

pH-Sensitive Nanoparticles for Colonic Delivery Anti-miR-301a in Mouse Models of Inflammatory Bowel Diseases

 , and
, and
Abstract
: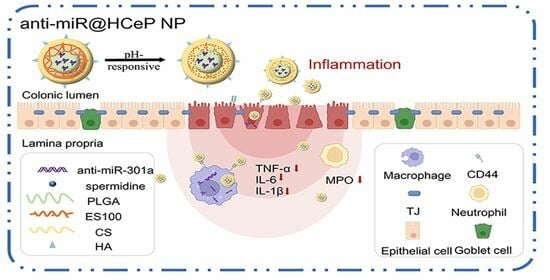
1. Introduction
2. Materials and Methods
2.1. Materials and Animals
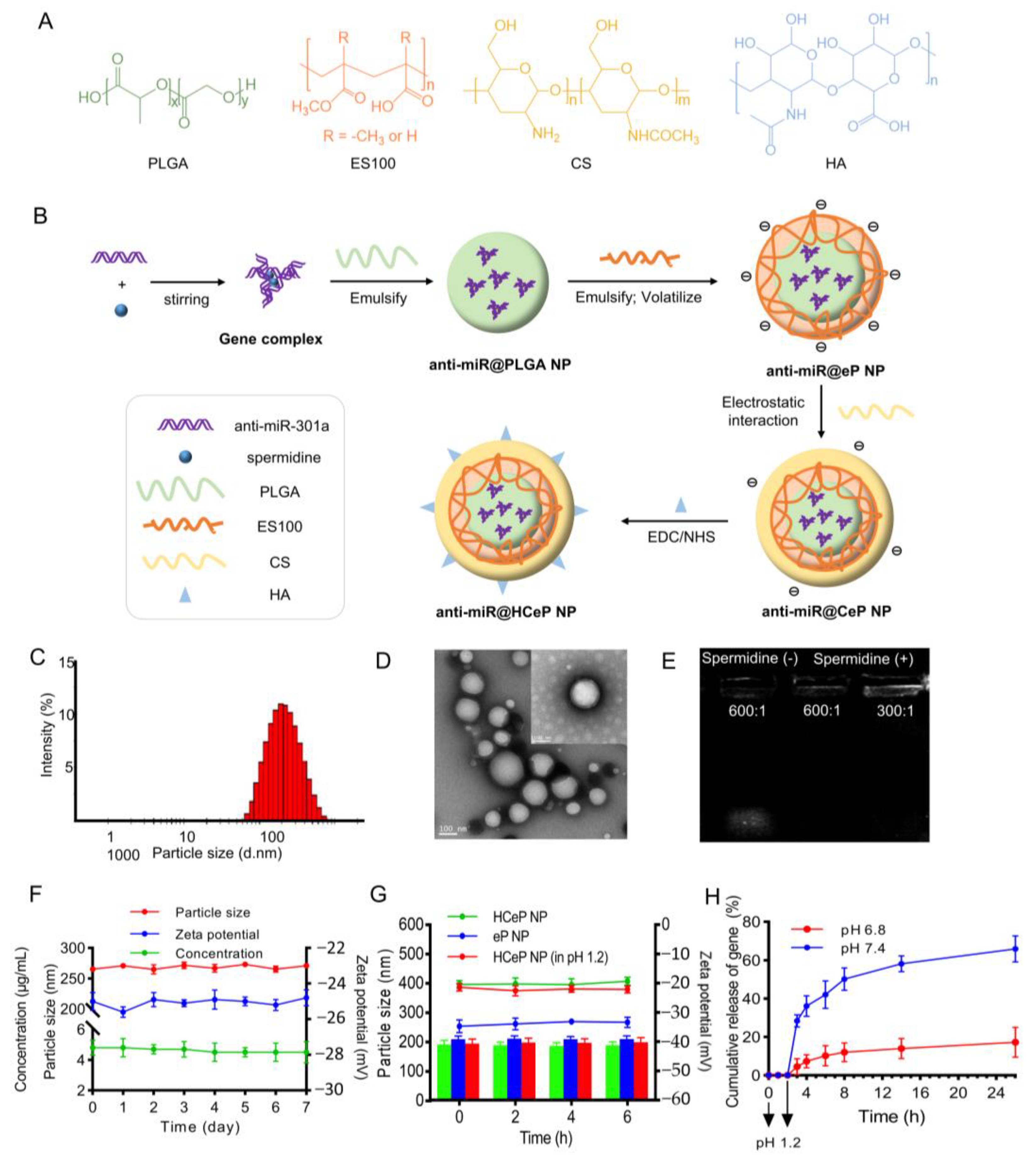
2.2. Preparation of Nanoparticles
2.3. Characterization of the Nanoparticles
2.4. Stability of Nanoparticles
2.5. Acid Resistance of Nanoparticles
2.6. In Vitro Drug Release
2.7. Cell Culture
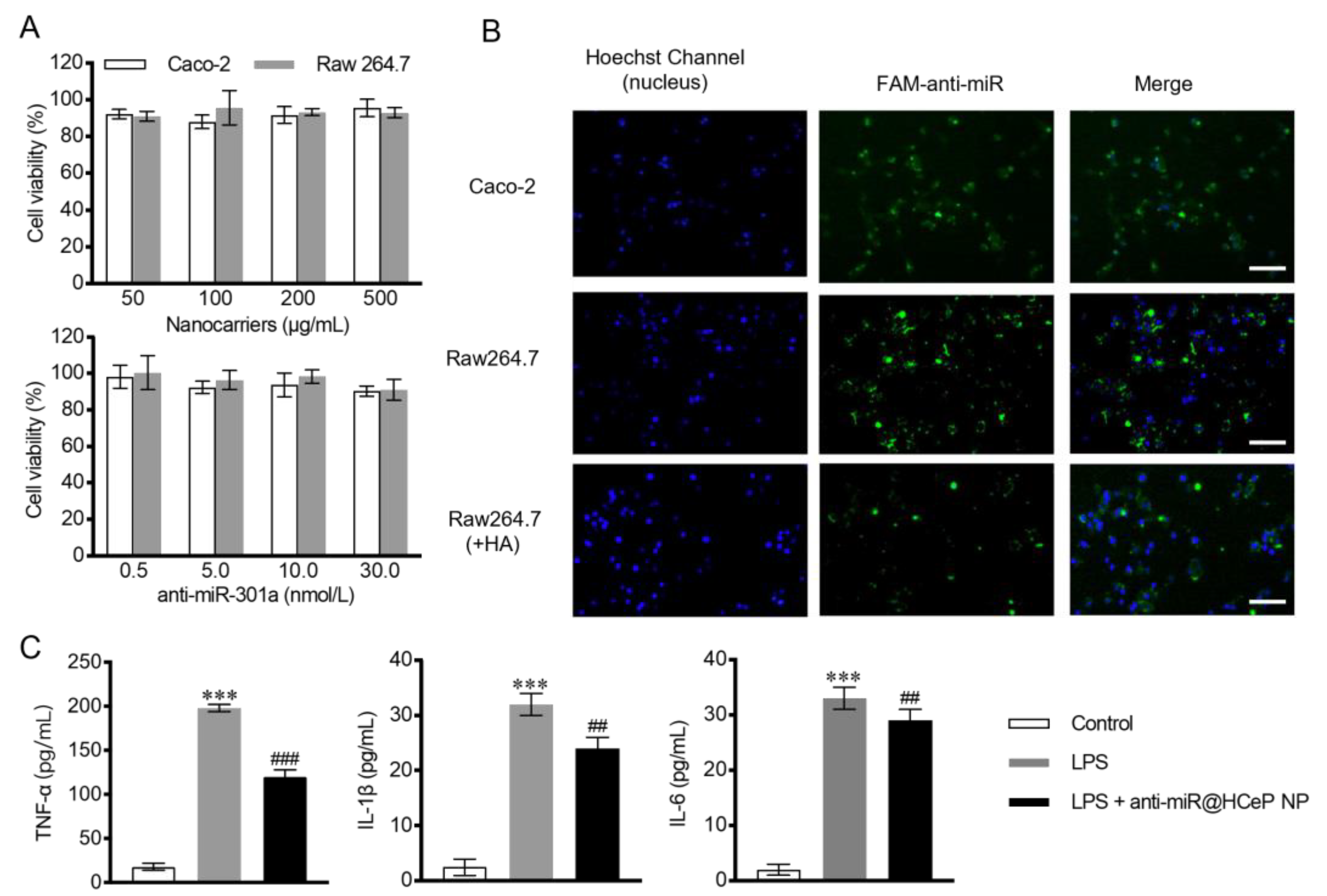
2.8. Cytotoxicity Assay
2.9. Cellular Uptake
2.10. In Vitro Cytokine Assay
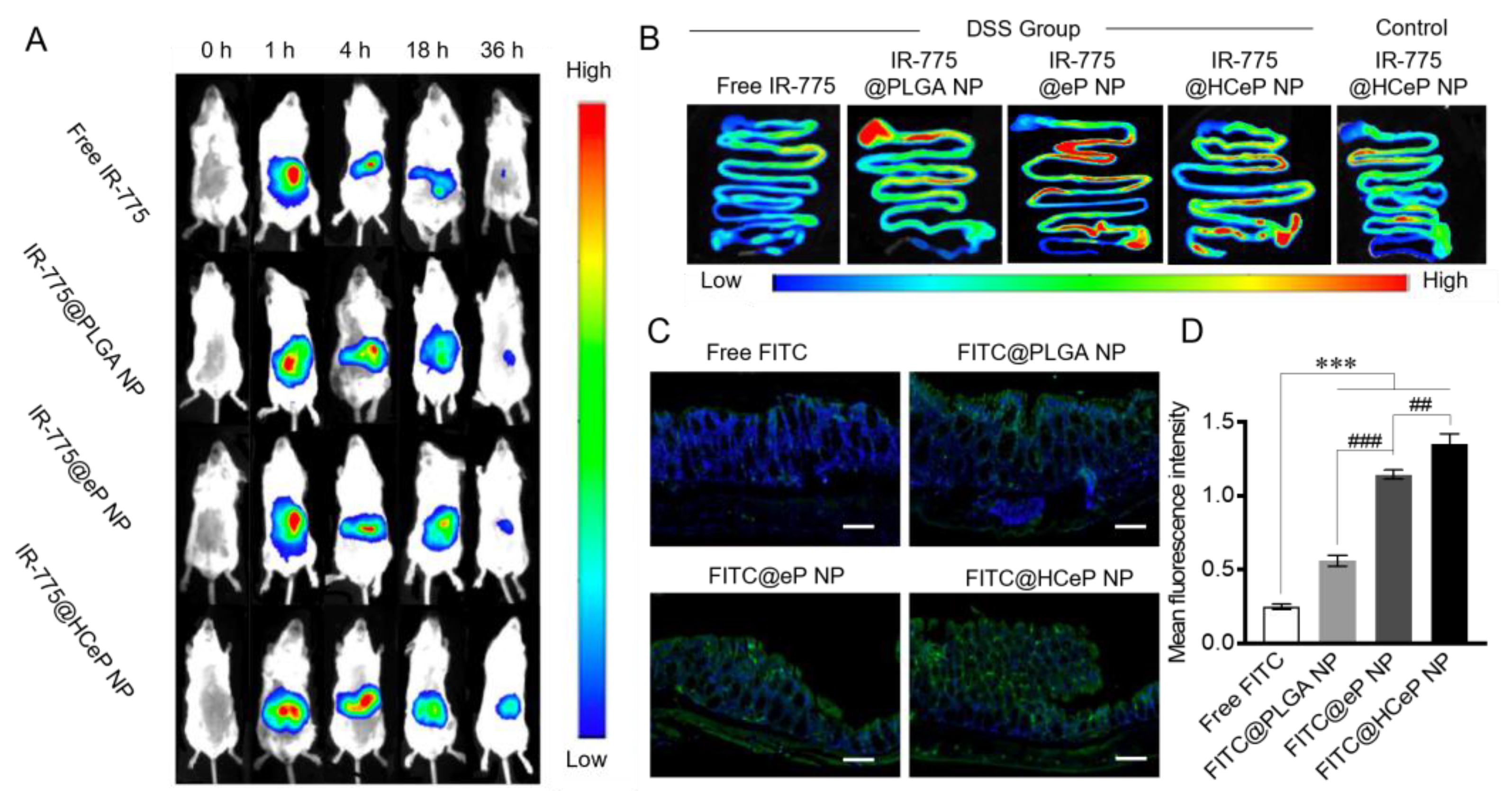
2.11. Biodistribution and Colon-Targeting Ability of Nanoparticles
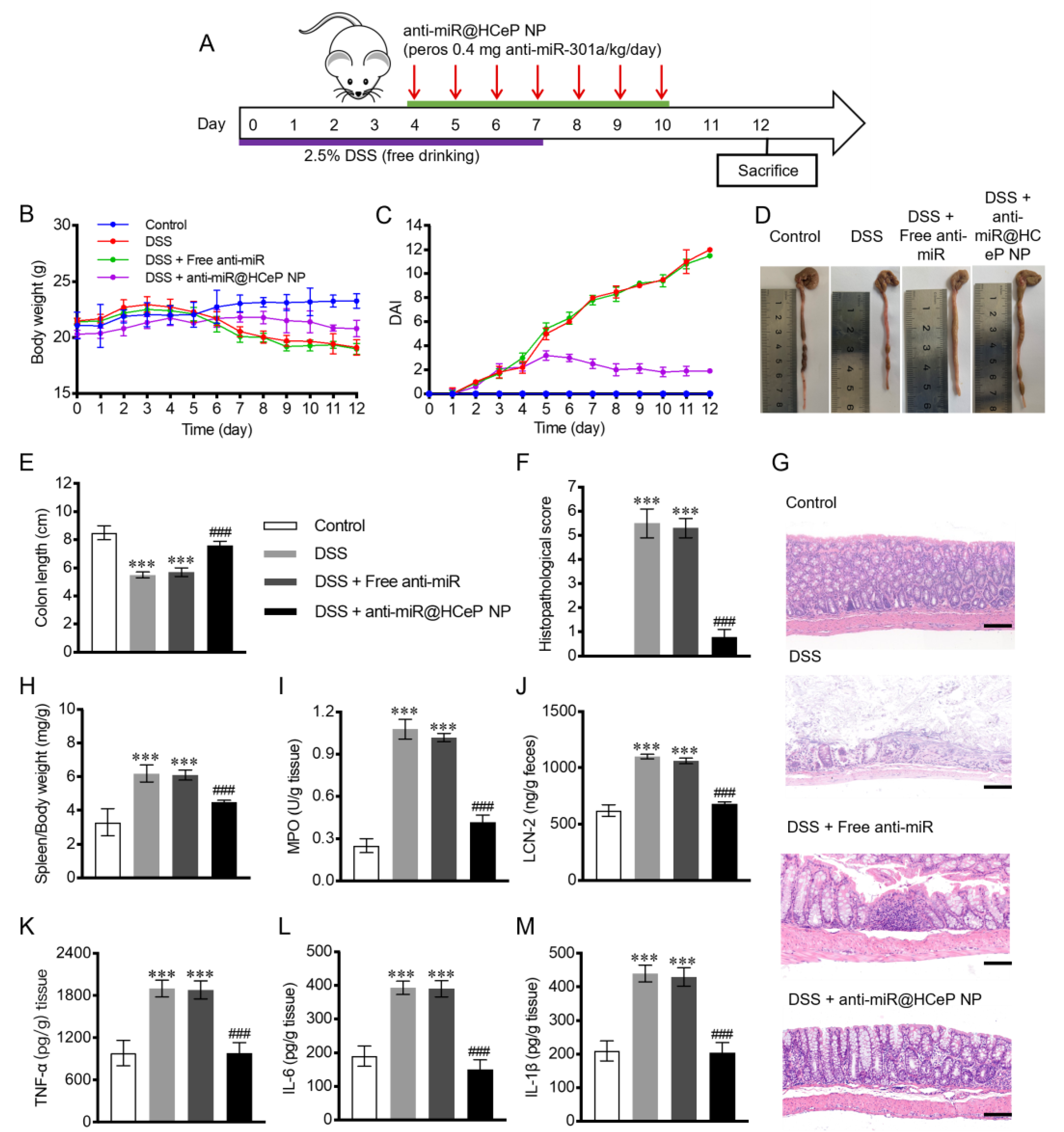
2.12. In Vivo Therapeutic Efficacy
2.13. Histology
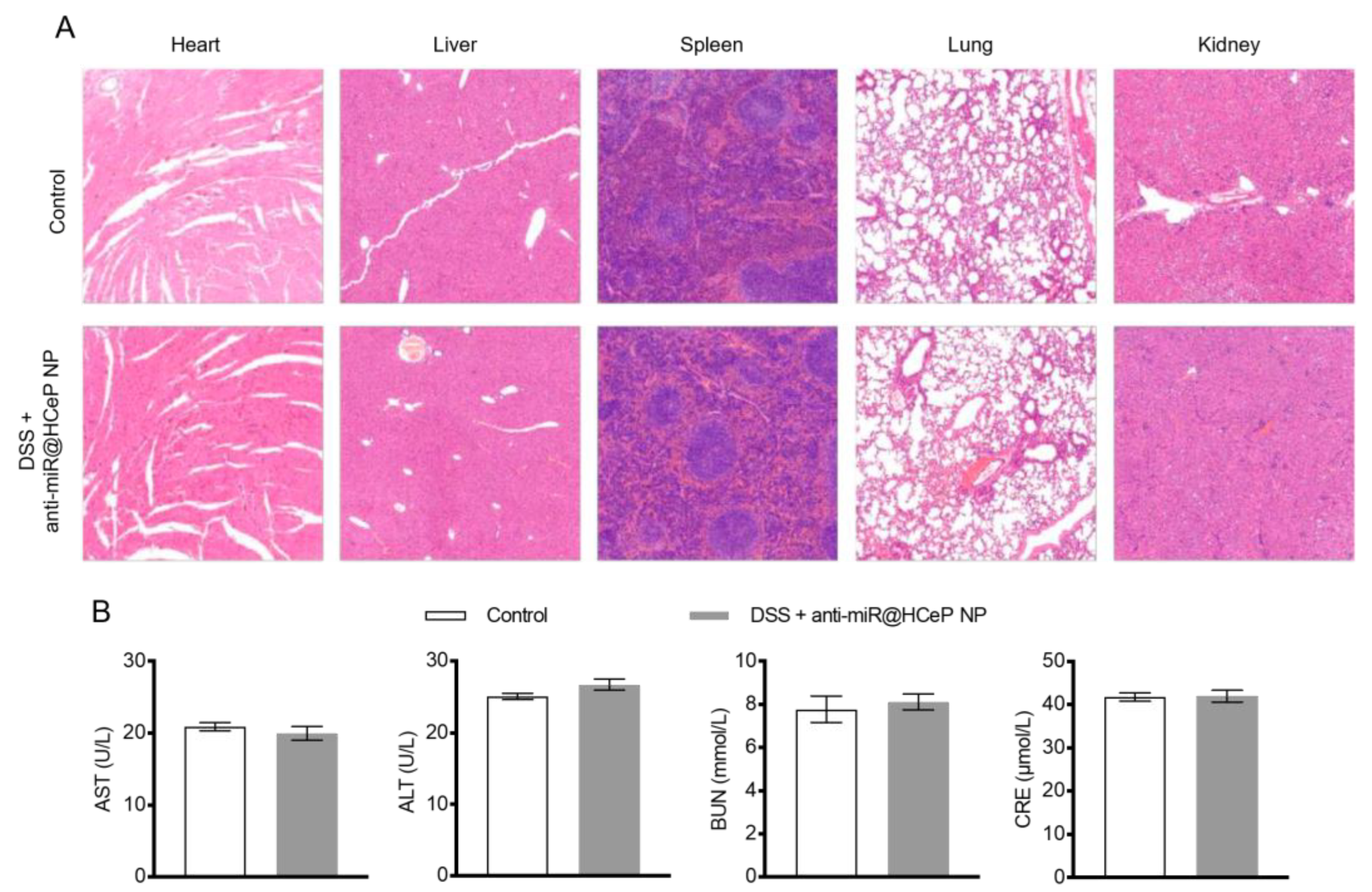
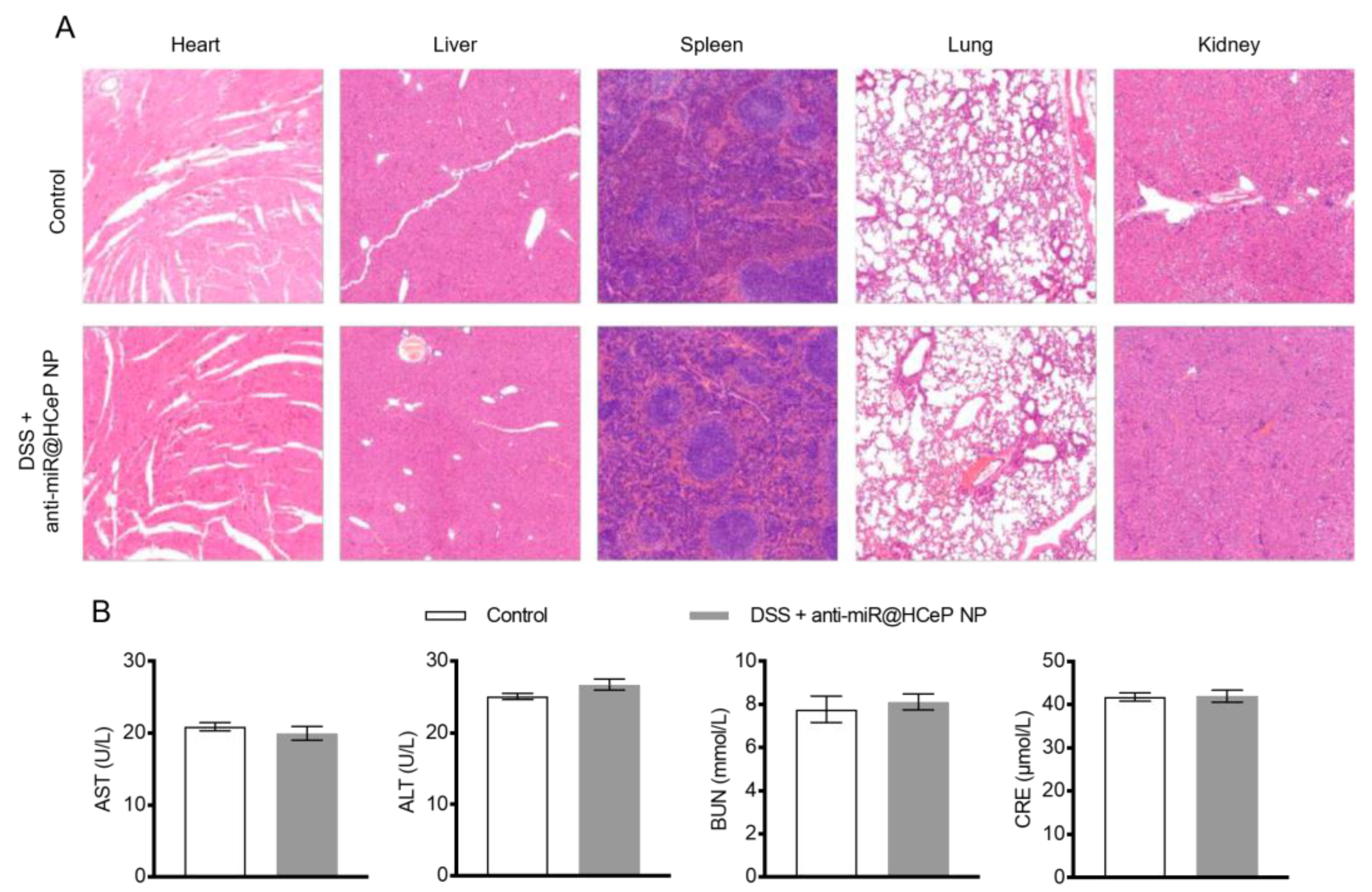
2.14. Safety Evaluation
2.15. Statistical Analysis
3. Results and Discussion
3.1. Characterization of the Nanoparticles
3.2. Storage Stability
3.3. Acid Resistance
3.4. In Vitro Drug Release
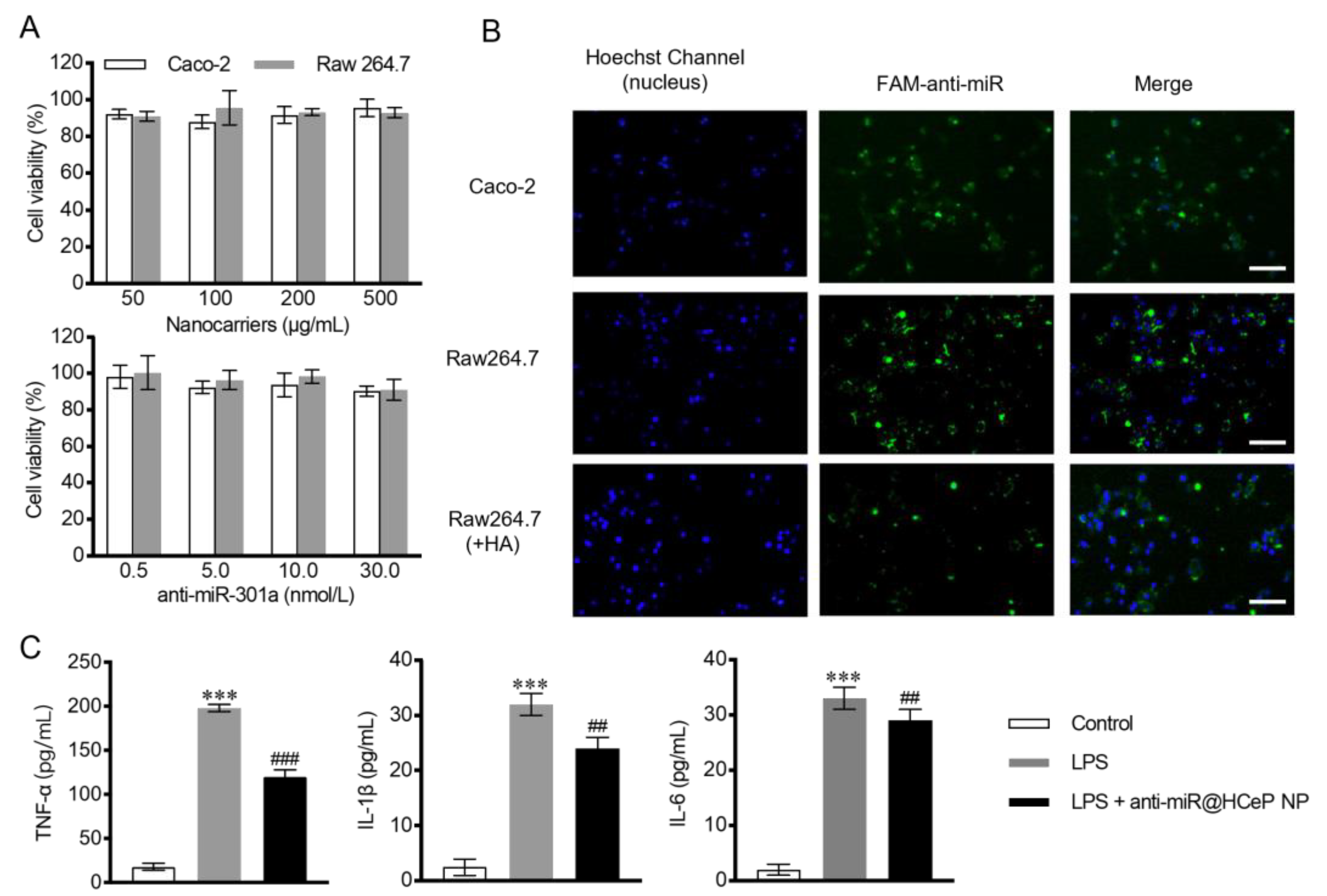
3.5. Cytotoxicity Assay
3.6. Cellular Uptake
3.7. In Vitro Cytokine Assay
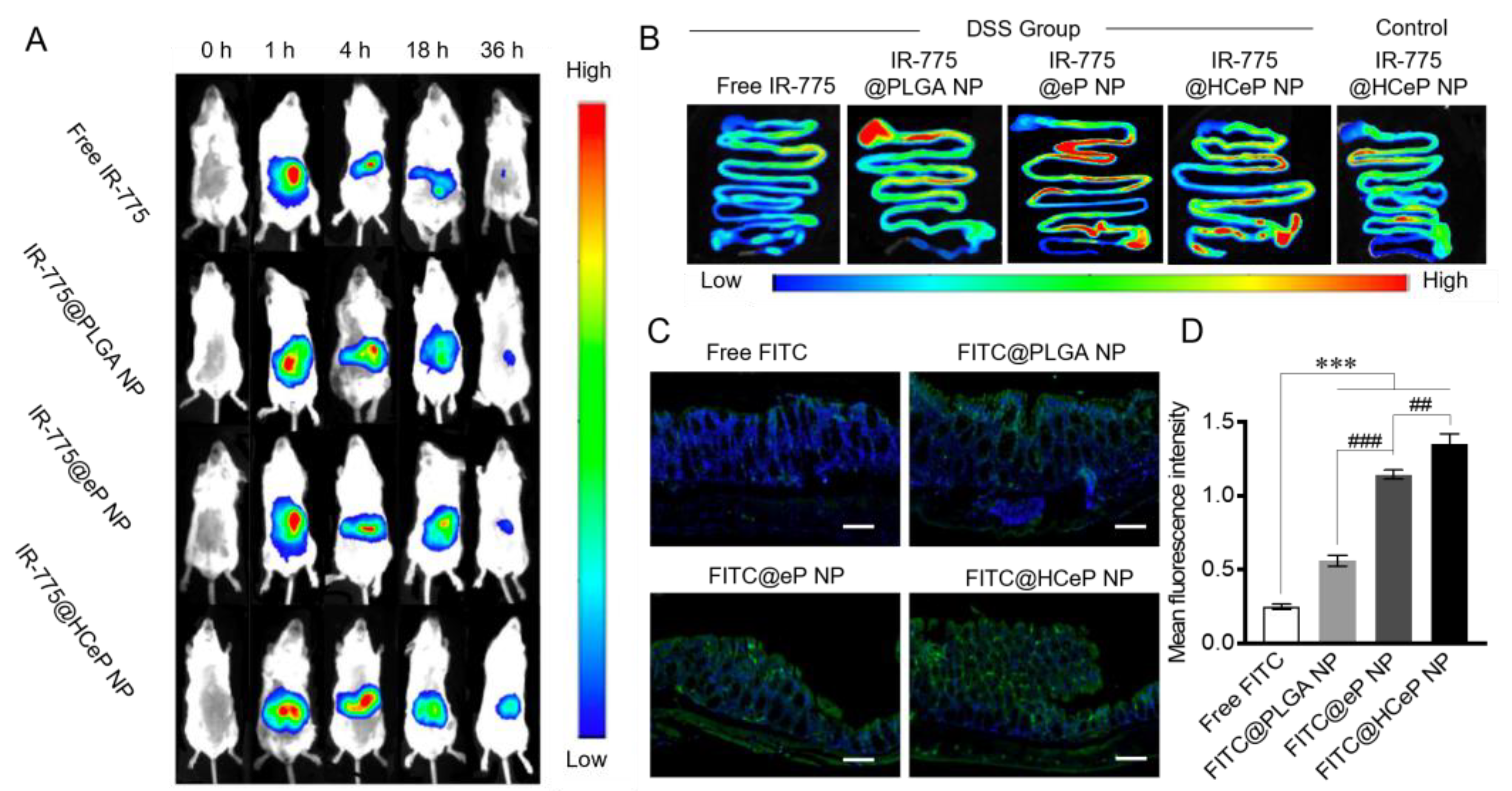
3.8. Biodistribution and Colon-Targeting Ability of Nanoparticles
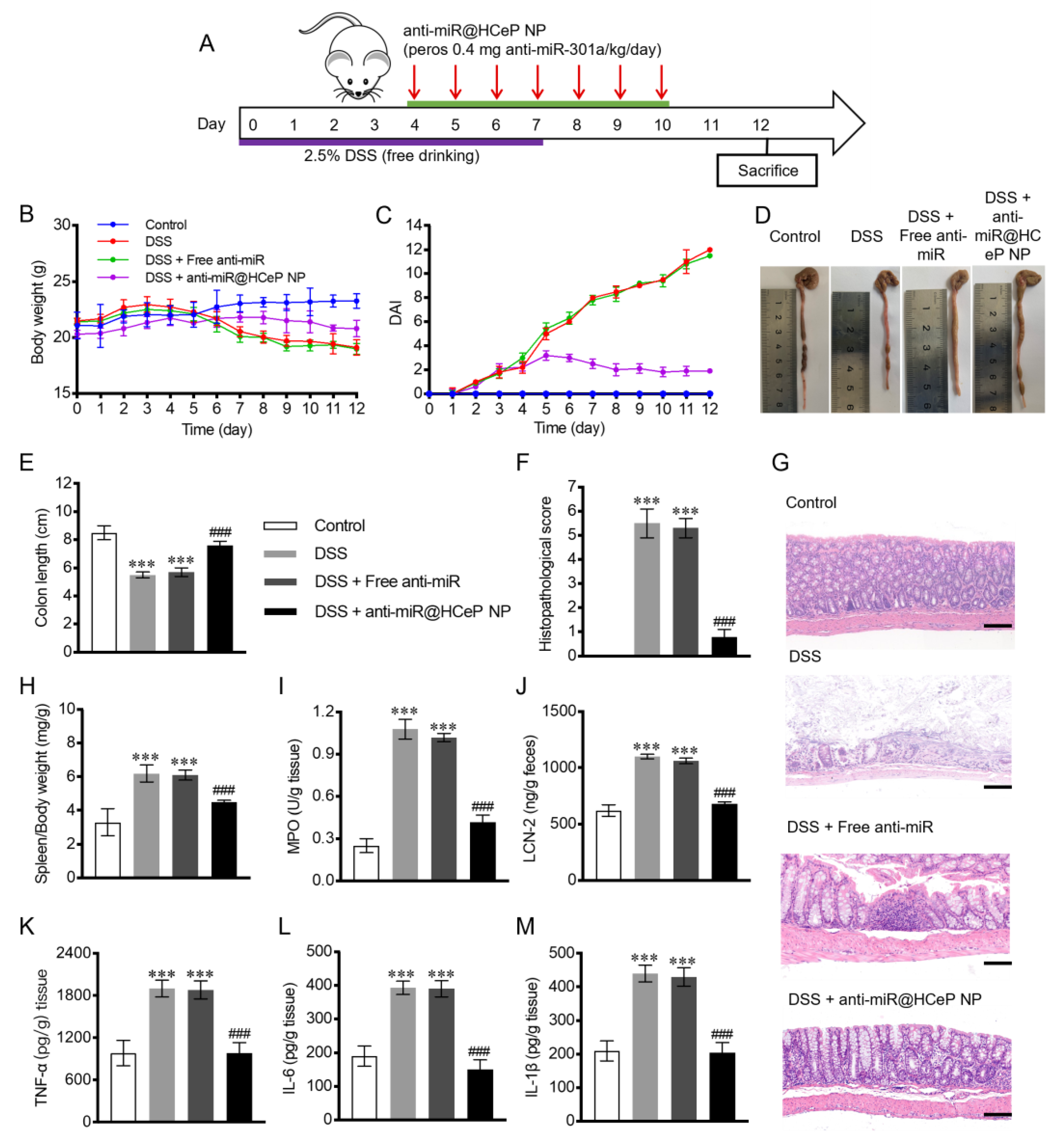
3.9. In Vivo Therapeutic Efficacy
3.10. Bodyweight and DAI
3.11. Morris Score and Colon Length
3.12. Histological Injury Assessment
3.13. Spleen Mass to Body Weight Ratio
3.14. Evaluation of MPO and LCN-2 Activity
3.15. Pro-Inflammatory Cytokines TNF-α, IL-6, and IL-1β Quantification
3.16. Safety Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaplan, G.G.; Ng, S.C. Globalisation of inflammatory bowel disease: Perspectives from the evolution of inflammatory bowel disease in the UK and China. Lancet Gastroenterol. Hepatol. 2016, 1, 307–316. [Google Scholar] [CrossRef]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Mulder, D.J.; Noble, A.J.; Justinich, C.J.; Duffin, J.M. A tale of two diseases: The history of inflammatory bowel disease. J. Crohns Colitis 2014, 8, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Pu, Z.; Yang, F.; Wang, L.; Diao, Y.; Chen, D. Advancements of compounds targeting Wnt and Notch signalling pathways in the treatment of inflammatory bowel disease and colon cancer. J. Drug Target. 2021, 29, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Kesisoglou, F.; Zimmermann, E.M. Novel drug delivery strategies for the treatment of inflammatory bowel disease. Expert. Opin. Drug Deliv. 2005, 2, 451–463. [Google Scholar] [CrossRef]
- Okamoto, R.; Watanabe, M. Role of epithelial cells in the pathogenesis and treatment of inflammatory bowel disease. J. Gastroenterol. 2016, 51, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.; Varum, F.; Bravo, R.; Furrer, E.; Bojic, D.; Basit, A.W. Inflammatory bowel disease: Exploring gut pathophysiology for novel therapeutic targets. Transl. Res. 2016, 176, 38–68. [Google Scholar] [CrossRef]
- Wright, E.K.; Ding, N.S.; Niewiadomski, O. Management of inflammatory bowel disease. Med. J. Aust. 2018, 209, 318–323. [Google Scholar] [CrossRef]
- Butnariu, M.; Quispe, C.; Herrera-Bravo, J.; Fernández-Ochoa, Á.; Emamzadeh-Yazdi, S.; Adetunji, C.O.; Memudu, A.E.; Otlewska, A.; Bogdan, P.; Antolak, H.; et al. A review on tradescantia: Phytochemical constituents, biological activities and health-promoting effects. Front. Biosci. (Landmark Ed.) 2022, 27, 197. [Google Scholar] [CrossRef]
- Butnariu, M.; Quispe, C.; Herrera-Bravo, J.; Helon, P.; Kukula-Koch, W.; López, V.; Les, F.; Vergara, C.V.; Alarcón-Zapata, P.; Alarcón-Zapata, B.; et al. The effects of thymoquinone on pancreatic cancer: Evidence from preclinical studies. Biomed. Pharmacother. 2022, 153, 113364. [Google Scholar] [CrossRef]
- Feng, J.; Wang, Y.; Lv, Y.; Fang, S.; Ren, M.; Yao, M.; Lan, M.; Zhao, Y.; Gao, F. Xa ph-responsive and colitis-targeted nanoparticle loaded with shikonin for the oral treatment of inflammatory bowel disease in mice. Mol. Pharm. 2022, 19, 4157–4170. [Google Scholar] [CrossRef] [PubMed]
- Fukata, T.; Mizushima, T.; Nishimura, J.; Okuzaki, D.; Wu, X.; Hirose, H.; Yokoyama, Y.; Kubota, Y.; Nagata, K.; Tsujimura, N.; et al. The supercarbonate apatite-microrna complex inhibits dextran sodium sulfate-induced colitis. Mol. Ther. Nucleic Acids 2018, 12, 658–671. [Google Scholar] [CrossRef] [PubMed]
- Coskun, M.; Bjerrum, J.T.; Seidelin, J.B.; Nielsen, O.H. MicroRNAs in inflammatory bowel disease--pathogenesis, diagnostics and therapeutics. World J. Gastroenterol. 2012, 18, 4629–4634. [Google Scholar] [CrossRef]
- Saliminejad, K.; Khorram Khorshid, H.R.; Soleymani Fard, S.; Ghaffari, S.H. An overview of micrornas: Biology, functions, therapeutics, and analysis methods. J. Cell. Physiol. 2019, 234, 5451–5465. [Google Scholar] [CrossRef]
- Van Meter, E.N.; Onyango, J.A.; Teske, K.A. A review of currently identified small molecule modulators of microrna function. Eur. J. Med. Chem. 2020, 188, 112008. [Google Scholar] [CrossRef] [PubMed]
- Paraskevi, A.; Theodoropoulos, G.; Papaconstantinou, I.; Mantzaris, G.; Nikiteas, N.; Gazouli, M. Circulating microrna in inflammatory bowel disease. J. Crohns Colitis 2012, 6, 900–904. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Eghtedarian, R.; Taheri, M. The crucial role of non-coding RNAs in the pathophysiology of inflammatory bowel disease. Biomed. Pharmacother. 2020, 129, 110507. [Google Scholar] [CrossRef]
- James, J.P.; Riis, L.B.; Malham, M.; Høgdall, E.; Langholz, E.; Nielsen, B.S. Microrna biomarkers in ibd-differential diagnosis and prediction of colitis-associated cancer. Int. J. Mol. Sci. 2020, 21, 7893. [Google Scholar] [CrossRef]
- Wu, F.; Guo, N.J.; Tian, H.; Marohn, M.; Gearhart, S.; Bayless, T.M.; Brant, S.R.; Kwon, J.H. Peripheral blood micrornas distinguish active ulcerative colitis and crohn’s disease. Inflamm. Bowel Dis. 2011, 17, 241–250. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, T.; Jin, R.; Zhao, H.; Hu, J.; Feng, B.; Zang, L.; Zheng, M.; Wang, M. MicroRNA-301a promotes migration and invasion by targeting TGFBR2 in human colorectal cancer. J. Exp. Clin. Cancer Res. 2014, 33, 113. [Google Scholar] [CrossRef]
- He, C.; Shi, Y.; Wu, R.; Sun, M.; Fang, L.; Wu, W.; Liu, C.; Tang, M.; Li, Z.; Wang, P.; et al. miR-301a promotes intestinal mucosal inflammation through induction of IL-17A and TNF-α in IBD. Gut 2016, 65, 1938–1950. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, X.; Zhong, M.; Wang, Y.; Zou, L.; Wang, M.; Gong, X.; Wang, X.; Zhou, C.; Ma, X.; et al. Mir-301a suppression within fibroblasts limits the progression of fibrosis through the tsc1/mtor pathway. Mol. Ther. Nucleic Acids 2020, 21, 217–228. [Google Scholar] [CrossRef]
- Hu, H.; Zhang, Q.; Chen, W.; Wu, T.; Liu, S.; Li, X.; Luo, B.; Zhang, T.; Yan, G.; Lu, H.; et al. MicroRNA-301a promotes pancreatic cancer invasion and metastasis through the JAK/STAT3 signaling pathway by targeting SOCS5. Carcinogenesis 2020, 41, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Yan, F.; Deng, Q.; Li, F.; Lu, Z.; Liu, M.; Wang, L.; Conklin, D.J.; McCracken, J.; Srivastava, S.; et al. Modulation of tumorigenesis by the pro-inflammatory microRNA miR-301a in mouse models of lung cancer and colorectal cancer. Cell Discov. 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Yu, T.; Shi, Y.; Ma, C.; Yang, W.; Fang, L.; Sun, M.; Wu, W.; Xiao, F.; Guo, F.; et al. Microrna 301a promotes intestinal inflammation and colitis-associated cancer development by inhibiting btg1. Gastroenterology 2017, 152, 1434–1448.e15. [Google Scholar] [CrossRef]
- Butnariu, M.; Quispe, C.; Koirala, N.; Khadka, S.; Salgado-Castillo, C.M.; Akram, M.; Anum, R.; Yeskaliyeva, B.; Cruz-Martins, N.; Martorell, M.; et al. Bioactive effects of curcumin in human immunodeficiency virus infection along with the most effective isolation techniques and type of nanoformulations. Int. J. Nanomed. 2022, 17, 3619–3632. [Google Scholar] [CrossRef]
- Aalinkeel, R.; Kutscher, H.L.; Singh, A.; Cwiklinski, K.; Khechen, N.; Schwartz, S.A.; Prasad, P.N.; Mahajan, S.D. Neuroprotective effects of a biodegradable poly(lactic-co-glycolic acid)-ginsenoside Rg3 nanoformulation: A potential nanotherapy for Alzheimer’s disease. J. Drug Target. 2018, 26, 182–193. [Google Scholar] [CrossRef]
- Ding, D.; Zhu, Q. Recent advances of PLGA micro/nanoparticles for the delivery of biomacromolecular therapeutics. Mater. Sci. Eng. C Mater. Biol. Appl. 2018, 92, 1041–1060. [Google Scholar] [CrossRef]
- Yuan, Z.; Ye, Y.; Gao, F.; Yuan, H.; Lan, M.; Lou, K.; Wang, W. Chitosan-graft-β-cyclodextrin nanoparticles as a carrier for controlled drug release. Int. J. Pharm. 2013, 446, 191–198. [Google Scholar] [CrossRef]
- He, M.; Zhong, C.; Hu, H.; Jin, Y.; Chen, Y.; Lou, K.; Gao, F. Cyclodextrin/chitosan nanoparticles for oral ovalbumin delivery: Preparation, characterization and intestinal mucosal immunity in mice. Asian J. Pharm. Sci. 2019, 14, 193–203. [Google Scholar] [CrossRef]
- Pavinatto, F.J.; Caseli, L.; Oliveira, O.N. Chitosan in nanostructured thin films. Biomacromolecules 2010, 11, 1897–1908. [Google Scholar] [CrossRef]
- Pramanik, S.; Sali, V. Connecting the dots in drug delivery: A tour d’horizon of chitosan-based nanocarriers system. Int. J. Biol. Macromol. 2021, 169, 103–121. [Google Scholar] [CrossRef]
- Proulx, J.; Joshi, C.; Vijayaraghavalu, S.; Saraswathy, M.; Labhasetwar, V.; Ghorpade, A.; Borgmann, K. Arginine-modified polymers facilitate poly (lactide-co-glycolide)-based nanoparticle gene delivery to primary human astrocytes. Int. J. Nanomed. 2020, 15, 3639–3647. [Google Scholar] [CrossRef]
- Kievit, F.M.; Veiseh, O.; Bhattarai, N.; Fang, C.; Gunn, J.W.; Lee, D.; Ellenbogen, R.G.; Olson, J.M.; Zhang, M. Pei–peg–chitosan-copolymer-coated iron oxide nanoparticles for safe gene delivery: Synthesis, complexation, and transfection. Adv. Funct. Mater. 2009, 19, 2244–2251. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, P.; Su, W.; Wang, S.; Liao, Z.; Niu, R.; Chang, J. PLGA/polymeric liposome for targeted drug and gene co-delivery. Biomaterials 2010, 31, 8741–8748. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; van Dijk, T.; Vepris, O.; Li, T.M.W.Y.; Schomann, T.; Baldazzi, F.; Kurita, R.; Nakamura, Y.; Grosveld, F.; Philipsen, S.; et al. Plga-nanoparticles for intracellular delivery of the crispr-complex to elevate fetal globin expression in erythroid cells. Biomaterials 2021, 268, 120580. [Google Scholar] [CrossRef]
- Alex, S.M.; Rekha, M.R.; Sharma, C.P. Spermine grafted galactosylated chitosan for improved nanoparticle mediated gene delivery. Int. J. Pharm. 2011, 410, 125–137. [Google Scholar] [CrossRef]
- Boca, S.; Gulei, D.; Zimta, A.-A.; Onaciu, A.; Magdo, L.; Tigu, A.B.; Ionescu, C.; Irimie, A.; Buiga, R.; Berindan-Neagoe, I. Nanoscale delivery systems for micrornas in cancer therapy. Cell. Mol. Life Sci. 2020, 77, 1059–1086. [Google Scholar] [CrossRef]
- Lv, Y.; Ren, M.; Yao, M.; Zou, J.; Fang, S.; Wang, Y.; Lan, M.; Zhao, Y.; Gao, F. Colon-specific delivery of methotrexate using hyaluronic acid modified pH-responsive nanocarrier for the therapy of colitis in mice. Int. J. Pharm. 2023, 635, 122741. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Ansari, M.M.; Mishra, R.K.; Kumar, A.; Vyawahare, A.; Verma, R.K.; Raza, S.S.; Khan, R. Enteric-coated gelatin nanoparticles mediated oral delivery of 5-aminosalicylic acid alleviates severity of DSS-induced ulcerative colitis. Mater. Sci. Eng. C Mater. Biol. Appl. 2021, 119, 111582. [Google Scholar] [CrossRef]
- Beloqui, A.; Coco, R.; Memvanga, P.B.; Ucakar, B.; des Rieux, A.; Préat, V. pH-sensitive nanoparticles for colonic delivery of curcumin in inflammatory bowel disease. Int. J. Pharm. 2014, 473, 203–212. [Google Scholar] [CrossRef]
- Xu, Y.-W.; Xing, R.-X.; Zhang, W.-H.; Li, L.; Wu, Y.; Hu, J.; Wang, C.; Luo, Q.-L.; Shen, J.-L.; Chen, X. Toxoplasma rop16i/iii ameliorated inflammatory bowel diseases via inducing m2 phenotype of macrophages. World J. Gastroenterol. 2019, 25, 6634–6652. [Google Scholar] [CrossRef]
- Ibrahim, S.; Zhu, X.; Luo, X.; Feng, Y.; Wang, J. Pik3r3 regulates zo-1 expression through the nf-kb pathway in inflammatory bowel disease. Int. Immunopharmacol. 2020, 85, 106610. [Google Scholar] [CrossRef] [PubMed]
- Stamenkovic, I.; Aruffo, A.; Amiot, M.; Seed, B. The hematopoietic and epithelial forms of CD44 are distinct polypeptides with different adhesion potentials for hyaluronate-bearing cells. EMBO J. 1991, 10, 343–348. [Google Scholar] [CrossRef]
- Rosenberg, W.M.C.; Jackson, D.G.; Trowell, J.M.; Bell, J.I.; Rosenberg, W.M.C.; Prince, C.; Chapman, R.W.; Trowell, J.M.; Jewel, D.P.; Kaklamanis, L.; et al. Increased expression of CD44v6 and CD44v3 in ulcerative colitis but not colonic Crohn’s disease. Lancet 1995, 345, 1205–1209. [Google Scholar] [CrossRef] [PubMed]
- Camacho, F.I.; Muñoz, C.; Sánchez-Verde, L.; Sáez, A.I.; Alcántara, M.; Rodríguez, R. CD44v6 expression in inflammatory bowel disease is associated with activity detected by endoscopy and pathological features. Histopathology 1999, 35, 144–149. [Google Scholar] [CrossRef]
- Farkas, S.; Hornung, M.; Sattler, C.; Anthuber, M.; Gunthert, U.; Herfarth, H.; Schlitt, H.J.; Geissler, E.K.; Wittig, B.M. Short-term treatment with anti-CD44v7 antibody, but not CD44v4, restores the gut mucosa in established chronic dextran sulphate sodium (DSS)-induced colitis in mice. Clin. Exp. Immunol. 2005, 142, 260–267. [Google Scholar] [CrossRef]
- Vasvani, S.; Kulkarni, P.; Rawtani, D. Hyaluronic acid: A review on its biology, aspects of drug delivery, route of administrations and a special emphasis on its approved marketed products and recent clinical studies. Int. J. Biol. Macromol. 2020, 151, 1012–1029. [Google Scholar] [CrossRef]
- Zhang, H.; Huang, S.; Yang, X.; Zhai, G. Current research on hyaluronic acid-drug bioconjugates. Eur. J. Med. Chem. 2014, 86, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Jiang, D.; Noble, P.W. Hyaluronan as a therapeutic target in human diseases. Adv. Drug Deliv. Rev. 2016, 97, 186–203. [Google Scholar] [CrossRef]
- He, M.; Huang, L.; Hou, X.; Zhong, C.; Bachir, Z.A.; Lan, M.; Chen, R.; Gao, F. Efficient ovalbumin delivery using a novel multifunctional micellar platform for targeted melanoma immunotherapy. Int. J. Pharm. 2019, 560, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Neudecker, V.; Haneklaus, M.; Jensen, O.; Khailova, L.; Masterson, J.C.; Tye, H.; Biette, K.; Jedlicka, P.; Brodsky, K.S.; Gerich, M.E.; et al. Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. J. Exp. Med. 2017, 214, 1737–1752. [Google Scholar] [CrossRef]
- Na, Y.R.; Stakenborg, M.; Seok, S.H.; Matteoli, G. Macrophages in intestinal inflammation and resolution: A potential therapeutic target in IBD. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.; Zhang, L.-Y.; He, W.; Li, W.-Y.; Zeng, Z.; Qian, B.; Wang, C.; Song, J.-L. Sinapic acid alleviated inflammation-induced intestinal epithelial barrier dysfunction in lipopolysaccharide- (lps-) treated caco-2 cells. Mediat. Inflamm. 2021, 2021, 5514075. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Zhang, Z.; Viennois, E.; Kang, Y.; Zhang, M.; Han, M.K.; Chen, J.; Merlin, D. Combination therapy for ulcerative colitis: Orally targeted nanoparticles prevent mucosal damage and relieve inflammation. Theranostics 2016, 6, 2250–2266. [Google Scholar] [CrossRef]
- Huang, L.; Liu, Y.; Wang, L.; Chen, R.; Ge, W.; Lin, Z.; Zhang, Y.; Liu, S.; Shan, Y.; Lin, Q.; et al. Down-regulation of miR-301a suppresses pro-inflammatory cytokines in Toll-like receptor-triggered macrophages. Immunology 2013, 140, 314–322. [Google Scholar] [CrossRef]
- Lautenschläger, C.; Schmidt, C.; Fischer, D.; Stallmach, A. Drug delivery strategies in the therapy of inflammatory bowel disease. Adv. Drug Deliv. Rev. 2014, 71, 58–76. [Google Scholar] [CrossRef]
- Lee, A.; De Mei, C.; Fereira, M.; Marotta, R.; Yoon, H.Y.; Kim, K.; Kwon, I.C.; Decuzzi, P. Dexamethasone-loaded polymeric nanoconstructs for monitoring and treating inflammatory bowel disease. Theranostics 2017, 7, 3653–3666. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Yang, C.; Yang, M.; Ma, Y.; Zhang, Y.; Zhang, Y.; Liu, C.; Xu, Q.; Tu, K.; Zhang, M. All-in-one theranostic nano-platform based on polymer nanoparticles for bret/fret-initiated bioluminescence imaging and synergistically anti-inflammatory therapy for ulcerative colitis. J. Nanobiotechnol. 2022, 20, 99. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanocarrier | PLGA:ES:CS:HA (mg/mg/mg/mg) | Particle Size (nm) | Zeta Potential (mV) | PDI |
|---|---|---|---|---|
| PLGA NP | 1/0/0/0 | 237.5 ± 1.4 | −6.1 ± 0.2 | 0.22 |
| eP NP | 2/1/0/0 | 272.5 ± 1.6 | −34.6 ± 0.1 | 0.22 |
| CeP NP | 2/1/0.1/0 | 266.2 ± 11.2 | −31.7 ± 0.7 | 0.33 |
| 2/1/0.2/0 | 256.1 ± 5.4 | −23.9 ± 0.7 | 0.28 | |
| 2/1/0.4/0 | 232.9 ± 2.7 | −10.9 ± 0.3 | 0.27 | |
| 2/1/0.6/0 | 212.7 ± 2.4 | −3.5 ± 0.2 | 0.27 | |
| 2/1/1/0 | __a | N/A | N/A | |
| HCeP NP | 2/1/0.4/0.08 | 237.2 ± 1.6 | −15.6 ± 0.4 | 0.24 |
| 2/1/0.4/0.2 | 261.1 ± 6.3 | −20.0 ± 0.2 | 0.26 | |
| 2/1/0.4/0.4 | 267.5 ± 8.9 | −17.5 ± 0.5 | 0.35 | |
| 2/1/0.4/0.8 | 268.3 ± 6.8 | −13.8 ± 1.3 | 0.39 |
| Group | Morris Score |
|---|---|
| Healthy | 0 |
| DSS | 6.8 ± 0.4 |
| DSS + Free anti-miR | 6.5 ± 0.3 |
| DSS + anti-miR@HCeP NPs | 1.3 ± 0.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Yao, M.; Zou, J.; Ding, W.; Sun, M.; Zhuge, Y.; Gao, F. pH-Sensitive Nanoparticles for Colonic Delivery Anti-miR-301a in Mouse Models of Inflammatory Bowel Diseases. Nanomaterials 2023, 13, 2797. https://doi.org/10.3390/nano13202797
Wang J, Yao M, Zou J, Ding W, Sun M, Zhuge Y, Gao F. pH-Sensitive Nanoparticles for Colonic Delivery Anti-miR-301a in Mouse Models of Inflammatory Bowel Diseases. Nanomaterials. 2023; 13(20):2797. https://doi.org/10.3390/nano13202797
Chicago/Turabian StyleWang, Junshan, Min Yao, Jiafeng Zou, Wenxing Ding, Mingyue Sun, Ying Zhuge, and Feng Gao. 2023. "pH-Sensitive Nanoparticles for Colonic Delivery Anti-miR-301a in Mouse Models of Inflammatory Bowel Diseases" Nanomaterials 13, no. 20: 2797. https://doi.org/10.3390/nano13202797
APA StyleWang, J., Yao, M., Zou, J., Ding, W., Sun, M., Zhuge, Y., & Gao, F. (2023). pH-Sensitive Nanoparticles for Colonic Delivery Anti-miR-301a in Mouse Models of Inflammatory Bowel Diseases. Nanomaterials, 13(20), 2797. https://doi.org/10.3390/nano13202797