
Identification and In Silico Characterization of a Conserved Peptide on Influenza Hemagglutinin Protein: A New Potential Antigen for Universal Influenza Vaccine Development

 , ,
, ,  ,
,
Abstract
:1. Introduction
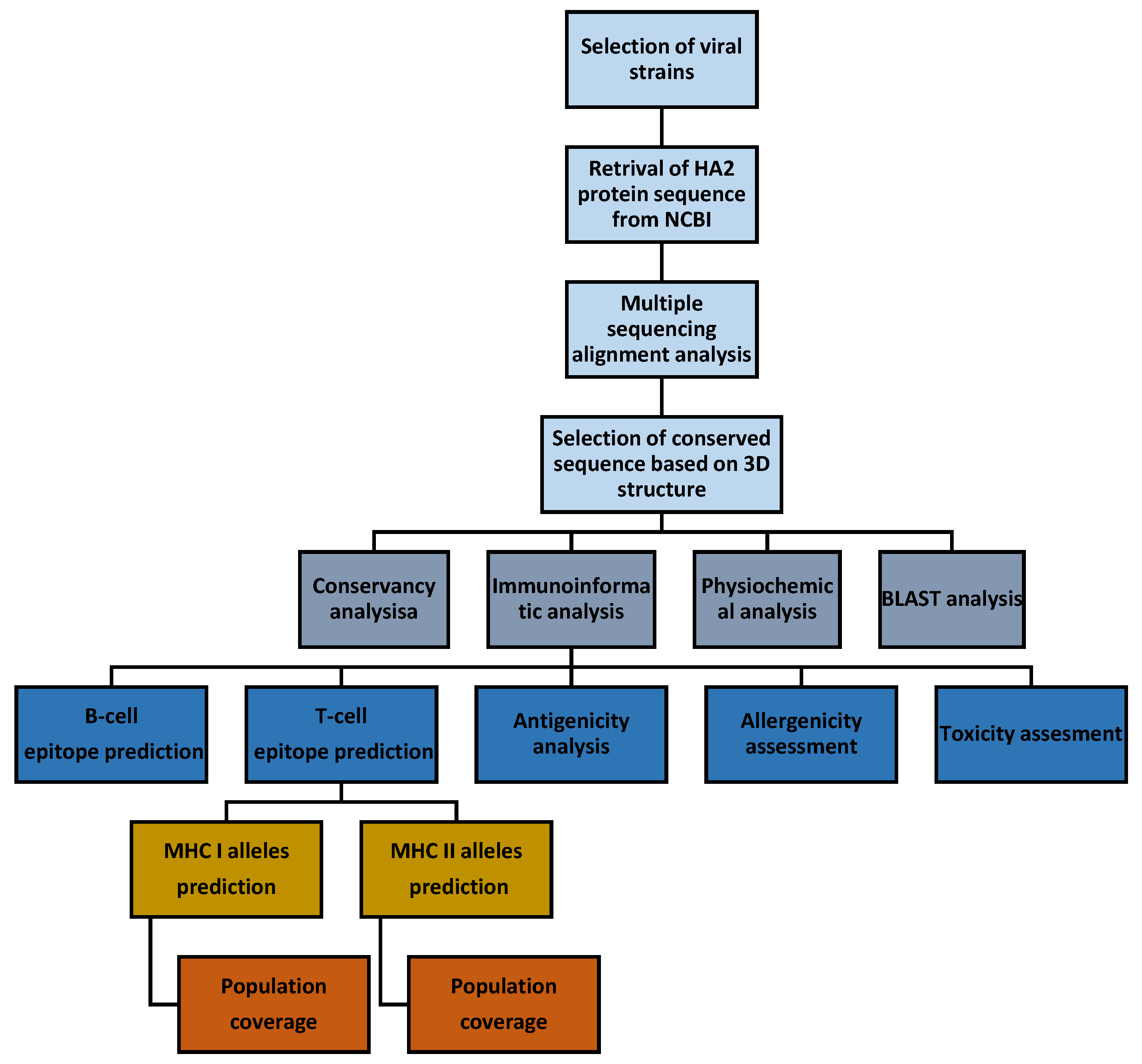
2. Materials and Methods
2.1. Multiple Sequence Alignment, Structure, and Conservancy Analysis
2.2. Identification of Linear B-Cell Epitopes
2.3. Identification of T-Cell Epitopes and Population Coverage Analysis
2.4. Cluster Analysis of the MHC-Restricted Alleles
2.5. Molecular Docking Analysis
2.6. Antigenicity, Allergenicity, and Toxicity Assessment
2.7. Assessment of Physicochemical Properties
3. Results
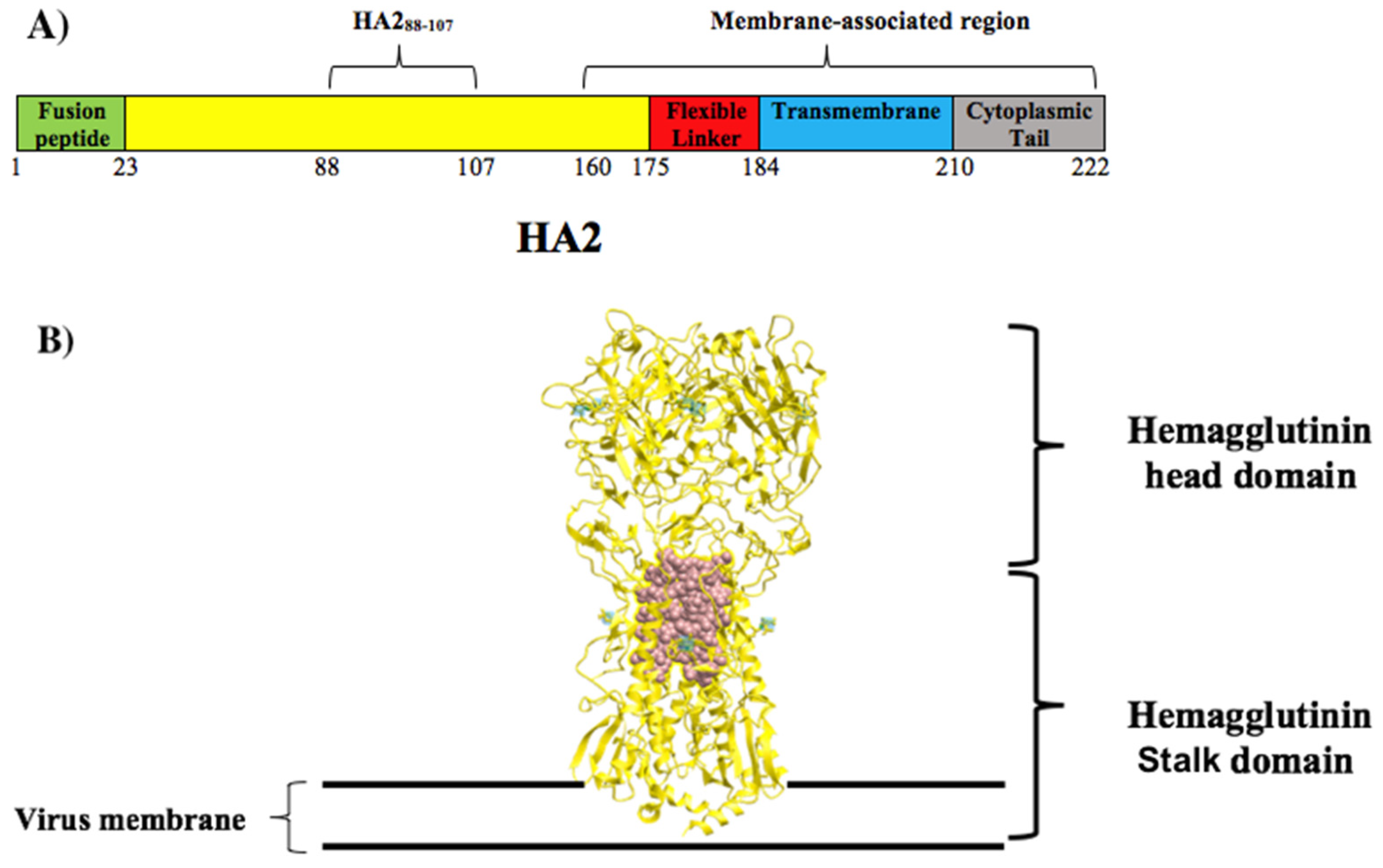
3.1. Selection of Conserved Peptide
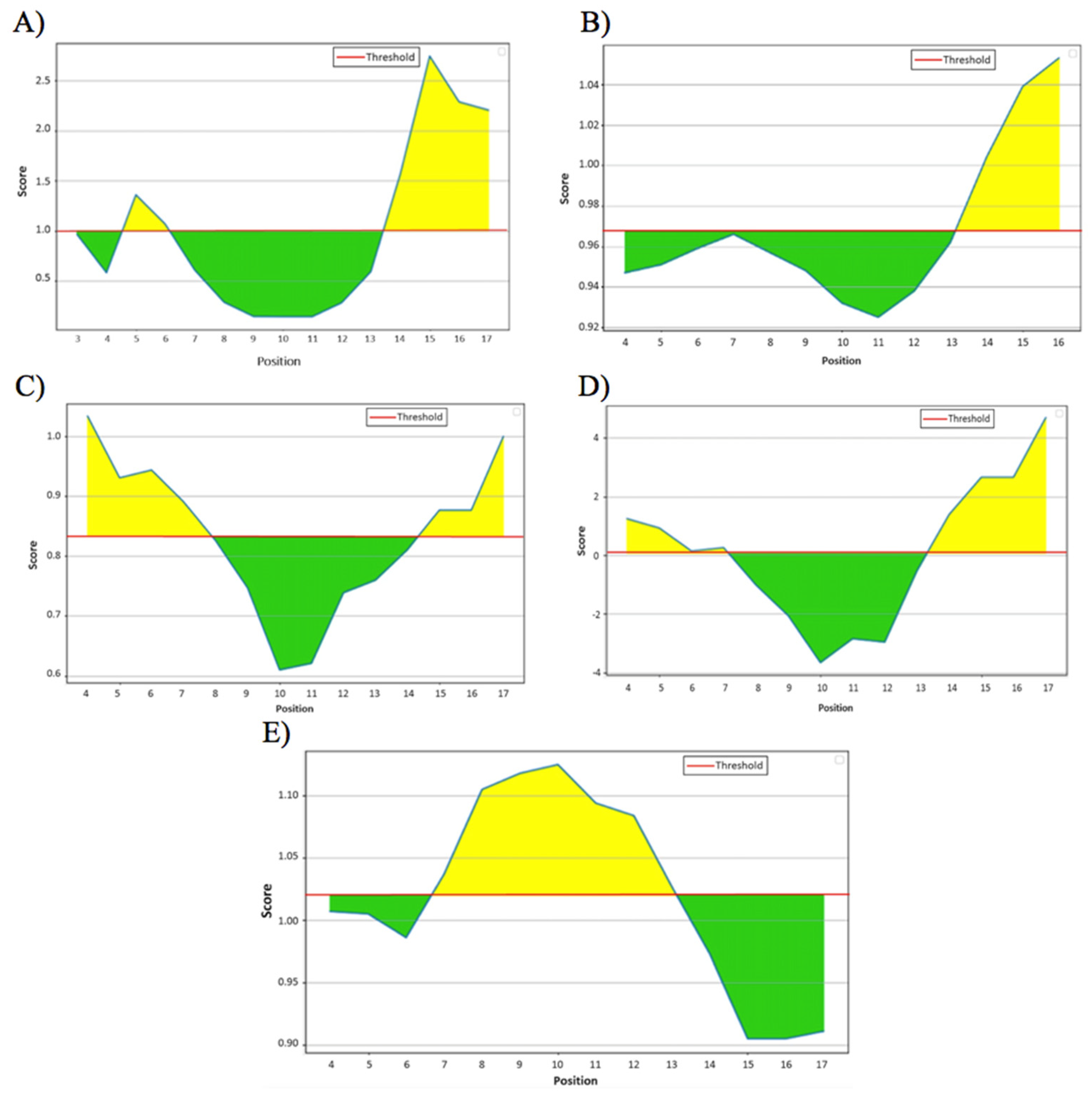
3.2. Linear B-Cell Epitopes of the HA288–107 Peptide
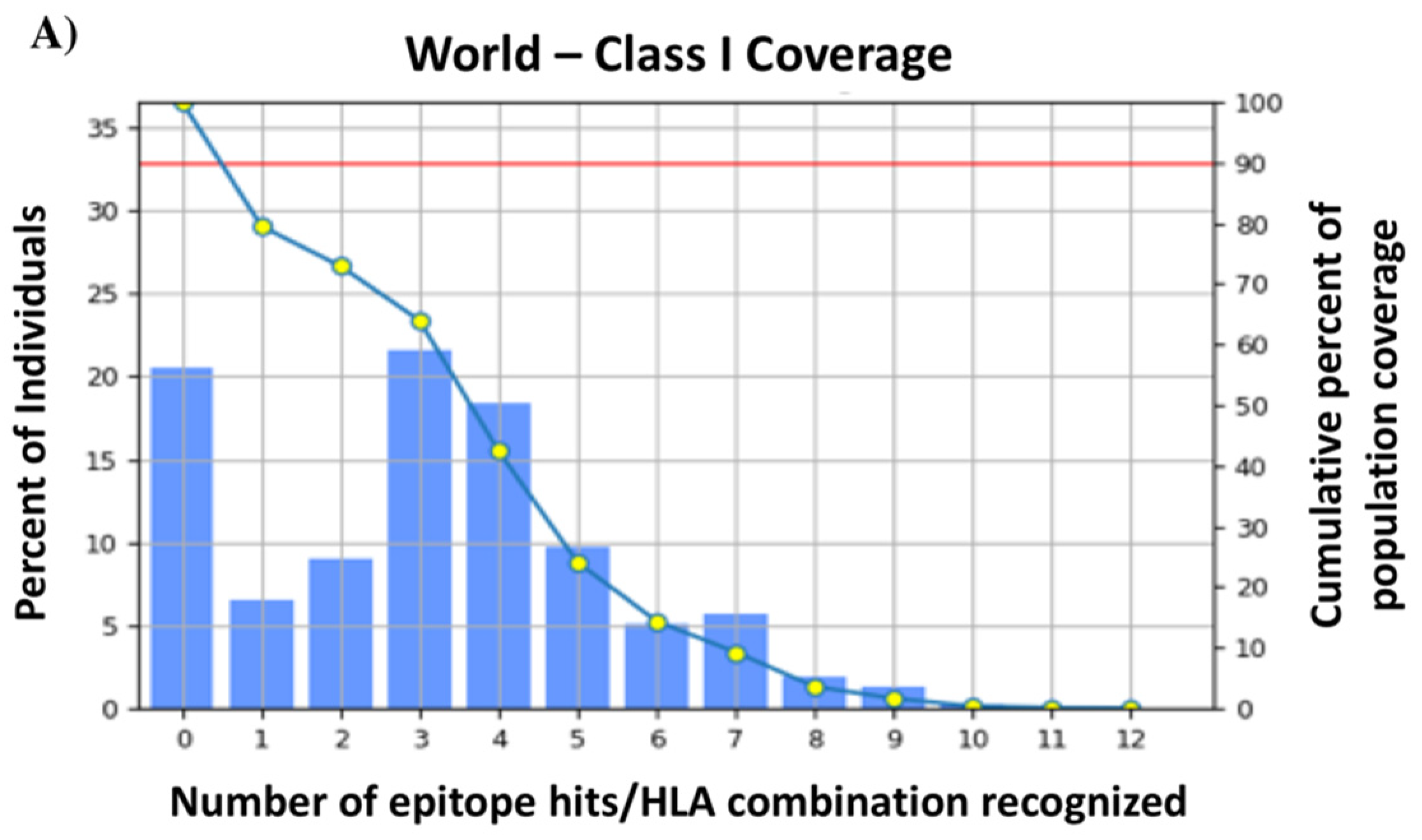
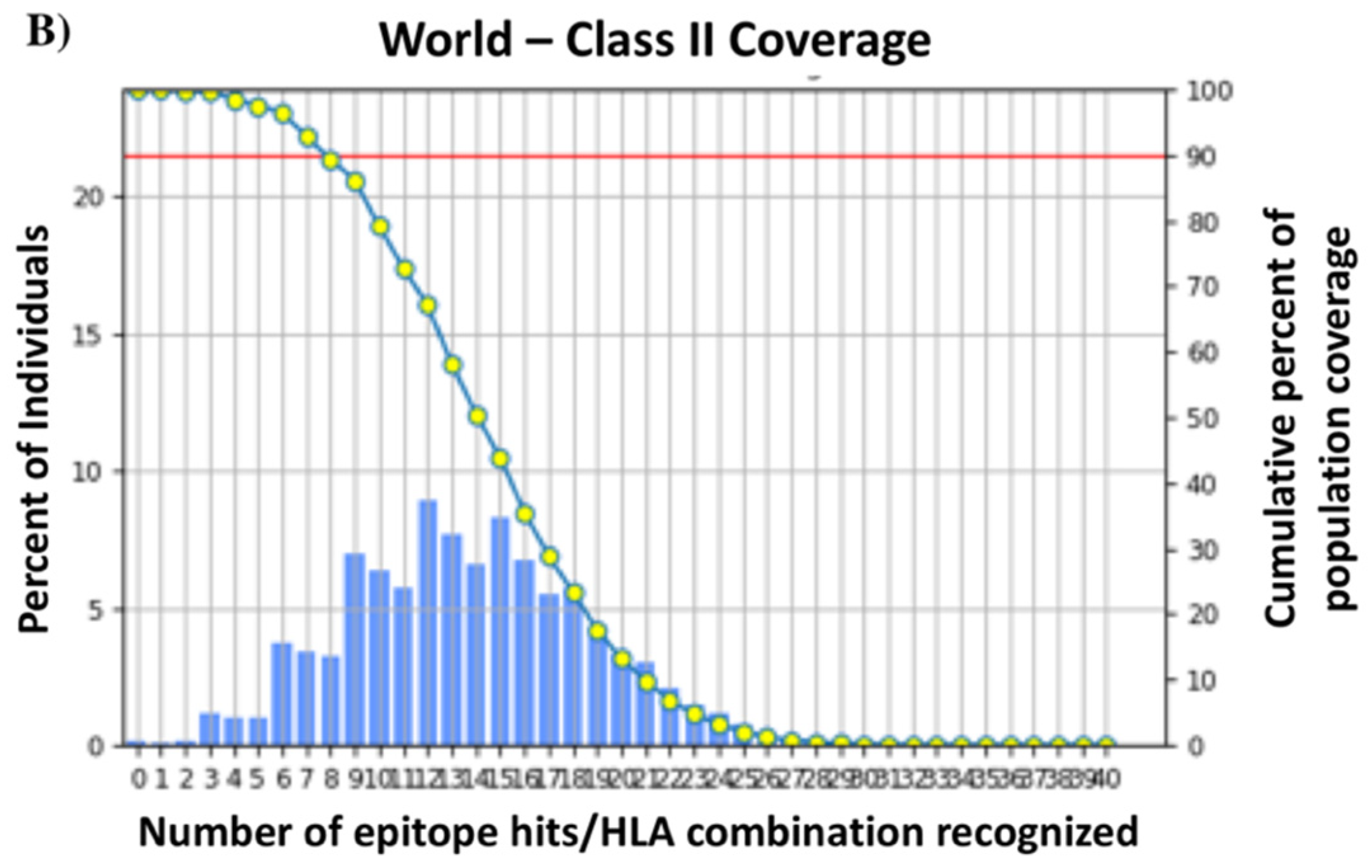
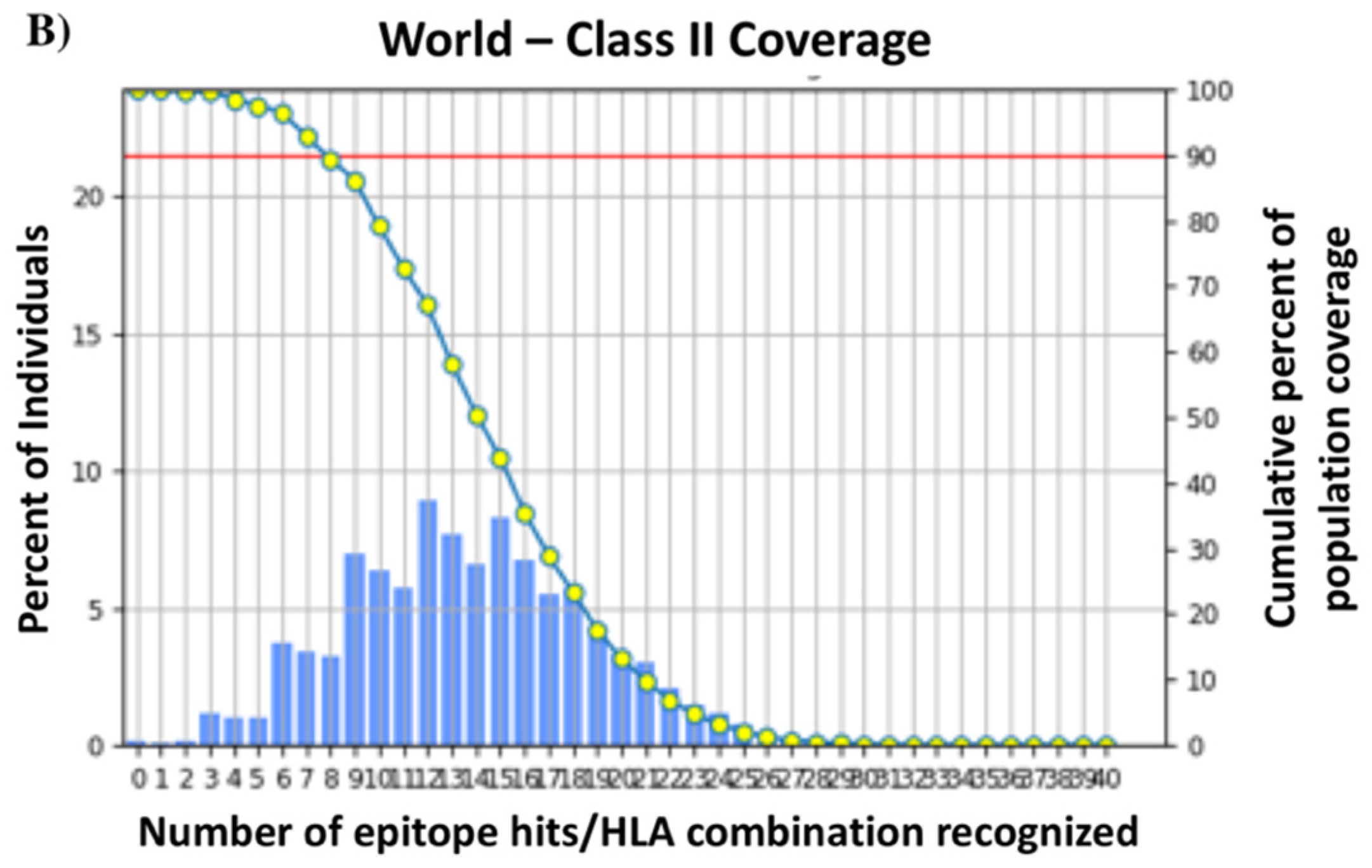
3.3. T-Cell Epitopes of the HA288–107 Peptide and Human Population Coverage
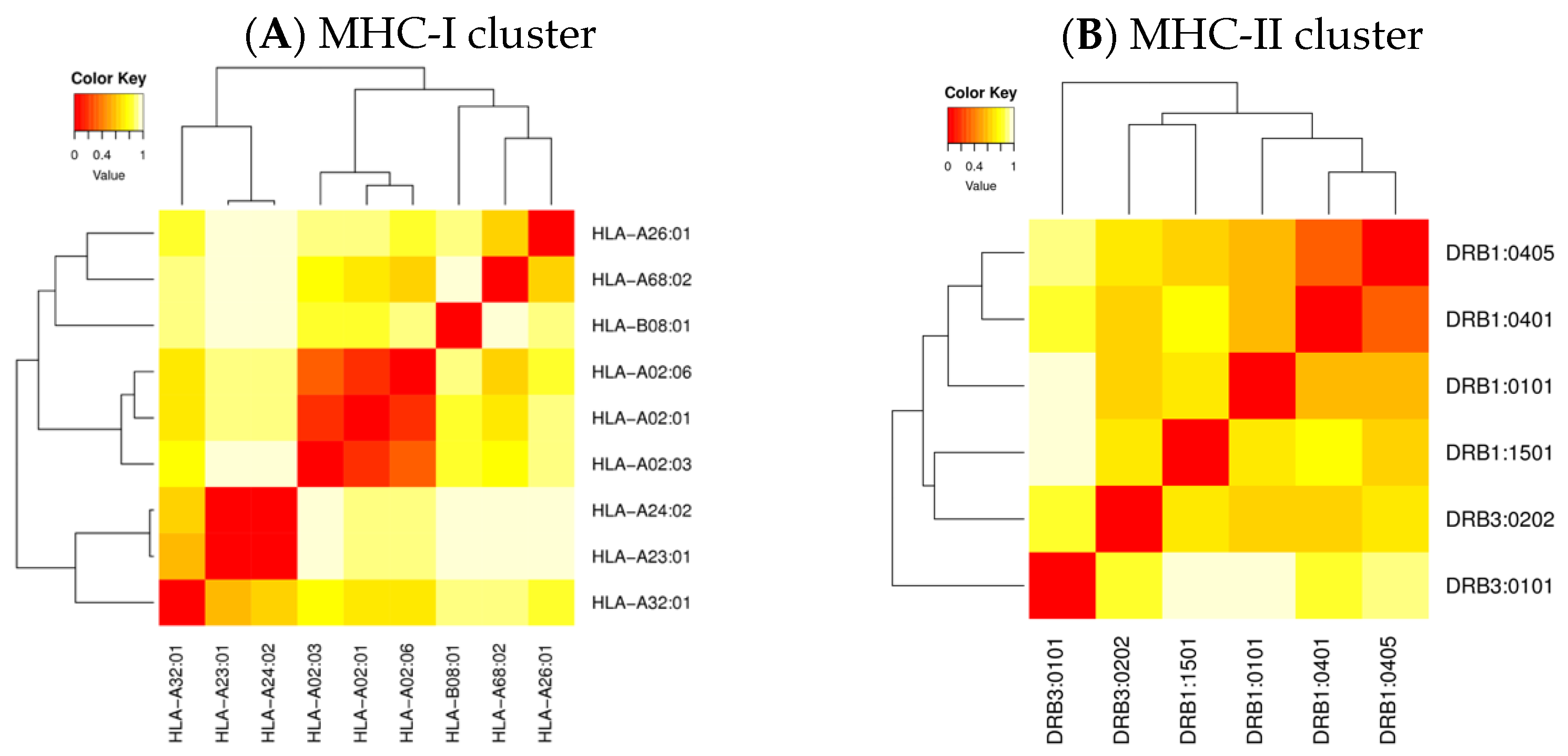
3.4. Cluster Analysis of the MHC-Restricted Alleles
3.5. Molecular Docking
3.6. Antigenicity and Safety of the HA288–107 Peptide
3.7. Physicochemical Properties of the HA288–107 Peptide
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. World Health Organization Factsheet, Influenza (Seasonal); World Health Organization: Geneva, Switzerland, 2018.
- Schild, G.C. The influenza virus: Antigenic composition and immune response. Postgrad. Med. J. 1979, 55, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC); National Center for Immunization and Respiratory Diseases (NCIRD). 2019. Available online: https://www.cdc.gov/flu/avianflu/influenza-a-virus-subtypes.htm (accessed on 1 June 2023).
- Peacock, T.P.; James, J.; Sealy, J.E.; Iqbal, M. A Global Perspective on H9N2 Avian Influenza Virus. Viruses 2019, 11, 620. [Google Scholar] [CrossRef]
- Wilson, I.A.; Skehel, J.J.; Wiley, D.C. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature 1981, 289, 366–373. [Google Scholar] [CrossRef]
- Xu, R.; Ekiert, D.C.; Krause, J.C.; Hai, R.; Crowe, J.E.; Wilson, I.A. Structural basis of preexisting immunity to the 2009 H1N1 pandemic influenza virus. Science 2010, 328, 357–360. Available online: https://science.sciencemag.org/content/328/5976/357 (accessed on 1 June 2023). [CrossRef] [PubMed]
- Subbarao, K.; Murphy, B.R.; Fauci, A.S. Development of Effective Vaccines against Pandemic Influenza. Immunity 2006, 24, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Skehel, J. An overview of influenza haemagglutinin and neuraminidase. Biologicals 2009, 37, 177–178. [Google Scholar] [CrossRef]
- Hayati, M.; Biller, P.; Colijn, C. Predicting the short-term success of human influenza virus variants with machine learning. bioRxiv 2020, 287, 20200319. [Google Scholar] [CrossRef] [PubMed]
- Blyth, C.C.; Macartney, K.K.; McRae, J.; Clark, J.E.; Marshall, H.S.; Buttery, J.; Francis, J.R.; Kotsimbos, T.; Kelly, P.M.; Cheng, A.C.; et al. Influenza Epidemiology, Vaccine Coverage and Vaccine Effectiveness in Children Admitted to Sentinel Australian Hospitals in 2017: Results from the PAEDS-FluCAN Collaboration. Clin. Infect. Dis. 2018, 68, ciy597. [Google Scholar] [CrossRef]
- Wiley, S.K. The 2009 influenza pandemic: 10 years later. Nursing 2020, 50, 17–20. Available online: https://journals.lww.com/nursing/Fulltext/2020/01000/The_2009_influenza_pandemic__10_years_later.6.aspx (accessed on 1 June 2023). [CrossRef]
- Erbelding, E.J.; Post, D.J.; Stemmy, E.J.; Roberts, P.C.; Augustine, A.D.; Ferguson, S.; Paules, C.I.; Graham, B.S.; Fauci, A.S. A Universal Influenza Vaccine: The Strategic Plan for the National Institute of Allergy and Infectious Diseases. J. Infect. Dis. 2018, 218, 347–354. [Google Scholar] [CrossRef]
- Sah, P.; Alfaro-Murillo, J.A.; Fitzpatrick, M.C.; Neuzil, K.M.; Meyers, L.A.; Singer, B.H.; Galvani, A.P. Future epidemiological and economic impacts of universal influenza vaccines. Proc. Natl. Acad. Sci. USA 2019, 116, 20786–20792. [Google Scholar] [CrossRef]
- Gori, A.; Longhi, R.; Peri, C.; Colombo, G. Peptides for immunological purposes: Design, strategies and applications. Amino Acids 2013, 45, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Moxon, R.; Reche, P.A.; Rappuoli, R. Editorial: Reverse Vaccinology. Front. Immunol. 2019, 10, 2776. [Google Scholar] [CrossRef]
- Wu, N.C.; Wilson, I.A. A Perspective on the Structural and Functional Constraints for Immune Evasion: Insights from Influenza Virus. J. Mol. Biol. 2017, 429, 2694–2709. [Google Scholar] [CrossRef]
- Bui, H.-H.; Sidney, J.; Li, W.; Fusseder, N.; Sette, A. Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinform. 2007, 8, 361. [Google Scholar] [CrossRef]
- Emini, E.A.; Hughes, J.V.; Perlow, D.S.; Boger, J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J. Virol. 1985, 55, 836–839. [Google Scholar] [CrossRef]
- Karplus, P.A.; Schulz, G.E. Prediction of chain flexibility in proteins. Sci. Nat. 1985, 72, 212–213. [Google Scholar] [CrossRef]
- Chou, P.Y.; Fasman, G.D. Prediction of the Secondary Structure of Proteins from their Amino Acid Sequence. Adv. Enzymol. Relat. Areas Mol. Biol. 1978, 47, 45–148. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.M.R.; Guo, D.; Hodges, R.S. New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: Correlation of predicted surface residues with antigenicity and x-ray-derived accessible sites. Biochemistry 1986, 25, 5425–5432. [Google Scholar] [CrossRef] [PubMed]
- Kolaskar, A.S.; Tongaonkar, P.C. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990, 276, 172–174. [Google Scholar] [CrossRef]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef]
- Sharma, S.; Kumari, V.; Kumbhar, B.V.; Mukherjee, A.; Pandey, R.; Kondabagil, K. Immunoinformatics approach for a novel multi-epitope subunit vaccine design against various subtypes of Influenza A virus. Immunobiology 2021, 226, 152053. [Google Scholar] [CrossRef]
- Li, L.; Dong, M.; Wang, X.G. The implication and significance of beta 2 microglobulin: A conservative multifunctional regulator. Chin. Med. J. 2016, 129, 448–455. [Google Scholar] [CrossRef]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v.2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, I.; Naneva, L.; Doytchinova, I.; Bangov, I. AllergenFP: Allergenicity prediction by descriptor fingerprints. Bioinformatics 2014, 30, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS ONE 2013, 8, 9. [Google Scholar] [CrossRef]
- Harnkit, N.; Khongsonthi, T.; Masuwan, N.; Prasartkul, P.; Noikaew, T.; Chumnanpuen, P. Virtual Screening for SARS-CoV-2 Main Protease Inhibitory Peptides from the Putative Hydrolyzed Peptidome of Rice Bran. Antibiotics 2022, 11, 1318. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Proteomics Protocols Handbook; Humana Press: Totowa, NJ, USA, 2005. [Google Scholar]
- Lear, S.; Cobb, S.L. Pep-Calc.com: A set of web utilities for the calculation of peptide and peptoid properties and automatic mass spectral peak assignment. J. Comput. Aided. Mol. Des. 2016, 30, 271–277. [Google Scholar] [CrossRef]
- Skehel, J.J.; Wiley, D.C. Receptor Binding and Membrane Fusion in Virus Entry: The Influenza Hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef]
- Thomsen, M.; Lundegaard, C.; Buus, S.; Lund, O.; Nielsen, M. MHCcluster, a method for functional clustering of MHC molecules. Immunogenetics 2013, 65, 655–665. [Google Scholar] [CrossRef]
- Kelsey, C.; Mhatre, V.M.; Ho, J.-A.L.; Martin, R.; Buchwald, S.L.; Ho, M.V.; Martin, K.C.; Craik, J.-A.L.; Manuscript, C.; Kantrowitz, A. NIH Public Access. Bone 2008, 23, 1–7. [Google Scholar]
- Knutson, K.L.; Schiffman, K.; Disis, M.L. Immunization with a HER-2/neu helper peptide vaccine generates HER-2/neu CD8 T-cell immunity in cancer patients. J. Clin. Investig. 2001, 107, 477–484. [Google Scholar] [CrossRef] [PubMed]
- López, J.A.; Weilenman, C.; Audran, R.; Roggero, M.A.; Bonelo, A.; Tiercy, J.; Spertini, F.; Corradin, G. A synthetic malaria vaccine elicits a potent CD8+ and CD4+ T lymphocyte immune response in humans. Implications for vaccination strategies. Eur. J. Immunol. 2001, 31, 1989–1998. [Google Scholar] [CrossRef] [PubMed]
- Nabel, G.J.; Fauci, A.S. Induction of unnatural immunity: Prospects for a broadly protective universal influenza vaccine. Nat. Med. 2010, 16, 1389–1391. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.G.; Govorkova, E.A. Continuing challenges in influenza. Ann. N. Y. Acad. Sci. 2014, 1323, 115–139. [Google Scholar] [CrossRef]
- Widdowson, M.-A.; Bresee, J.S.; Jernigan, D.B. The Global Threat of Animal Influenza Viruses of Zoonotic Concern: Then and Now. J. Infect. Dis. 2017, 216 (Suppl. 4), S493–S498. Available online: https://pubmed.ncbi.nlm.nih.gov/28934463 (accessed on 1 June 2023). [CrossRef]
- Stepanova, L.A.; Sergeeva, M.V.; Shuklina, M.A.; Shaldzhyan, A.A.; Potapchuk, M.V.; Korotkov, A.V.; Tsybalova, L.M. A fusion protein based on the second subunit of hemagglutinin of influenza A/H2N2 viruses provides cross immunity. Acta Nat. 2016, 8, 116–126. [Google Scholar] [CrossRef]
- Maginnis, M.S. Virus–Receptor Interactions: The Key to Cellular Invasion. J. Mol. Biol. 2018, 430, 2590–2611. [Google Scholar] [CrossRef]
- Eickhoff, C.S.; Terry, F.E.; Peng, L.; Meza, K.A.; Sakala, I.G.; Van Aartsen, D.; Moise, L.; Martin, W.D.; Schriewer, J.; Buller, R.M.; et al. Highly conserved influenza T cell epitopes induce broadly protective immunity. Vaccine 2019, 37, 5371–5381. [Google Scholar] [CrossRef]
- Sette, A.; Rappuoli, R. Reverse Vaccinology: Developing Vaccines in the Era of Genomics. Immunity 2010, 33, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, S.; Goraya, M.U.; Maarouf, M.; Huang, S.; Chen, J.-L. Host Immune Response to Influenza A Virus Infection. Front. Immunol. 2018, 9, 320. [Google Scholar] [CrossRef] [PubMed]
- Paul, W.E. Fundamental Immunology Wolters Kluwer Health; Fundamental Immunology. 2012. Available online: https://books.google.com.my/books?id=x0gpapT-Y2kC (accessed on 1 June 2023).
- Jespersen, M.C.; Mahajan, S.; Peters, B.; Nielsen, M.; Marcatili, P. Antibody Specific B-Cell Epitope Predictions: Leveraging Information From Antibody-Antigen Protein Complexes. Front. Immunol. 2019, 10, 298. Available online: https://www.frontiersin.org/article/10.3389/fimmu.2019.00298 (accessed on 1 June 2023). [CrossRef]
- Sanchez-Trincado, J.L.; Gomez-Perosanz, M.; Reche, P.A. Fundamentals and Methods for T- and B-Cell Epitope Prediction. J. Immunol. Res. 2017, 2017, 2680160. [Google Scholar] [CrossRef]
- Gori, A.; Peri, C.; Quilici, G.; Nithichanon, A.; Gaudesi, D.; Longhi, R.; Gourlay, L.; Bolognesi, M.; Lertmemongkolchai, G.; Musco, G.; et al. Flexible vs Rigid Epitope Conformations for Diagnostic- and Vaccine-Oriented Applications: Novel Insights from the Burkholderia pseudomallei BPSL2765 Pal3 Epitope. ACS Infect. Dis. 2016, 2, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Pellequer, J.-L.; Westhof, E.; Van Regenmortel, M.H. Correlation between the location of antigenic sites and the prediction of turns in proteins. Immunol. Lett. 1993, 36, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Malonis, R.J.; Lai, J.R.; Vergnolle, O. Peptide-Based Vaccines: Current Progress and Future Challenges. Chem Rev. 2020, 120, 3210–3229. [Google Scholar] [CrossRef]
- Igietseme, J.U.; Eko, F.O.; He, Q.; Black, C.M. Antibody regulation of T-cell immunity: Implications for vaccine strategies against intracellular pathogens. Expert Rev. Vaccines 2004, 3, 23–34. [Google Scholar] [CrossRef]
- Bacchetta, R.; Gregori, S.; Roncarolo, M.-G. CD4+ regulatory T cells: Mechanisms of induction and effector function. Autoimmun. Rev. 2005, 4, 491–496. [Google Scholar] [CrossRef]
- Patronov, A.; Doytchinova, I. T-cell epitope vaccine design by immunoinformatics. Open Biol. 2013, 3, 120139. Available online: http://www.ncbi.nlm.nih.gov/pubmed/23303307%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC3603454 (accessed on 1 June 2023). [CrossRef]
- Knapp, B.; van der Merwe, P.A.; Dushek, O.; Deane, C.M. MHC binding affects the dynamics of different T-cell receptors in different ways. PLoS Comput. Biol. 2019, 15, e1007338. [Google Scholar] [CrossRef]
- McMichael, A.J.; Gotch, F.M.; Noble, G.R.; Beare, P.A.S. Cytotoxic T-Cell Immunity to Influenza. N. Engl. J. Med. 1983, 309, 13–17. [Google Scholar] [CrossRef]
- Hoft, D.F.; Babusis, E.; Worku, S.; Spencer, C.T.; Lottenbach, K.; Truscott, S.M.; Abate, G.; Sakala, I.G.; Edwards, K.M.; Creech, C.B.; et al. Live and Inactivated Influenza Vaccines Induce Similar Humoral Responses, but Only Live Vaccines Induce Diverse T-Cell Responses in Young Children. J. Infect. Dis. 2011, 204, 845–853. [Google Scholar] [CrossRef]
- Gustiananda, M. Immunoinformatics analysis of H5N1 proteome for designing an epitope-derived vaccine and predicting the prevalence of pre-existing cellular-mediated immunity toward bird flu virus in Indonesian population. Immunome Res. 2011, 7, 1. [Google Scholar]
- Moutaftsi, M.; Peters, B.; Pasquetto, V.; Tscharke, D.C.; Sidney, J.; Bui, H.-H.; Grey, H.; Sette, A. A consensus epitope prediction approach identifies the breadth of murine TCD8+-cell responses to vaccinia virus. Nat. Biotechnol. 2006, 24, 817–819. [Google Scholar] [CrossRef] [PubMed]
- Maleki, A.; Russo, G.; Palumbo, G.A.P.; Pappalardo, F. In silico design of recombinant multi-epitope vaccine against influenza A virus. BMC Bioinform. 2022, 22, 617. [Google Scholar] [CrossRef] [PubMed]
- Lokki, M.; Paakkanen, R. The complexity and diversity of major histocompatibility complex challenge disease association studies. HLA 2019, 93, 3–15. [Google Scholar] [CrossRef]
- Reche, P.A.; Reinherz, E.L. Sequence Variability Analysis of Human Class I and Class II MHC Molecules: Functional and Structural Correlates of Amino Acid Polymorphisms. J. Mol. Biol. 2003, 331, 623–641. [Google Scholar] [CrossRef]
- Bui, H.-H.; Sidney, J.; Dinh, K.; Southwood, S.; Newman, M.J.; Sette, A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 2006, 7, 477–484. [Google Scholar] [CrossRef]
- González-Galarza, F.F.; Takeshita, L.Y.; Santos, E.J.; Kempson, F.; Maia, M.H.T.; Da Silva, A.L.S.; Silva, A.L.T.E.; Ghattaoraya, G.S.; Alfirevic, A.; Jones, A.R.; et al. Allele frequency net 2015 update: New features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res. 2015, 43, D784–D788. [Google Scholar] [CrossRef]
- Larsen, M.E.; Kloverpris, H.; Stryhn, A.; Koofhethile, C.K.; Sims, S.; Ndung’u, T.; Goulder, P.; Buus, S.; Nielsen, M. HLArestrictor—A tool for patient-specific predictions of HLA restriction elements and optimal epitopes within peptides. Immunogenetics 2010, 63, 43–55. [Google Scholar] [CrossRef]
- McConkey, B.J.; Sobolev, V.; Edelman, M. The performance of current methods in ligand–protein docking. Curr. Sci. 2002, 83, 845–856. Available online: http://www.jstor.org/stable/24107087 (accessed on 1 June 2023).
- Zahroh, H.; Ma’rup, A.; Tambunan, U.S.F.; Parikesit, A.A. Immunoinformatics approach in designing epitopebased vaccine against meningitis-inducing bacteria (Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae type b). Drug Target Insights 2016, 10, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, S.A.; Venturi, V.; Dang, T.H.Y.; Bird, N.L.; Doherty, P.C.; Turner, S.J.; Davenport, M.P.; Kedzierska, K. Early Priming Minimizes the Age-Related Immune Compromise of CD8+ T Cell Diversity and Function. PLoS Pathog. 2012, 8, e1002544. Available online: https://pubmed.ncbi.nlm.nih.gov/22383879 (accessed on 1 June 2023). [CrossRef]
- Khalaj-Hedayati, A.; Chua, C.L.L.; Smooker, P.; Lee, K.W. Nanoparticles in influenza subunit vaccine development: Immunogenicity enhancement. Influ. Other Respir. Viruses 2019, 14, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Khalaj-Hedayati, A.; Chua, C.L.L.; Smooker, P.; Lee, K.W. Universal influenza vaccine technologies and recombinant virosome production. In Methods in Recombinant Protein Production, 1st ed.; Elsevier Ltd.: Amsterdam, The Netherlands, 2022; pp. 45–89. [Google Scholar] [CrossRef]
- Odelram, H.; Granström, M.; Hedenskog, S.; Duchén, K.; Björkstén, B. Immunogloblin E and G responses to pertussis toxin after booster immunization in relation to atopy, local reactions and aluminium content of the vaccines. Pediatr. Allergy Immunol. 1994, 5, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.K.; Arora, S.; Gupta, S.; Sanyal, R.K. Studies of adrenergic mechanisms in relation to histamine sensitivity in children immunized with Bordetella pertussis vaccine. J. Allergy Clin. Immunol. 1974, 54, 25–31. [Google Scholar] [CrossRef]
- McKeever, T.M.; Lewis, S.A.; Smith, C.; Hubbard, R. Vaccination and Allergic Disease: A Birth Cohort Study. Am. J. Public Health 2004, 94, 985–989. [Google Scholar] [CrossRef]
- Robles-Loaiza, A.A.; Pinos-Tamayo, E.A.; Mendes, B.; Ortega-Pila, J.A.; Proaño-Bolaños, C.; Plisson, F.; Teixeira, C.; Gomes, P.; Almeida, J.R. Traditional and Computational Screening of Non-Toxic Peptides and Approaches to Improving Selectivity. Pharmaceuticals 2022, 15, 323. [Google Scholar] [CrossRef]
- Guruprasad, K.; Reddy, B.; Pandit, M.W. Correlation between stability of a protein and its dipeptide composition: A novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng. Des. Sel. 1990, 4, 155–161. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar] [CrossRef]
- Zayas, J.F. (Ed.) Solubility of Proteins BT—Functionality of Proteins in Food; Springer: Berlin/Heidelberg, Germany, 1997; pp. 6–75. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Subtype | NCBI Accession Number | FASTA Format of HA2 Amino Acid Sequence |
|---|---|---|---|
| 1 | H1N1 | YP009121767 | >YP_009121767.1 FGAIAGFIEGGWTGMVDGWYGYHHQNEQGSGYAADLKSTQNAIDEITNKVNSVIEKMNTQFTAVGKEFNHLEKRIENLNKKVDDGFLDIWTYNAELLVLLENERTLDYHDSNVKNLYEKVRSQLKNNAKEIGNGCFEFYHKCDNTCMESVKNGTYDYPKYSEEAKLNREEIDGVKLESTRIYQILAIYSTVASSLVLVVSLGAISFWMCSNGSLQCRICI |
| 2 | H3N2 | ASV62273 | >ASV62273.1 FGAIAGFIENGWEGMVDGWYGFRHQNSEGTGQAADLKSTQAAINQITGKLNRIIKKTNEKFHQIEKEFSEVEGRIQDLEKYVEDTKVDLWSYNAELLVALENQHTIDLTDSEMSKLFERTRRQLRENAEDMGNGCFKIYHKCDNACIGSIRNGTYDHDIYRDEALNNRFQIKGVQLKSGYKDWILWISFAISCFLLCVVLLGFIMWACQKGNIRCNICI |
| 3 | H5N1 | ACI06178 | >ACI06178.1 FGAIAGFIEGGWQGMVDGWYGYHHSNEQGSGYAADKESTQKAIDGVTNKVNSIIDKMNTQFEAVGREFNNLERRIENLNKKMEDGFLDVWTYNAELLVLMENERTLDFHDSNVKNLYDKVRLQLRDNAKELGNGCFEFYHKCDNECMESVRNGTYDYPQYSEEARLKREEISGVKLESIGIYQILSIYSTVASSLALAIMVAGLSLWMCSNGSLQCK |
| 4 | H7N9 | AGI60292 | >AGI60292.1 FGAIAGFIENGWEGLIDGWYGFRHQNAQGEGTAADYKSTQSAIDQITGKLNRLIEKTNQQFELIDNEFNEVEKQIGNVINWTRDSITEVWSYNAELLVAMENQHTIDLADSEMDKLYERVKRQLRENAEEDGTGCFEIFHKCDDDCMASIRNNTYDHSKYREEAMQNRIQIDPVKLSSGYKDVILWFSFGASCFILLAIVMGLVFICVKNGNMRCTICI |
| 5 | H9N2 | CAB95857 | >CAB95857.1 FGAIAGFIEGGWPGLVAGWYGFQHSNDQGVGMAADRDSTQKAIDKITSKVNNIVDKMNKQYEIIDHEFSEVETRLNMINNKIDDQIQDVWAYNAELLVLLENQKTLDEHDANVNNLYNKVKRALGSNAMEDGKGCFELYHKCDDQCMETIRNGTYNRRKYREESRLERQKIEGVKLESEGAYKILTIYSTVASSLVLAMGFAAFLFWAMSNGSCRCNICI |
| 6 | H2N2 | AAY28987 | >AAY28987.1 FGAIAGFIEGGWQGMVDGWYGYHHSNDQGSGYAADKESTQKAFDGITNKVNSVIEKMNTQFEAVGKEFSNLERRLENLNKKMEDGFLDVWTYNAELLVLMENERTLDFHDSNVKNLYDKVRMQLRDNVKELGNGCFEFYHKCDDECMNSVKNGTYDYPKYEEESKLNRNEIKGVKLSSMGVYQILAIYATVAGSLSLAIMMAGISFWMCSNGSLQCRICI |
| HA288–107 Sequence | Peptide Identity among the Strains | |||||
|---|---|---|---|---|---|---|
| H1N1 | H3N2 | H5N1 | H7N9 | H9N2 | H2N2 | |
| DVWTYNAELLVLMENERTLD | 90% | 65% | 100% | 70% | 80% | 100% |
| Average: 84.16% | ||||||
| No. | Methods | Epitope Sequence | Start Position | End Position | Score Value | Threshold Value |
|---|---|---|---|---|---|---|
| 1 | Emini Surface Accessibility | MENERT | 13 | 18 | 2.745 | 1.000 |
| 2 | Karplus and Schulz Flexibility | MENERTL | 13 | 19 | 1.053 | 0.968 |
| 3 | Chou and Fasman Beta-turn | DVWTYNA | 1 | 7 | 1.034 | 0.834 |
| 4 | Parker Hydrophilicity | ENERTLD | 14 | 20 | 4.686 | 0.064 |
| 5 | Kolaskar and Tongaonkar Antigenicity | AELLVLM | 7 | 13 | 1.125 | 1.020 |
| No. | Epitopes | MHC I Allele | Percentile Rank < 1 | Binding Energy (kcal/mol) | No. | Epitopes | MHC II Allele | Percentile Rank < 10 |
|---|---|---|---|---|---|---|---|---|
| 1 | DVWTYNAEL | HLA-A*68:02 HLA-A*26:01 | 0.29 0.55 | −155.5 | 1 | DVWTYNAELLVLMEN | HLA-DQA1*03:01/DQB1*03:02 HLA-DQA1*04:01/DQB1*04:02 HLA-DQA1*05:01/DQB1*02:01 HLA-DQA1*01:01/DQB1*05:01 HLA-DQA1*01:02/DQB1*06:02 HLA-DPA1*01:03/DPB1*04:01 HLA-DPA1*01:03/DPB1*02:01 HLA-DPA1*02:01/DPB1*01:01 HLA-DPA1*03:01/DPB1*04:02 HLA-DRB3*01:01 HLA-DRB3*02:02 | 3.7 4.6 4.8 6.3 9.4 3.8 4.4 5.2 7.8 7.4 8.2 |
| 2 | TYNAELLVL | HLA-A*24:02 HLA-A*23:01 | 0.08 0.15 | −103.2 | 2 | NAELLVLMENERTLD | HLA-DRB1*04:05 HLA-DQA1*03:01/DQB1*03:02 HLA-DRB1*04:01 HLA-DRB1*01:01 HLA-DRB1*15:01 | 1.9 2.2 3.7 4.9 6.7 |
| 3 | TYNAELLVLM | HLA-A*24:02 HLA-A*23:01 | 0.43 0.48 | −115 | 3 | TYNAELLVLMENERT | HLA-DQA1*03:01/DQB1*03:02 HLA-DRB1*04:05 HLA-DQA1*04:01/DQB1*04:02 HLA-DRB1*04:01 HLA-DQA1*01:02/DQB1*06:02 HLA-DRB1*15:01 | 1.5 1.9 3.6 3.7 6.5 8.5 |
| 4 | VLMENERTL | HLA-A*02:01 HLA-A*02:03 HLA-A*02:06 HLA-A*32:01 HLA-B*08:01 | 0.1 0.15 0.3 0.46 0.51 | −160.8 | 4 | VWTYNAELLVLMENE | HLA-DQA1*03:01/DQB1*03:02 HLA-DQA1*04:01/DQB1*04:02 HLA-DQA1*01:02/DQB1*06:02 HLA-DPA1*01:03/DPB1*02:01 HLA-DQA1*05:01/DQB1*02:01 HLA-DPA1*01:03/DPB1*04:01 HLA-DPA1*02:01/DPB1*01:01 HLA-DPA1*03:01/DPB1*04:02 HLA-DRB3*01:01 | 1.3 3.1 4.2 4.4 4.4 4.6 5.8 7.4 9.6 |
| 5 | VWTYNAELL | HLA-A*24:02 HLA-A*23:01 | 0.54 0.81 | −133.8 | 5 | WTYNAELLVLMENER | HLA-DQA1*03:01/DQB1*03:02 HLA-DQA1*04:01/DQB1*04:02 HLA-DQA1*01:02/DQB1*06:02 HLA-DQA1*05:01/DQB1*02:01 HLA-DPA1*02:01/DPB1*01:01 HLA-DPA1*01:03/DPB1*04:01 HLA-DPA1*01:03/DPB1*02:01 | 1.5 3.5 4.6 4.7 7.9 8.6 10 |
| 6 | WTYNAELLV | HLA-A*68:02 | 0.38 | −857.9 | 6 | YNAELLVLMENERTL | HLA-DRB1*04:05 HLA-DQA1*03:01/DQB1*03:02 HLA-DRB1*04:01 HLA-DQA1*04:01/DQB1*04:02 HLA-DRB1*01:01 HLA-DRB1*15:01 | 1.9 2 3.7 5.4 5.4 7.9 |
| Population/Area | MHC Class I | MHC Class II | ||||
|---|---|---|---|---|---|---|
| Coverage a | Average-Hit b | PC90 c | Coverage a | Average-Hit b | PC90 c | |
| World | 79.52% | 3.12 | 0.49 | 99.88% | 13.79 | 7.81 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalaj-Hedayati, A.; Moosavi, S.; Manta, O.; Helal, M.H.; Ibrahim, M.M.; El-Bahy, Z.M.; Supriyanto, G. Identification and In Silico Characterization of a Conserved Peptide on Influenza Hemagglutinin Protein: A New Potential Antigen for Universal Influenza Vaccine Development. Nanomaterials 2023, 13, 2796. https://doi.org/10.3390/nano13202796
Khalaj-Hedayati A, Moosavi S, Manta O, Helal MH, Ibrahim MM, El-Bahy ZM, Supriyanto G. Identification and In Silico Characterization of a Conserved Peptide on Influenza Hemagglutinin Protein: A New Potential Antigen for Universal Influenza Vaccine Development. Nanomaterials. 2023; 13(20):2796. https://doi.org/10.3390/nano13202796
Chicago/Turabian StyleKhalaj-Hedayati, Atin, Seyedehmaryam Moosavi, Otilia Manta, Mohamed H. Helal, Mohamed M. Ibrahim, Zeinhom M. El-Bahy, and Ganden Supriyanto. 2023. "Identification and In Silico Characterization of a Conserved Peptide on Influenza Hemagglutinin Protein: A New Potential Antigen for Universal Influenza Vaccine Development" Nanomaterials 13, no. 20: 2796. https://doi.org/10.3390/nano13202796
APA StyleKhalaj-Hedayati, A., Moosavi, S., Manta, O., Helal, M. H., Ibrahim, M. M., El-Bahy, Z. M., & Supriyanto, G. (2023). Identification and In Silico Characterization of a Conserved Peptide on Influenza Hemagglutinin Protein: A New Potential Antigen for Universal Influenza Vaccine Development. Nanomaterials, 13(20), 2796. https://doi.org/10.3390/nano13202796