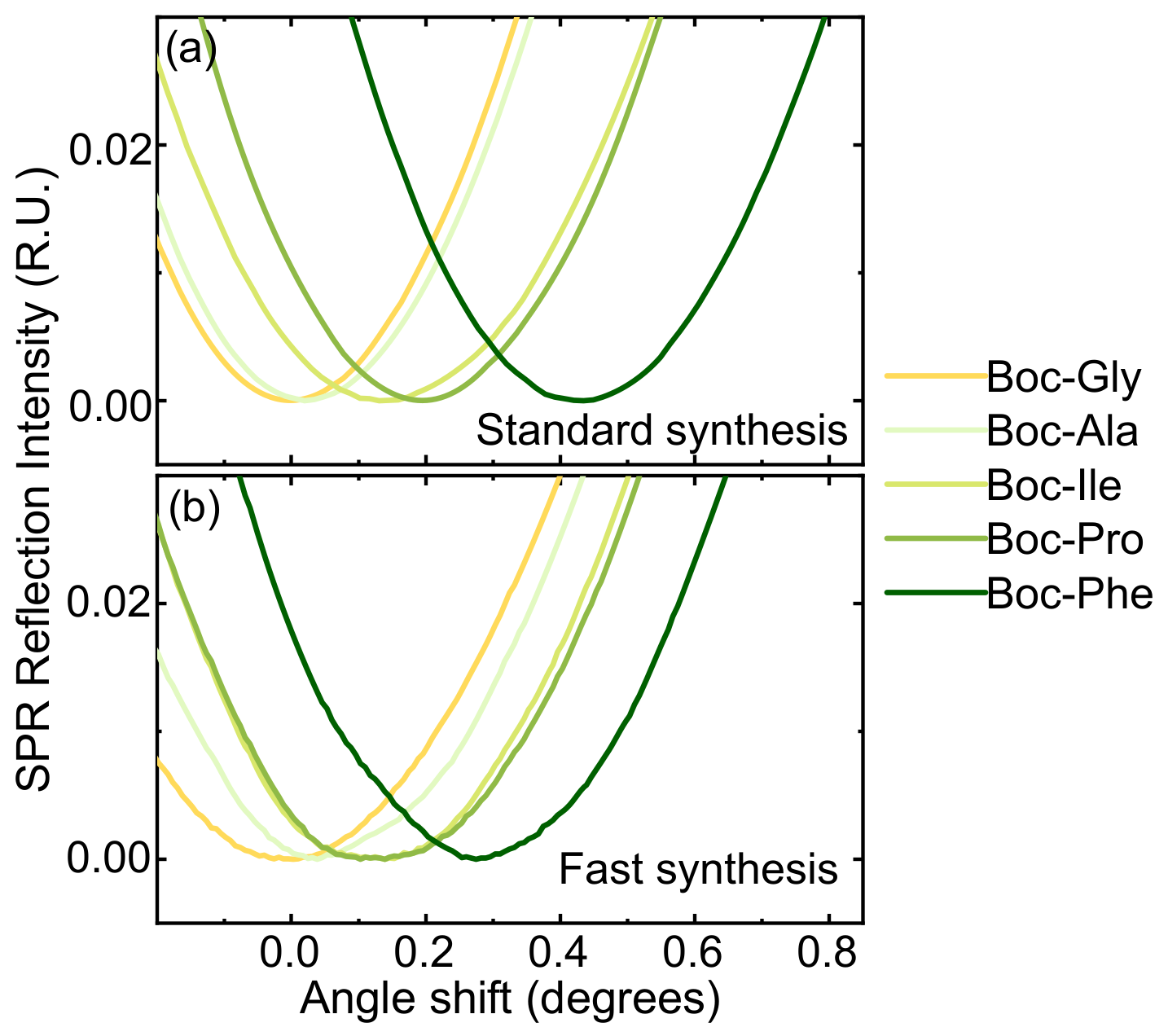
3.5. Translation and Optimization to Flat SiO2 Surfaces
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References and Note
- Syu, Y.-C.; Hsu, W.-E.; Lin, C.-T. Review—Field-Effect Transistor Biosensing: Devices and Clinical Applications. ECS J. Solid State Sci. Technol. 2018, 7, Q3196. [Google Scholar] [CrossRef]
- Rothberg, J.M.; Hinz, W.; Rearick, T.M.; Schultz, J.; Mileski, W.; Davey, M.; Leamon, J.H.; Johnson, K.; Milgrew, M.J.; Edwards, M.; et al. An Integrated Semiconductor Device Enabling Non-Optical Genome Sequencing. Nature 2011, 475, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Beyer, M.; Felgenhauer, T.; Ralf Bischoff, F.; Breitling, F.; Stadler, V. A Novel Glass Slide-Based Peptide Array Support with High Functionality Resisting Non-Specific Protein Adsorption. Biomaterials 2006, 27, 3505–3514. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Carney, R.P.; Liu, R.; Fan, J.; Zhao, S.; Chen, Y.; Lam, K.S.; Pan, T. Microfluidic Print-to-Synthesis Platform for Efficient Preparation and Screening of Combinatorial Peptide Microarrays. Anal. Chem. 2018, 90, 5833–5840. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.D.; Heider, P.L.; Adamo, A.; Vinogradov, A.A.; Mong, S.K.; Li, X.; Berger, T.; Policarpo, R.L.; Zhang, C.; Zou, Y.; et al. Rapid Flow-Based Peptide Synthesis. ChemBioChem 2014, 15, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Truex, N.L.; Holden, R.L.; Wang, B.Y.; Chen, P.G.; Hanna, S.; Hu, Z.; Shetty, K.; Olive, O.; Neuberg, D.; Hacohen, N.; et al. Automated Flow Synthesis of Tumor Neoantigen Peptides for Personalized Immunotherapy. Sci. Rep. 2020, 10, 723. [Google Scholar] [CrossRef] [PubMed]
- Hartrampf, N.; Saebi, A.; Poskus, M.; Gates, Z.P.; Callahan, A.J.; Cowfer, A.E.; Hanna, S.; Antilla, S.; Schissel, C.K.; Quartararo, A.J.; et al. Synthesis of Proteins by Automated Flow Chemistry. Science 2020, 368, 980–987. [Google Scholar] [CrossRef]
- Mijalis, A.J.; Thomas, D.A.; Simon, M.D.; Adamo, A.; Beaumont, R.; Jensen, K.F.; Pentelute, B.L. A Fully Automated Flow-Based Approach for Accelerated Peptide Synthesis. Nat. Chem. Biol. 2017, 13, 464–466. [Google Scholar] [CrossRef]
- Welden, R.; Schöning, M.J.; Wagner, P.H.; Wagner, T. Light-Addressable Electrodes for Dynamic and Flexible Addressing of Biological Systems and Electrochemical Reactions. Sensors 2020, 20, 1680. [Google Scholar] [CrossRef]
- Price, J.V.; Tangsombatvisit, S.; Xu, G.; Yu, J.; Levy, D.; Baechler, E.C.; Gozani, O.; Varma, M.; Utz, P.J.; Liu, C.L. On Silico Peptide Microarrays for High-Resolution Mapping of Antibody Epitopes and Diverse Protein-Protein Interactions. Nat. Med. 2012, 18, 1434–1440. [Google Scholar] [CrossRef]
- Egeland, R.D.; Southern, E.M. Electrochemically Directed Synthesis of Oligonucleotides for DNA Microarray Fabrication. Nucleic Acids Res. 2005, 33, e125. [Google Scholar] [CrossRef] [PubMed]
- Maurer, K.; Cooper, J.; Caraballo, M.; Crye, J.; Suciu, D.; Ghindilis, A.; Leonetti, J.A.; Wang, W.; Rossi, F.M.; Stöver, A.G.; et al. Electrochemically Generated Acid and Its Containment to 100 Micron Reaction Areas for the Production of DNA Microarrays. PLoS ONE 2006, 1, e34. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, D.; Lamblin, G.; Thomann, J.S.; Van Den Berg, A.; Olthuis, W.; Pascual-García, C. Electrochemical Control of PH in Nanoliter Volumes. Nano Lett. 2018, 18, 2807–2815. [Google Scholar] [CrossRef] [PubMed]
- El Maiss, J.; Balakrishnan, D.; Pascual, C. Universal Control of Protons Concentration Using Electrochemically Generated Acid Compatible with Miniaturization. Nanoscale Adv. 2022, 4, 3233–3242. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, D.; El Maiss, J.; Olthuis, W.; Pascual García, C. Miniaturized Control of Acidity in Multiplexed Microreactors. ACS Omega 2023, 8, 7587–7594. [Google Scholar] [CrossRef]
- Shukla, R.P.; Bomer, J.G.; Wijnperle, D.; Kumar, N.; Georgiev, V.P.; Singh, A.C.; Krishnamoorthy, S.; Pascual García, C.; Pud, S.; Olthuis, W. Planar Junctionless Field-Effect Transistor for Detecting Biomolecular Interactions. Sensors 2022, 22, 5783. [Google Scholar] [CrossRef] [PubMed]
- Rollo, S.; Rani, D.; Leturcq, R.; Olthuis, W.; Pascual García, C. A High Aspect Ratio Fin-Ion Sensitive Field Effect Transistor: Compromises towards Better Electrochemical Bio-Sensing. Nano Lett. 2019, 19, 2879–2887. [Google Scholar] [CrossRef]
- Walsh, M.K.; Wang, X.; Weimer, B.C. Optimizing the Immobilization of Single-Stranded DNA onto Glass Beads. J. Biochem. Biophys. Methods 2001, 47, 221–231. [Google Scholar] [CrossRef]
- Sheng, H.; Ye, B.C. Different Strategies of Covalent Attachment of Oligonucleotide Probe onto Glass Beads and the Hybridization Properties. Appl. Biochem. Biotechnol. 2009, 152, 54–65. [Google Scholar] [CrossRef]
- Rollo, S.; Rani, D.; Olthuis, W.; Pascual García, C. High Performance Fin-FET Electrochemical Sensor with High-k Dielectric Materials. Sens. Actuators B Chem. 2020, 303, 127215. [Google Scholar] [CrossRef]
- Rani, D.; Rollo, S.; Olthuis, W.; Krishnamoorthy, S.; García, C.P. Combining Chemical Functionalization and Finfet Geometry for Field Effect Sensors as Accessible Technology to Optimize PH Sensing. Chemosensors 2021, 9, 20. [Google Scholar] [CrossRef]
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Barany, G.; Merrifield, R.B. Solid Phase Peptide Synthesis. In The Peptides; Gross, E., Meienhofer, J., Eds.; Academic Press: New York, NY, USA, 1979; Volume 2, pp. 1–284. [Google Scholar]
- Fields, G.B.; Noble, R.L. Solid Phase Peptide Synthesis Utilizing 9-fluorenylmethoxycarbonyl Amino Acids. Int. J. Pept. Protein Res. 1990, 35, 161–214. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, X.F.; Chen, Y.; Zhang, L.H.; Yang, Z.J. A Solid-Phase Method for Peptide-SiRNA Covalent Conjugates Based on Click Chemistry. MedChemComm 2012, 3, 506–511. [Google Scholar] [CrossRef]
- Rink, H. Solid-Phase Synthesis of Protected Peptide Fragments Using a Trialkoxy-Diphenyl-Methylester Resin. Tetrahedron Lett. 1987, 28, 3787–3790. [Google Scholar] [CrossRef]
- Bernatowicz, M.S.; Daniels, S.B.; Köster, H. A Comparison of Acid Labile Linkage Agents for the Synthesis of Peptide C-Terminal Amides. Tetrahedron Lett. 1989, 30, 4645–4648. [Google Scholar] [CrossRef]
- Hansen, J.; Diness, F.; Meldal, M. C-Terminally Modified Peptides via Cleavage of the HMBA Linker by O-, N- or S-Nucleophiles. Org. Biomol. Chem. 2016, 14, 3238–3245. [Google Scholar] [CrossRef]
- In this work the IUPAC-IUB one- and three-letter codes for amino acid residues (Pure & Appl. Chem., Vol. 56, No. 5, pp. 595–624, 1984) are used, though not strictly interchangeably: To denote individual residues (e.g., Leu), the three letter code is preferred; for longer sequences, the shorter, one-letter notation is adopted.
- Geiger, R.; König, W. Amine Protecting Groups. In The Peptides; Gross, E., Meienhofer, J., Eds.; Academic Press: New York, NY, USA, 1981; Volume 3, pp. 70–80. [Google Scholar]
- Mthembu, S.N.; Sharma, A.; Albericio, F.; de la Torre, B.G. Breaking a Couple: Disulfide Reducing Agents. ChemBioChem 2020, 21, 1947–1954. [Google Scholar] [CrossRef]
- Yamashiro, D.; Li, C.H. Protection of Tyrosine in Solid Phase Peptide Synthesis. J. Org. Chem. 1973, 38, 591–592. [Google Scholar]
- De Feijter, J.A.; Benjamins, J.; Veer, F.A. Ellipsometry as a Tool to Study the Adsorption Behavior of Synthetic and Biopolymers at the Air–Water Interface. Biopolymers 1978, 17, 1759–1772. [Google Scholar] [CrossRef]
- Zhao, H.; Brown, P.H.; Schuck, P. On the Distribution of Protein Refractive Index Increments. Biophys. J. 2011, 100, 2309–2317. [Google Scholar] [CrossRef]
- Harpaz, Y.; Gerstein, M.; Chothia, C. Volume Changes on Protein Folding. Structure 1994, 2, 641–649. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Acid | Solvent | Time | Boc Removal * |
|---|---|---|---|---|
| 1 | 50% TFA | DCM | 5 min | 93.9% (78%) |
| 2 | 50% TFA | DCM | 25 min | 100% (100%) |
| 3 | 4 M HCl/dioxane | dioxane | 5 min | 96.8% (88%) |
| 4 | 4 M HCl/dioxane | dioxane | 25 min | 100% (100%) |
| Entry | Sequence | Side Chain Protection a | Solvent b | Time b | Deprotection Time c | Yield d |
|---|---|---|---|---|---|---|
| 1 | FPLAG | none | DMF | 1 h (×2) | 45 min (×2) | 98% |
| 2 | FPIAG | none | DMF | 1 h (×2) | 45 min (×2) | 95% |
| 3 | FPVAG | none | DMF | 1 h (×2) | 45 min (×2) | 94% |
| 4 | FPDAG | OFm | DMF | 1 h (×2) | 45 min (×2) | 96% |
| 5 | FPEAG | OFm | DMF | 1 h (×2) | 45 min (×2) | 95% |
| 6 | FPEAG | OFm | MeCN | 1 h (×2) | 45 min (×2) | 95% |
| 7 | FPKAG | Fmoc | DMF | 1 h (×2) | 45 min (×2) | 86% |
| 8 | FPNAG | none | DMF | 30 min (×2) | 30 min | 80% |
| 9 | FPQAG | none | DMF | 30 min (×2) | 30 min | 96% |
| 10 | FPWAG | none | DMF | 30 min (×2) | 30 min | 96% |
| 11 | FPHAG | none | DMF | 30 min (×2) | 30 min | 0% |
| 12 | FPHAG | Dnp | DMF | 30 min (×2) | 30 min (×2) | 74% |
| 13 | FPHAG | Trt | DMF | 30 min (×2) | 3 h | 91% |
| 14 | FPMAG | none | DMF | 30 min (×2) | 30 min | 76% |
| 15 | FPCAG | none | DMF | 30 min (×2) | 30 min | 0% |
| 16 | FPCAG | Fm | DMF | 30 min (×2) | 30 min | 58% |
| 17 | FPCAG | Fm/DODt | DMF | 30 min (×2) | 30 min | 75% |
| 18 | FPSAG | none | DMF | 25 min (×2) | 45 min | 77% |
| 19 | FPSAG | tBu | DMF | 25 min (×2) | 45 min | 82% |
| 20 | FPTAG | none | DMF | 25 min (×2) | 45 min | 81% |
| 21 | FPTAG | tBu | DMF | 25 min (×2) | 45 min | 81% |
| 22 | FPYAG | none | DMF | 25 min (×2) | 45 min | 56% |
| 23 | FPYAG | BrZ | DMF | 25 min (×2) | 45 min | 95% |
| Entry | Sequence | Side Chain Protection | Coupling Time a | Yield b |
|---|---|---|---|---|
| 1 | FGAFPIAG | none | 30 min (×2) | 94% |
| 2 | FGAFPVAG | none | 30 min (×2) | 91% |
| 3 | FGAFPEAG | OFm | 30 min (×2) | 97% |
| 4 | GAFGAFPVAG | none | 30 min (×2) | 90% |
| 5 | EAFGAFPEAG | OFm × 2 | 25 min (×2) | 89% |
| 6 | GDEAFGAFPEAG | OFm × 3 | 25 min (×2) | 80% |
| 7 | FGAFGAFGAFPIAG | none | 25 min (×2) | 76% |
| 8 | DYKG | OFm, none and Fmoc | 25 min (×2) | 60% |
| 9 | DYKG | OFm, 2-Br-Z and Fmoc | 25 min (×2) | 77% |
| 10 | DYKGG | OFm, none and Fmoc | 25 min (×2) | 62% |
| 11 | DYKGG | OFm, 2-Br-Z and Fmoc | 25 min (×2) | 72% |
| 12 | DYKD | OFm, none, Fmoc and OFm | 25 min (×2) | 63% |
| 13 | DYKDD | OFm, none, Fmoc and OFm (×2) | 25 min (×2) | 63% |
| 14 | DYKK | OFm, none, Fmoc (×3) | 25 min (×2) | 57% |
| APTES Functionalization | HMBA-Linker Attachment | C-Terminal Anchoring | AA Coupling | Deprotection | |
|---|---|---|---|---|---|
| Standard | 12 h | 120 min | 5 h | 60 min | 30 min |
| Fast | 3 h | 5 min | 3 × 5 min | 2 × 5 min | 5 min |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cristóbal-Lecina, E.; El-Maiss, J.; Figueras, E.; Singh, A.C.; Krishnamoorthy, S.; Østerbye, T.; Pascual García, C.; Andreu, D. Acid-Modulated Peptide Synthesis for Application on Oxide Biosensor Interfaces. Nanomaterials 2023, 13, 3092. https://doi.org/10.3390/nano13243092
Cristóbal-Lecina E, El-Maiss J, Figueras E, Singh AC, Krishnamoorthy S, Østerbye T, Pascual García C, Andreu D. Acid-Modulated Peptide Synthesis for Application on Oxide Biosensor Interfaces. Nanomaterials. 2023; 13(24):3092. https://doi.org/10.3390/nano13243092
Chicago/Turabian StyleCristóbal-Lecina, Edgar, Janwa El-Maiss, Eduard Figueras, Aruna Chandra Singh, Sivashankar Krishnamoorthy, Thomas Østerbye, César Pascual García, and David Andreu. 2023. "Acid-Modulated Peptide Synthesis for Application on Oxide Biosensor Interfaces" Nanomaterials 13, no. 24: 3092. https://doi.org/10.3390/nano13243092
APA StyleCristóbal-Lecina, E., El-Maiss, J., Figueras, E., Singh, A. C., Krishnamoorthy, S., Østerbye, T., Pascual García, C., & Andreu, D. (2023). Acid-Modulated Peptide Synthesis for Application on Oxide Biosensor Interfaces. Nanomaterials, 13(24), 3092. https://doi.org/10.3390/nano13243092