
Transition Metal Phosphides (TMP) as a Versatile Class of Catalysts for the Hydrodeoxygenation Reaction (HDO) of Oil-Derived Compounds

 , and
, and 
Abstract
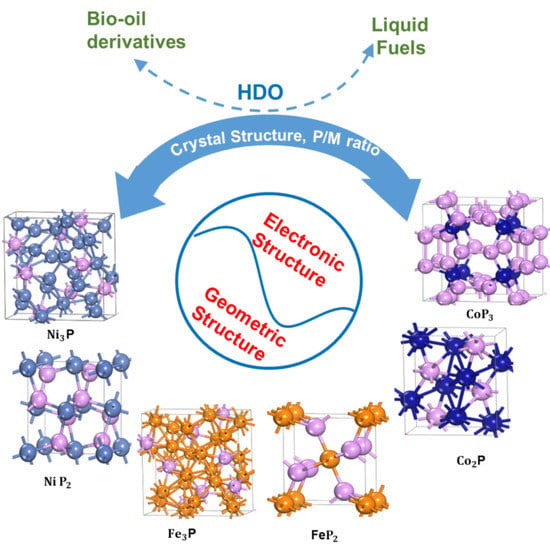
:
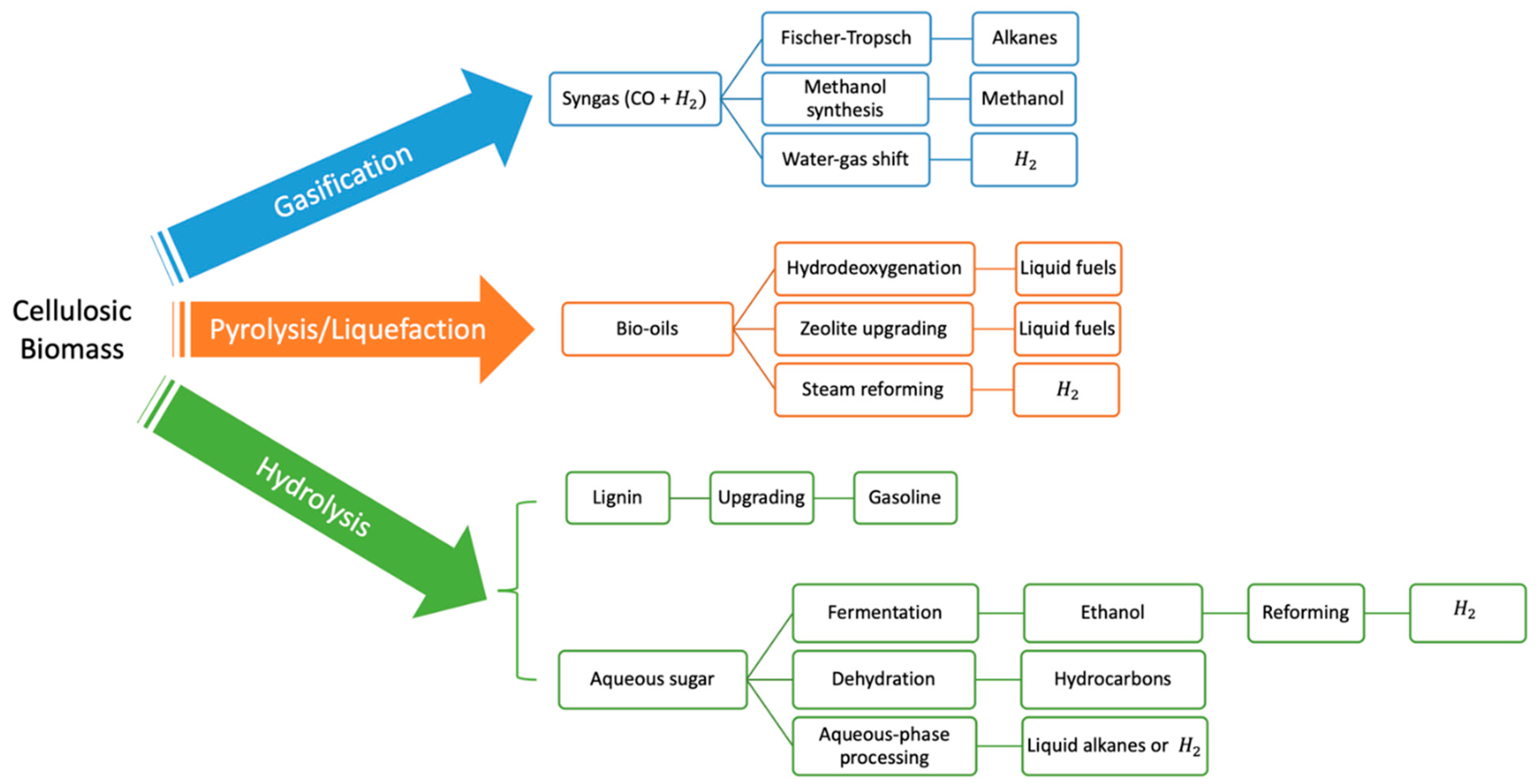
1. Introduction
2. A Brief on the Transition Metal Phosphide (TMP)
2.1. Structural Concepts
2.2. Functionality (Acidity) of TMPs
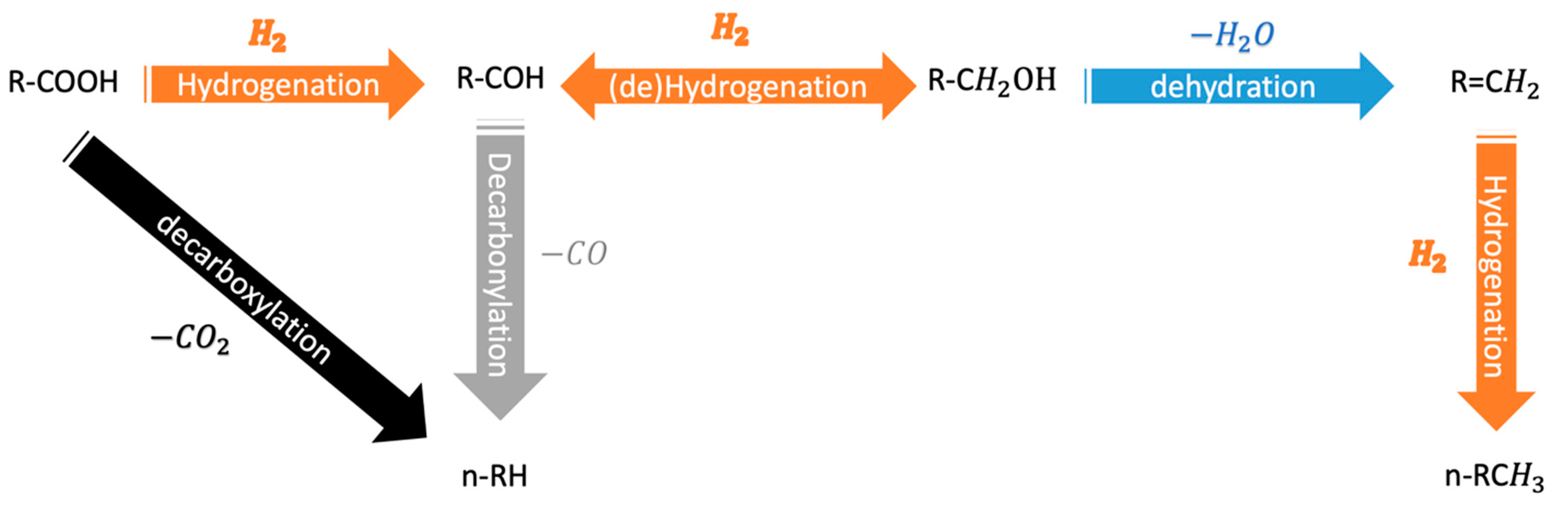
2.3. A Close Look on the TMP Active Sites for the HDO Reaction
2.4. TMPs Preparation Strategies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Reactants | Reaction Conditions | Coversion (%) | Selectivtiy (%) | Reference | |
|---|---|---|---|---|---|---|
| P (bar) | ||||||
| Phenol | 220 | 20 | ~100 | ~92 Cyclohexane | [104] | |
| 2-methyltetrahydrofuran | 275 | 1 | 15 | 14 Pentane 70 Pentenes 7 Pentadienes | [105] | |
| 11 | 40 Pentane 15 Pentenes 25 C14 | |||||
| 12 | 67 pentane 2 pentanone 15 C14 | [106] | ||||
| dibenzofuran | 275 | 30 | ~90 | ~72 Bicyclohexane | [107] | |
| guaiacol | 300 | 1 | 92.9 | 14.1 Phenol 58.7 Benzene | [108] | |
| 93.9 | 31.5 Phenol 34.9 Benzene | |||||
| 99.8 | 23.9 Phenol 22.8 Benzene | |||||
| / pillared -ZSM-5 | 260 | 40 | 78 | ~82 cyclohexane | [109] | |
| FeMoP | Anisole | 400 | 21 | >99 | 90 Benzene 10 Cyclohexane | [110] |
| methyl laurate | 340 | 20 | ~98 | ~96 C11 + C12~12 C11/C12 | [111] | |
| , MCM-41 | 340 | 20-30 | 97-99 | ~100 C11 + C12 | [112] | |
| / SBA-15 | methyl oleate | 270 | 30 | ~84 | ~45 C18/(C17+C18) | [113] |
| methyl laurate | 340 | 30 | ~97 | ~87 C11 + ~12 C12 | [114] | |
| 340 | 30 | ~90 | ~4 C11 + ~80 C12 | |||
| 340 | 30 | ~98 | ~51 C11 + ~49 C12 | |||
| /activated carbon | palmitic acid | 350 | 1 | 100 | 57 C15 7 C11-C14 21 alkenes | [115] |
| /HZSM-22 | 350 | 1 | 99.6 | - | [116] | |
| 2-furyl methyl ketone | 400 | 1 | 100 | ~100 Methyl Cyclopentane | [117] | |
3. Characterization of TMP Features towards Unveiling Outstanding Performance
3.1. Structural Features (XRD, Raman, TEM, SAED)
3.2. Dispersion of the TMPs over a Support
3.3. Surface Studies and Coordination Environment (XPS, XANES, NMR)
4. TMPs as Catalysts for the Upgrading of Bio-Oil through HDO Reaction
4.1. Lignin Phenolic Derivatives
4.2. Vegetable Oils
4.3. Cellullose and Semi-Cellullose Furan Derivatives
4.4. On the HDO Activity of the Multi-Elemental TMPs
5. Deactivation during the HDO Reaction
6. Computational Approaches to Study the HDO Reaction over TMPs
7. Future Perspectives and Outlook
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Alvarez-Galvan, M.C.; Campos-Martin, J.M.; Fierro, J.L.G. Transition Metal Phosphides for the Catalytic Hydrodeoxygenation of Waste Oils into Green Diesel. Catalysts 2019, 9, 293. [Google Scholar] [CrossRef] [Green Version]
- Jovičić, N.; Antonović, A.; Matin, A.; Antolović, S.; Kalambura, S.; Krička, T. Biomass Valorization of Walnut Shell for Liquefaction Efficiency. Energies 2022, 15, 495. [Google Scholar] [CrossRef]
- Moretti, L.; Arpino, F.; Cortellessa, G.; Di Fraia, S.; Di Palma, M.; Vanoli, L. Reliability of Equilibrium Gasification Models for Selected Biomass Types and Compositions: An Overview. Energies 2022, 15, 61. [Google Scholar] [CrossRef]
- Shi, Y.; Xing, E.; Wu, K.; Wang, J.; Yang, M.; Wu, Y. Recent progress on upgrading of bio-oil to hydrocarbons over metal/zeolite bifunctional catalysts. Catal. Sci. Technol. 2017, 7, 2385–2415. [Google Scholar] [CrossRef]
- Kumar, R.; Strezov, V.; Lovell, E.; Kan, T.; Weldekidan, H.; He, J.; Dastjerdi, B.; Scott, J. Bio-oil upgrading with catalytic pyrolysis of biomass using Copper/zeolite-Nickel/zeolite and Copper-Nickel/zeolite catalysts. Bioresour. Technol. 2019, 279, 404–409. [Google Scholar] [CrossRef]
- López-Renau, L.M.; García-Pina, L.; Hernando, H.; Gómez-Pozuelo, G.; Botas, J.A.; Serrano, D.P. Enhanced bio-oil upgrading in biomass catalytic pyrolysis using KH-ZSM-5 zeolite with acid-base properties. Biomass Convers. Biorefin. 2021, 11, 2311–2323. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Siakavelas, G.; Tzounis, L.; Dou, B.; Sebastian, V.; Hinder, S.J.; Baker, M.A.; Polychronopoulou, K.; Goula, M.A. Ni/Y2O3–ZrO2 catalyst for hydrogen production through the glycerol steam reforming reaction. Int. J. Hydrogen Energy 2020, 45, 10442. [Google Scholar] [CrossRef]
- Polychronopoulou, K.; Charisiou, N.D.; Siakavelas, G.I.; AlKhoori, A.A.; Sebastian, V.; Hinder, S.J.; Baker, M.A.; Goula, M.A. Ce–Sm–x Cu cost-efficient catalysts for H2 production through the glycerol steam reforming reaction. Sustain. Energy Fuels 2019, 3, 673–691. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Siakavelas, G.I.; Dou, B.; Sebastian, V.; Hinder, S.J.; Baker, M.A.; Polychronopoulou, K.; Goula, M.A. Nickel supported on AlCeO3 as a highly selective and stable catalyst for hydrogen production via the glycerol steam reforming reaction. Catalysts 2019, 9, 411. [Google Scholar] [CrossRef] [Green Version]
- Polychronopoulou, K.; Bakandritsos, A.; Tzitzios, V.; Fierro, J.L.G.; Efstathiou, A.M. Absorption-enhanced reforming of phenol by steam over supported Fe catalysts. J. Catal. 2006, 241, 132–148. [Google Scholar] [CrossRef]
- Polychronopoulou, K.; Fierro, J.L.G.; Efstathiou, A.M. The phenol steam reforming reaction over MgO-based supported Rh catalysts. J. Catal. 2004, 228, 417–432. [Google Scholar] [CrossRef]
- Musso, M.; Veiga, S.; Perdomo, F.; Rodríguez, T.; Mazzei, N.; Decarlini, B.; Portugau, P.; Bussi, J. Hydrogen production via steam reforming of small organic compounds present in the aqueous fraction of bio-oil over Ni-La-Me catalysts (Me = Ce, Ti, Zr). Biomass Convers. Biorefin. 2022, 1–17. [Google Scholar] [CrossRef]
- Papageridis, K.N.; Charisiou, N.D.; Douvartzides, S.L.; Sebastian, V.; Hinder, S.J.; Baker, M.A.; Al Khoori, S.; Polychronopoulou, K.; Goula, M.A. Effect of operating parameters on the selective catalytic deoxygenation of palm oil to produce renewable diesel over Ni supported on Al2O3, ZrO2 and SiO2 catalysts. Fuel Process. Technol. 2020, 209, 106547. [Google Scholar] [CrossRef]
- Papageridis, K.N.; Charisiou, N.D.; Douvartzides, S.; Sebastian, V.; Hinder, S.J.; Baker, M.A.; AlKhoori, A.A.; AlKhoori, S.I.; Polychronopoulou, K.; Goula, M.A. Continuous selective deoxygenation of palm oil for renewable diesel production over Ni catalysts supported on Al2O3 and La2O3–Al2O3. RSC Adv. 2021, 11, 8569–8584. [Google Scholar] [CrossRef]
- Zhang, Q.; Chang, J.; Wang, T.; Xu, Y. Review of Biomass Pyrolysis Oil Properties and Upgrading Research. Energy Convers. Manag. 2007, 48, 87–92. [Google Scholar] [CrossRef]
- Czernik, S.; Bridgwater, A.V. Overview of Applications of Biomass Fast Pyrolysis Oil. Energy Fuels 2004, 18, 590–598. [Google Scholar] [CrossRef]
- Furimsky, E. Catalytic hydrodeoxygenation. Appl. Catal. A Gen. 2000, 199, 147–190. [Google Scholar] [CrossRef]
- Sunarno, S.; Zahrina, I.; Nanda, W.R.; Amri, A. Upgrading of pyrolysis oil via catalytic co-pyrolysis of treated palm oil empty fruit bunch and plastic waste. Biomass Convers. Biorefin. 2022, 1–9. [Google Scholar] [CrossRef]
- Yun, G.; Takagaki, A.; Kikuchia, R.; Oyama, S.T. Hydrodeoxygenation of gamma-valerolactone on transition metal phosphide catalysts. Catal. Sci. Technol. 2017, 7, 281–292. [Google Scholar] [CrossRef]
- Chen, J.; Xu, Q. Hydrodeoxygenation of biodiesel-related fatty acid methyl esters to diesel-range alkanes over zeolite-supported ruthenium catalysts. Catal. Sci. Technol. 2016, 6, 7239–7251. [Google Scholar] [CrossRef]
- Satyarthi, J.K.; Chiranjeevi, T.; Gokak, D.T.; Viswanathan, P.S. An overview of catalytic conversion of vegetable oils/fats into middle distillates. Catal. Sci. Technol. 2013, 3, 70–80. [Google Scholar] [CrossRef]
- Şenol, O.İ.; Ryymin, E.M.; Viljava, T.R.; Krause, A.O.I. Effect of hydrogen sulphide on the hydrodeoxygenation of aromatic and aliphatic oxygenates on sulphided catalysts. J. Mol. Catal. A Chem. 2007, 277, 107–112. [Google Scholar] [CrossRef]
- Ryymin, E.M.; Honkela, M.L.; Viljava, T.R.; Krause, A.O.I. Insight to sulfur species in the hydrodeoxygenation of aliphatic esters over sulfided NiMo/γ-Al2O3 catalyst. Appl. Catal. A Gen. 2009, 358, 42–48. [Google Scholar] [CrossRef]
- Dhandapani, B.; Clair, T.S.; Oyama, S.T. Simultaneous hydrodesulfurization, hydrodeoxygenation, and hydrogenation with molybdenum carbide. Appl. Catal. A Gen. 1998, 168, 219–228. [Google Scholar] [CrossRef]
- Elliott, D.C.; Neuenschwander, G.G.; Hart, T.R.; Hu, J.; Solana, A.E.; Cao, C. Hydrogenation of Bio-Oil for Chemicals and Fuels Production; Pacific Northwest National Lab. (PNNL), Environmental Molecular Sciences Lab. (EMSL): Richland, WA, USA, 2006. [Google Scholar]
- Wang, J.-J.; Li, X.-P.; Cui, B.-F.; Zhang, Z.; Hu, X.-F.; Ding, J.; Deng, Y.-D.; Han, X.-P.; Hu, W.-B. A review of non-noble metal-based electrocatalysts for CO2 electroreduction. Rare Met. 2021, 40, 3019–3037. [Google Scholar] [CrossRef]
- Lin, Z.; Chen, R.; Qu, Z.; Chen, J.G. Hydrodeoxygenation of biomass-derived oxygenates over metal carbides: From model surfaces to powder catalysts. Green Chem. 2018, 20, 2679–2696. [Google Scholar] [CrossRef]
- Boullosa-Eiras, S.; Lodeng, R.; Bergem, H.; Stocker, M.; Hannevold, L.; Blekkana, E.A. Catalytic hydrodeoxygenation (HDO) of phenol over supported molybdenum carbide, nitride, phosphide and oxide catalysts. Catal. Today 2014, 223, 44–53. [Google Scholar] [CrossRef]
- Wang, W.; Yang, Y.; Bao, J.; Luo, H. Characterization and catalytic properties of Ni–Mo–B amorphous catalysts for phenol hydrodeoxygenation. Catal. Commun. 2009, 11, 100–105. [Google Scholar] [CrossRef]
- Chen, X.; Liang, C. Transition metal silicides: Fundamentals, preparation and catalytic applications. Catal. Sci. Technol. 2019, 9, 4785–4820. [Google Scholar] [CrossRef]
- Wen, C.; Chen, L.; Xie, Y.; Bai, J.; Liang, X. MOF-derived carbon-containing Fe doped porous CoP nanosheets towards hydrogen evolution reaction and hydrodesulfurization. Int. J. Hydrogen Energy 2021, 46, 33420–33428. [Google Scholar] [CrossRef]
- Oyama, S.T.; Wang, X.; Lee, Y.-K.; Bando, K.; Requejo, F.G. Effect of Phosphorus Content in Nickel Phosphide Catalysts Studied by XAFS and Other Techniques. J. Catal. 2002, 210, 207–217. [Google Scholar] [CrossRef]
- Abu, I.I.; Smith, K.J. HDN and HDS of model compounds and light gas oil derived from Athabasca bitumen using supported metal phosphide catalysts. Appl. Catal. A Gen. 2007, 328, 58–67. [Google Scholar] [CrossRef]
- Li, W.; Dhandapani, B.; Oyama, S.T. Molybdenum Phosphide: A Novel Catalyst for Hydrodenitrogenation. Chem. Lett. 1998, 27, 207–208. [Google Scholar] [CrossRef]
- Yin, P.; Yang, Y.S.; Chen, L.F.; Xu, M.; Chen, C.Y.; Zhao, X.J.; Zhang, X.; Yan, H.; Wei, M. DFT Study on the Mechanism of the Water Gas Shift Reaction over NixPy Catalysts: The Role of P. J. Phys. Chem. C 2020, 124, 6598–6610. [Google Scholar] [CrossRef]
- Hughes, M. Silica Supported Transition Metal Phosphides-Alternative Materials for the Water–Gas Shift Reaction. PhD. Thesis, University of Glasgow, Glasgow, UK, 2014. [Google Scholar]
- Zhang, F.; Zhang, J.; Li, J.; Jin, X.; Li, Y.; Wu, M.; Kang, X.; Hu, T.; Wang, X.; Ren, W.; et al. Modulating charge transfer dynamics for g-C3N4 through a dimension and interface engineered transition metal phosphide co-catalyst for efficient visible-light photocatalytic hydrogen generation. J. Mater. Chem. A 2019, 7, 6939–6945. [Google Scholar] [CrossRef]
- Zeng, D.; Lu, Z.; Gao, X.; Wu, B.; Ong, W. Hierarchical flower-like ZnIn2S4 anchored with well-dispersed Ni12P5 nanoparticles for high-quantum-yield photocatalytic H2 evolution under visible light. Catal. Sci. Technol. 2019, 9, 4010–4016. [Google Scholar] [CrossRef]
- Liu, S.; Huang, J.; Su, H.; Tang, G.; Liu, Q.; Sun, J.; Xu, J. Multiphase phosphide cocatalyst for boosting efficient photocatalytic H2 production and enhancing the stability. Ceram. Int. 2021, 47, 1414–1420. [Google Scholar] [CrossRef]
- Boakye, F.O.; Fan, M.; Zhang, F.; Tang, H.; Zhang, R.; Zhang, H. Growth of branched heterostructure of nickel and iron phosphides on carbon cloth as electrode for hydrogen evolution reaction under wide pH ranges. J. Solid State Electrochem. 2022, 1–11. [Google Scholar] [CrossRef]
- Kolny-Olesiak, J. Recent Advances in the colloidal synthesis of ternarty transition metal phosphides. Z. Naturforsch. 2019, 74, 709–711. [Google Scholar] [CrossRef]
- Liyanage, D.R.; Li, D.; Cheek, Q.B.; Baydoun, H.; Brock, S.L. Synthesis and oxygen evolution reaction (OER) catalytic performance of Ni2−xRux P nanocrystals: Enhancing activity by dilution of the noble metal. J. Mater. Chem. A 2017, 5, 17609–17618. [Google Scholar] [CrossRef]
- Mutinda, S.I.; Li, D.; Kay, J.; Brock, S.L. Synthesis and characterization of Co2−x RhxP nanoparticles and their catalytic activity towards the oxygen evolution reaction. J. Mater. Chem. A 2018, 6, 12142–12152. [Google Scholar] [CrossRef]
- Ren, T.; Li, M.; Chu, Y.; Chen, J. Thioetherification of Isoprene and Butanethiol on Transition Metal Phosphides. J. Energy Chem. 2018, 27, 930–939. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.K.; Oyama, S.T. Bifunctional nature of a SiO2-supported Ni2P catalyst for hydrotreating: EXAFS and FTIR studies. J. Catal. 2006, 239, 376–389. [Google Scholar] [CrossRef]
- Golubevaa, M.A.; Zakharyana, E.M.; Maximov, A.L. Transition Metal Phosphides (Ni, Co, Mo, W) for Hydrodeoxygenation of Biorefinery Products (a Review). Pet. Chem. 2020, 60, 1109–1128. [Google Scholar] [CrossRef]
- Oyama, S.T. Novel catalysts for advanced hydroprocessing: Transition metal phosphides. J. Catal. 2003, 216, 343. [Google Scholar] [CrossRef]
- Brock, S.L.; Senevirathne, K. Recent developments in synthetic approaches to transition metal phosphide nanoparticles for magnetic and catalytic applications. J. Solid State Chem. 2008, 181, 1552–1559. [Google Scholar] [CrossRef]
- Zhu, Y.; Lu, P.; Li, F.; Ding, Y.; Chen, Y. Metal-Rich Porous Copper Cobalt Phosphide Nanoplates as a High-Rate and Stable Battery-Type Cathode Material for Battery–Supercapacitor Hybrid Devices. ACS Appl. Energy Mater. 2021, 4, 3962–3974. [Google Scholar] [CrossRef]
- Alexander, A.M.; Hargreaves, J.S. Alternative catalytic materials: Carbides, nitrides, phosphides and amorphous boron alloys. Chem. Soc. Rev. 2010, 39, 4388–4401. [Google Scholar] [CrossRef]
- Chen, J.-H.; Whitmire, K.H. A Structural Survey of the Binary Transition Metal Phosphides and Arsenides of the D-Block Elements. Coord. Chem. Rev. 2018, 355, 271–327. [Google Scholar] [CrossRef]
- Bussell, M.E. New methods for the preparation of nanoscale nickel phosphide catalysts for heteroatom removal reactions. React. Chem. Eng. 2017, 2, 628–635. [Google Scholar] [CrossRef]
- Pu, Z.; Liu, T.; Amiinu, I.S.; Cheng, R.; Wang, P.; Zhang, C.; Ji, P.; Hu, W.; Liu, J.; Mu, S. Transition-Metal Phosphides: Activity Origin, Energy-Related Electrocatalysis Applications, and Synthetic Strategies. Adv. Funct. Mater. 2020, 30, 2004009. [Google Scholar] [CrossRef]
- Shi, Y.; Li, M.; Yu, Y.; Zhang, B. Recent advantages in nanostructured transition metal phospides: Synthesis and energy-related applications. Energy Environ. Sci. 2020, 13, 4564–4582. [Google Scholar] [CrossRef]
- Jun RE, N.; Wang, J.G.; Li, J.F.; Li, Y.W. Density functional theory study on crystal nickel phosphides. J. Fuel Chem. Technol. 2007, 35, 458–464. [Google Scholar]
- Pei, Y.; Cheng, Y.; Chen, J.; Smith, W.; Dong, P.; Ajayan, P.M.; Ye, M.; Shen, J. Recent developments of transition metal phosphides as catalysts in the energy conversion field. J. Mater. Chem. A 2018, 6, 23220. [Google Scholar] [CrossRef]
- Yu, S.H.; Chua, D.H. Toward high-performance and low-cost hydrogen evolution reaction electrocatalysts: Nanostructuring cobalt phosphide (CoP) particles on carbon fiber paper. ACS Appl. Mater. Interfaces 2018, 10, 14777–14785. [Google Scholar] [CrossRef]
- Wu, C.; Yang, Y.; Dong, D.; Zhang, Y.; Li, J. In situ coupling of CoP polyhedrons and carbon nanotubes as highly efficient hydrogen evolution reaction electrocatalyst. Small 2017, 13, 1602873. [Google Scholar] [CrossRef]
- Zeng, Y.; Wang, Y.; Huang, G.; Chen, C.; Huang, L.; Chen, R.; Wang, S. Porous CoP nanosheets converted from layered double hydroxides with superior electrochemical activity for hydrogen evolution reactions at wide pH ranges. Chem. Commun. 2018, 54, 1465–1468. [Google Scholar] [CrossRef] [Green Version]
- Ye, C.; Wang, M.Q.; Chen, G.; Deng, Y.H.; Li, L.J.; Luo, H.Q.; Li, N.B. One-step CVD synthesis of carbon framework wrapped Co 2 P as a flexible electrocatalyst for efficient hydrogen evolution. J. Mater. Chem. A 2017, 5, 7791–7795. [Google Scholar] [CrossRef]
- Huang, H.B.; Luo, S.H.; Liu, C.L.; Yi, T.F.; Zhai, Y.C. High-Surface-Area and Porous Co2P Nanosheets as Cost-Effective Cathode Catalysts for Li–O2 Batteries. ACS Appl. Mater. Interfaces 2018, 10, 21281–21290. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, R.; Zhang, L.; Liu, D.; Hao, S.; Du, G.; Asiri, A.M.; Kong, R.; Sun, X. Energy-efficient electrolytic hydrogen generation using a Cu3P nanoarray as a bifunctional catalyst for hydrazine oxidation and water reduction. Inorg. Chem. Front. 2017, 4, 420–423. [Google Scholar] [CrossRef]
- Fan, M.; Chen, Y.; Xie, Y.; Yang, T.; Shen, X.; Xu, N.; Yu, H.; Yan, C. Half-cell and full-cell applications of highly stable and binder-free sodium ion batteries based on Cu3P nanowire anodes. Adv. Funct. Mater. 2016, 26, 5019–5027. [Google Scholar] [CrossRef]
- Jiang, Y.; Lu, Y.; Lin, J.; Wang, X.; Shen, Z. A hierarchical MoP nanoflake array supported on Ni foam: A bifunctional electrocatalyst for overall water splitting. Small Methods 2018, 2, 1700369. [Google Scholar] [CrossRef]
- Lan, K.; Wang, X.; Yang, H.; Iqbal, K.; Zhu, Y.; Jiang, P.; Tang, Y.; Yang, Y.; Gao, W.; Li, R. Ultrafine MoP Nanoparticles Well Embedded in Carbon Nanosheets as Electrocatalyst with High Active Site Density for Hydrogen Evolution. ChemElectroChem 2018, 5, 2256–2262. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, X.; Yu, S.; Wen, T.; Zhu, X.; Yang, F.; Sun, X.; Wang, X.; Hu, W. Ternary NiCo2Px nanowires as pH-universal electrocatalysts for highly efficient hydrogen evolution reaction. Adv. Mater. 2017, 29, 1605502. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, G.; Yu, L.; Qu, J.; Liu, H. Oxygen doping to optimize atomic hydrogen binding energy on NiCoP for highly efficient hydrogen evolution. Small 2018, 14, 1800421. [Google Scholar] [CrossRef]
- Xuan, C.; Wang, J.; Xia, W.; Peng, Z.; Wu, Z.; Lei, W.; Xia, K.; Xin, H.L.; Wang, D. Porous structured Ni–Fe–P nanocubes derived from a prussian blue analogue as an electrocatalyst for efficient overall water splitting. ACS Appl. Mater. Interfaces 2017, 9, 26134–26142. [Google Scholar] [CrossRef]
- Ma, Y.Y.; Wu, C.X.; Feng, X.J.; Tan, H.Q.; Yan, L.K.; Liu, Y.; Kang, Z.-H.; Wang, E.-B.; Li, Y.G. Highly efficient hydrogen evolution from seawater by a low-cost and stable CoMoP@ C electrocatalyst superior to Pt/C. Energy Environ. Sci. 2017, 10, 788–798. [Google Scholar] [CrossRef]
- Asnavandi, M.; Suryanto, B.H.R.; Yang, W.; Bo, X.; Zhao, C. Dynamic Hydrogen Bubble Templated NiCu Phosphide Electrodes for pH-Insensitive Hydrogen Evolution Reactions. ACS Sustain. Chem. Eng. 2018, 6, 2866–2871. [Google Scholar] [CrossRef]
- Park, J.; Koo, B.; Yoon, K.Y.; Hwang, Y.; Kang, M.; Park, J.G.; Hyeon, T. Generalized synthesis of metal phosphide nanorods via thermal decomposition of continuously delivered metal−phosphine complexes using a syringe pump. J. Am. Chem. Soc. 2005, 127, 8433–8440. [Google Scholar] [CrossRef]
- Guan, Q.; Sun, C.; Li, R.; Li, W. The synthesis and investigation of ruthenium phosphide catalysts. Catal. Commun. 2011, 14, 114–117. [Google Scholar] [CrossRef]
- Berenguer, A.; Sankaranarayanan, T.M.; Gómez, G.; Moreno, I.; Coronado, J.M.; Pizarro, P.; Serrano, D.P. Evaluation of transition metal phosphides supported on ordered mesoporous materials as catalysts for phenol hydrodeoxygenation. Green Chem. 2016, 18, 1938–1951. [Google Scholar] [CrossRef]
- Shu, Y.; Oyama, S.T. Synthesis, characterization, and hydrotreating activity of carbon-supported transition metal phosphides. Carbon 2005, 43, 1517–1532. [Google Scholar] [CrossRef]
- Korányi, T.I.; Vít, Z.; Poduval, D.G.; Ryoo, R.; Kim, H.S.; Hensen, E.J.M. SBA-15-supported nickel phosphide hydrotreating catalysts. J. Catal. 2008, 253, 119–131. [Google Scholar] [CrossRef]
- D’Aquino, A.I.; Danforth, S.J.; Clinkingbeard, T.R.; Ilic, B.; Pullan, L.; Reynolds, M.A.; Murray, B.D.; Bussel, B.E. Highly-active nickel phosphide hydrotreating catalysts prepared in situ using nickel hypophosphite precursors. J. Catal. 2016, 335, 204–214. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.-K.; Lai, P.-C.; Lin, Y.-C.; Wan, H.-P.; Lee, H.-T.; Chang, Y.-H. Atmospheric Hydrodeoxygenation of Guaiacol over Alumina-, Zirconia-, and Silica-Supported Nickel Phosphide Catalysts. ACS Sustain. Chem. Eng. 2013, 1, 349–358. [Google Scholar] [CrossRef]
- Gonçalves, V.O.O.; de Souza, P.M.; Cabioc’h, T.; da Silva, V.T.; Noronha, F.B.; Richard, F. Effect of P/Ni Ratio on the Performance of Nickel Phosphide Phases Supported on Zirconia for the Hydrodeoxygenation of m-Cresol. Catal. Commun. 2019, 119, 33–38. [Google Scholar] [CrossRef]
- Moon, J.-S.; Lee, Y.-K. Support Effects of Ni2P Catalysts on the Hydrodeoxygenation of Guaiacol: In Situ XAFS Studies. Top. Catal. 2015, 58, 211–218. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, F.; Han, H.; Xiao, L.; Wu, W. Metal Phosphide:A Highly Efficient Catalyst for the Selective Hydrodeoxygenation of Furfural to 2-Methylfuran. ChemistrySelect 2018, 3, 7926–7933. [Google Scholar] [CrossRef]
- Oyama, S.T.; Gott, T.; Zhao, H.; Lee, Y.K. Transition metal phosphide hydroprocessing catalysts: A review. Catal. Today 2009, 143, 94–107. [Google Scholar] [CrossRef]
- Prins, R.; Bussell, M.E. Metal phosphides: Preparation, characterization and catalytic reactivity. Catal. Lett. 2012, 142, 1413–1436. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Kim, J.Y.; Hanson, J.C.; Sawhill, S.J.; Bussell, M.E. Physical and chemical properties of MoP, Ni2P, and MoNiP hydrodesulfurization catalysts: Time-resolved X-ray diffraction, density functional, and hydrodesulfurization activity studies. J. Phys. Chem. B 2003, 107, 6276–6285. [Google Scholar] [CrossRef]
- Liu, P.; Rodriguez, J.A.; Asakura, T.; Gomes, J.; Nakamura, K. Desulfurization reactions on Ni2P (001) and α-Mo2C (001) surfaces: Complex role of P and C sites. J. Phys. Chem. B 2005, 109, 4575–4583. [Google Scholar] [CrossRef]
- Zhou, R.; Zhang, J.; Chen, Z.; Han, X.; Zhong, C.; Hu, W.; Deng, Y. Phase and Composition Controllable Synthesis of Nickel Phosphide-Based Nanoparticles via a Low-Temperature Process for Efficient Electrocatalytic Hydrogen Evolution. Electrochim. Acta 2017, 258, 866–875. [Google Scholar] [CrossRef]
- Liu, P.; Rodriguez, J.A. Catalysts for hydrogen evolution from the [NiFe] hydrogenase to the Ni2P (001) surface: The importance of ensemble effect. J. Am. Chem. Soc. 2005, 127, 14871–14878. [Google Scholar] [CrossRef] [PubMed]
- Oyama, S.T.; Lee, Y.K. Mechanism of hydrodenitrogenation on phosphides and sulfides. J. Phys. Chem. B 2005, 109, 2109–2119. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.M.; Hensley, J.E.; Medlin, J.W. Bifunctional Catalysts for Upgrading of Biomass-Derived Oxygenates: A Review. ACS Catal. 2016, 6, 5026–5043. [Google Scholar] [CrossRef]
- Lu, X.; Baker, M.A.; Anjum, D.H.; Basina, G.; Hinder, S.J.; Papawassiliou, W.; Pell, A.J.; Karagianni, M.; Papavassiliou, G.; Shetty, D.; et al. Ni2P Nanoparticles Embedded in Mesoporous SiO2 for Catalytic Hydrogenation of SO2 to Elemental S. ACS Appl. Nano Mater. 2021, 4, 5665–5676. [Google Scholar] [CrossRef]
- Lu, X.; Baker, M.A.; Anjum, D.H.; Papawassiliou, W.; Pell, A.J.; Fardis, M.; Papavassiliou, G.; Hinder, S.J.; Gaber, S.A.A.; Gaber, D.A.A.; et al. Nickel Phosphide Nanoparticles for Selective Hydrogenation of SO2 to H2S. ACS Appl. Nano Mater. 2021, 4, 6568–6582. [Google Scholar] [CrossRef]
- Yao, Z.W.; Wang, L.; Dong, H. A new approach to the synthesis of molybdenum phosphide via internal oxidation and reduction route. J. Alloys Compd. 2009, 473, L10–L12. [Google Scholar] [CrossRef]
- Aitken, J.A.; Ganzha-Hazen, V.; Brock, S.L. Solvothermal syntheses of Cu3P via reactions of amorphous red phosphorus with a variety of copper sources. J. Solid State Chem. 2005, 178, 970–975. [Google Scholar] [CrossRef]
- Cecilia, J.A.; Infantes-Molina, A.; Rodríguez-Castellón, E.; Jiménez-López, A. Dibenzothiophene hydrodesulfurization over cobalt phosphide catalysts prepared through a new synthetic approach: Effect of the support. Appl. Catal. B Environ. 2009, 92, 100–113. [Google Scholar] [CrossRef]
- Zhang, G.; Li, B.; Liu, M.; Yuan, S.; Niu, L. Liquid Phase Synthesis of CoP Nanoparticles with High Electrical Conductivity for Advanced Energy Storage. J. Nanomater. 2017, 2017, 9728591. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, C.; Li, W.; Yang, Q. One-Pot Synthesis of Carbon-Coated Ni5P4 Nanoparticles and CoP Nanorods for High-Rate and High-Stability Lithium-ion Batteries. J. Mater. Chem. A 2015, 3, 23345–23351. [Google Scholar] [CrossRef]
- Ha, D.; Moreau, L.M.; Bealing, C.R.; Zhang, H.; Hennig, R.G.; Robinson, R.D. The structural evolution and diffusion during the chemical transformation from cobalt to cobalt phosphide nanoparticles. J. Mater. Chem. 2011, 21, 11498–11510. [Google Scholar] [CrossRef] [Green Version]
- D’Accriscio, F.; Schrader, E.; Sassoye, C.; Selmane, M.; André, R.F.; Lamaison, S.; Wakerley, D.; Fontecave, M.; Mougel, V.; le Corre, G.; et al. A Single Molecular Stoichiometric P-Source for Phase-Selective Synthesis of Crystalline and Amorphous Iron Phosphide Nanocatalysts. ChemNanoMat 2020, 6, 1208–1219. [Google Scholar] [CrossRef]
- Li, H.; Lu, S.; Sun, J.; Pei, J.; Liu, D.; Xue, Y.; Mao, J.; Zhu, W.; Zhuang, Z. Phase controlled synthesis of nickel phosphide nanocrystals and their electro-catalytic performance for hydrogen evolution reaction. Chem. Eur. J. 2018, 24, 11748–11754. [Google Scholar] [CrossRef]
- Sun, S.; Zhou, X.; Cong, B.; Hong, W.; Chen, G. Tailoring the d-Band Centers Endows (NixFe1-x)2P Nanosheets with Efficient Oxygen Evolution Catalysis. ACS Catal. 2020, 10, 9086–9097. [Google Scholar] [CrossRef]
- Ye, E.; Zhang, S.Y.; Lim, S.H.; Bosman, M.; Zhang, Z.; Win, K.Y.; Han, M.Y. Ternary cobalt–iron phosphide nanocrystals with controlled compositions, properties, and morphologies from nanorods and nanorice to split nanostructures. Chem.–A Eur. J. 2011, 17, 5982–5988. [Google Scholar] [CrossRef]
- Li, D.; Arachchige, M.P.; Kulikowski, B.; Lawes, G.; Seda, T.; Brock, S.L. Control of Composition and Size in Discrete Co x Fe2–x P Nanoparticles: Consequences for Magnetic Properties. Chem. Mater. 2016, 28, 3920–3927. [Google Scholar] [CrossRef]
- Mendoza-Garcia, A.; Zhu, H.; Yu, Y.; Li, Q.; Zhou, L.; Su, D.; Kramer, M.J.; Sun, S. Controlled Anisotropic Growth of Co-Fe-P from Co-Fe-O Nanoparticles. Angew. Chem. 2015, 127, 9778–9781. [Google Scholar] [CrossRef]
- Perera, S.C.; Tsoi, G.; Wenger, L.E.; Brock, S.L. Synthesis of MnP nanocrystals by treatment of metal carbonyl complexes with phosphines: A new, versatile route to nanoscale transition metal phosphides. J. Am. Chem. Soc. 2003, 125, 13960–13961. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, X.; Zhu, L.; Zhang, H.; Chen, B. Hydrodeoxygenation of phenol as a bio-oil model compound over intimate contact noble metal–Ni2P/SiO2 catalysts. RSC Adv. 2015, 5, 80388–80396. [Google Scholar] [CrossRef]
- Bui, P.; Cecilia, J.A.; Oyama, S.T.; Takagaki, A.; Infantes-Molina, A.; Zhao, H. Dan Li, Rodríguez-Castellón, E.; López, A.J. Studies of the synthesis of transition metal phosphides and their activity in the hydrodeoxygenation of a biofuel model compound. J. Catal. 2012, 294, 184–198. [Google Scholar] [CrossRef]
- Koike, N.; Hosokai, S.; Takagaki, A.; Nishimura, S.; Kikuchi, R.; Ebitani, K.; Suzuki, Y.; Oyama, S.T. Upgrading of pyrolysis bio-oil using nickel phosphide catalysts. J. Catal. 2016, 333, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Aguado, E.; Infantes-Molina, A.; Cecilia, J.A.; Ballesteros-Plata, D.; López-Olmo, R.; Rodríguez-Castellón, E. CoxPy Catalysts in HDO of Phenol and Dibenzofuran: Effect of P Content. Top. Catal. 2017, 60, 1094–1107. [Google Scholar] [CrossRef]
- Wu, S.-K.; Lai, P.-C.; Lin, Y.-C. Atmospheric Hydrodeoxygenation of Guaiacol over Nickel Phosphide Catalysts: Effect of Phosphorus Composition. Catal. Lett. 2014, 144, 878–889. [Google Scholar] [CrossRef]
- Gutiérrez-Rubio, S.; Berenguer, A.; Přech, J.; Opanasenko, M.; Ochoa-Hernández, C.; Pizarro, P.; Čejka, J.; Serrano, D.P.; Coronado, J.M.; Moreno, I. Guaiacol Hydrodeoxygenation over Ni2P Supported on 2D-Zeolites. Catal. Today 2020, 345, 48–58. [Google Scholar] [CrossRef]
- Rensel, D.J.; Rouvimov, S.; Gin, M.E.; Hicks, J.C. Highly Selective Bimetallic FeMoP Catalyst for C–O Bond Cleavage of Aryl Ethers. J. Catal. 2013, 305, 256–263. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, M.; Chen, J. Effects of P/Ni ratio and Ni content on performance of γ-Al2O3-supported nickel phosphides for deoxygenation of methyl laurate to hydrocarbons. Appl. Surf. Sci. 2016, 360, 353–364. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, J.; Shi, H. Deoxygenation of Methyl Laurate as a Model Compound to Hydrocarbons on Ni2P/SiO2, Ni2P/MCM-41, and Ni2P/SBA-15 Catalysts with Different Dispersions. Energy Fuels 2013, 27, 3400–3409. [Google Scholar] [CrossRef]
- Yang, Y.; Ochoa-Hernández, C.; de la Peña O’Shea, V.A.; Coronado, J.M.; Serrano, D.P. Ni2P/SBA-15 As a Hydrodeoxygenation Catalyst with Enhanced Selectivity for the Conversion of Methyl Oleate Into n-Octadecane. ACS Catal. 2012, 2, 592–598. [Google Scholar] [CrossRef]
- Chen, J.; Yang, Y.; Shi, H.; Li, M.; Chu, Y.; Pan, Z.; Yu, X. Regulating Product Distribution in Deoxygenation of Methyl Laurate on Silica-Supported Ni–Mo Phosphides: Effect of Ni/Mo Ratio. Fuel 2014, 129, 1–10. [Google Scholar] [CrossRef]
- Xin, H.; Guo, K.; Li, D.; Yang, H.; Hu, C. Production of high-grade diesel from palmitic acid over activated carbon-supported nickel phosphide catalysts. Appl. Catal. B Environ. 2016, 187, 375–385. [Google Scholar] [CrossRef]
- Liu, Y.; Yao, L.; Xin, H.; Wang, G.; Li, D.; Hu, C. The production of diesel-like hydrocarbons from palmitic acid over HZSM-22 supported nickel phosphide catalysts. Appl. Catal. B Environ. 2015, 504–514. [Google Scholar] [CrossRef]
- Le, T.A.; Ly, H.V.; Kim, J.; Kim, S.S.; Choi, J.H.; Woo, H.C.; Othman, M.R. Hydrodeoxygenation of 2-furyl methyl ketone as a model compound in bio-oil from pyrolysis of Saccharina Japonica Alga in fixed-bed reactor. Chem. Eng. J. 2014, 250, 157–163. [Google Scholar] [CrossRef]
- Inocencio, C.V.M.; de Souza, P.M.; Rabelo-Neto, R.C.; da Silva, V.T.; Noronha, F.B. A systematic study of the synthesis of transition metal phosphides and their activity for hydrodeoxygenation of phenol. Catal. Today 2021, 381, 133–142. [Google Scholar] [CrossRef]
- Peroni, M. Transition Metal Phosphides Catalysts for the Hydroprocessing of Triglycerides to Green Fuel. Ph.D. Thesis, Technical University of Munich, Munich, Germany, 2017. [Google Scholar]
- Vereshchagin, O.S.; Pankin, D.V.; Smirnov, M.B.; Vlasenko, N.S.; Shilovskikh, V.V.; Britvin, S.N. Raman spectroscopy: A promising tool for the characterization of transition metal phosphides. J. Alloys Compd. 2021, 853, 156468. [Google Scholar] [CrossRef]
- Layman, K.A.; Bussell, M.E. Infrared Spectroscopic Investigation of CO Adsorption on Silica-Supported Nickel Phosphide Catalysts. J. Phys. Chem. B 2004, 108, 10930–10941. [Google Scholar] [CrossRef]
- Ballinger, T.H.; Yates, J.T., Jr. IR spectroscopic detection of Lewis acid sites on alumina using adsorbed carbon monoxide. Correlation with aluminum-hydroxyl group removal. Langmuir 1991, 7, 3041–3045. [Google Scholar] [CrossRef]
- Wang, X.; Clark, P.; Oyama, S.T. Synthesis, characterization, and hydrotreating activity of several iron group transition metal phosphides. J. Catal. 2002, 208, 321–331. [Google Scholar] [CrossRef]
- Feng, Z.; Liang, C.; Wu, W.; Wu, Z.; van Santen, R.A.; Li, C. Carbon monoxide adsorption on molybdenum phosphides: Fourier transform infrared spectroscopic and density functional theory studies. J. Phys. Chem. B 2003, 107, 13698–13702. [Google Scholar] [CrossRef]
- Singh, K.P.; Bae, E.J.; Yu, J.S. Fe–P: A new class of electroactive catalyst for oxygen reduction reaction. J. Am. Chem. Soc. 2015, 137, 3165–3168. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Gu, S.; Liu, R.; Li, C.M. Highly active and inexpensive iron phosphide nanorods electrocatalyst towards hydrogen evolution reaction. Int. J. Hydrogen Energy 2015, 40, 14272–14278. [Google Scholar] [CrossRef]
- Burns, A.W.; Layman, K.A.; Bale, D.H.; Bussell, M.E. Understanding the relationship between composition and hydrodesulfurization properties for cobalt phosphide catalysts. Appl. Catal. A Gen. 2008, 343, 68–76. [Google Scholar] [CrossRef]
- Cecilia, J.A.; Infantes-Molina, A.; Rodríguez-Castellón, E.; Jiménez-López, A. A novel method for preparing an active nickel phosphide catalyst for HDS of dibenzothiophene. J. Catal. 2009, 263, 4–15. [Google Scholar] [CrossRef]
- Chen, J.; Shi, H.; Li, L.; Li, K. Deoxygenation of methyl laurate as a model compound to hydrocarbons on transition metal phosphide catalysts. Appl. Catal. B Environ. 2014, 144, 870–884. [Google Scholar] [CrossRef]
- Bekaert, E.; Bernardi, J.; Boyanov, S.; Monconduit, L.; Doublet, M.L.; Ménétrier, M. Direct Correlation between the 31P MAS NMR Response and the Electronic Structure of Some Transition Metal Phosphides. J. Phys. Chem. C 2008, 112, 20481–20490. [Google Scholar] [CrossRef]
- Zuzaniuk, V.; Prins, R. Synthesis and characterization of silica-supported transition-metal phosphides as HDN catalysts. J. Catal. 2003, 219, 85–96. [Google Scholar] [CrossRef]
- Papawassiliou, W.; Carvalho, J.P.; Panopoulos, N.; al Wahedi, Y.; Wadi, V.K.S.; Lu, X.; Polychronopoulou, K.; Lee, J.B.; Lee, S.; Kim, C.Y.; et al. Crystal and electronic facet analysis of ultrafine Ni2P particles by solid-state NMR nanocrystallography. Nat. Commun. 2021, 12, 4334. [Google Scholar] [CrossRef]
- Wanmolee, W.; Sosa, N.; Junkaew, A.; Youngjan, S.; Geantet, C.; Afanasiev, P.; Puzenat, E.; Laurenti, D.; Faungnawakij, K.; Khemthong, P. Phase speciation and surface analysis of copper phosphate on high surface area silica support by in situ XAS/XRD and DFT: Assessment for guaiacol hydrodeoxygenation. Appl. Surf. Sci. 2022, 574, 151577. [Google Scholar] [CrossRef]
- Infantes-Molina, A.; Gralberg, E.; Cecilia, J.; Finocchio, E.; Rodríguez-Castellón, E. Nickel and cobalt phosphides as effective catalysts for oxygen removal of dibenzofuran: Role of contact time, hydrogen pressure and hydrogen/feed molar ratio. Catal. Sci. Technol. 2015, 5, 3403–3415. [Google Scholar] [CrossRef]
- Mendes, M.J.; Santos, O.A.A.; Jordão, E.; Silva, A.M. Hydrogenation of oleic acid over ruthenium catalysts. Appl. Catal. A Gen. 2001, 217, 253–262. [Google Scholar] [CrossRef]
- Pestman, R.; Koster, R.M.; Pieterse, J.A.Z.; Ponec, V. Reactions of Carboxylic Acids on Oxides: 1. Selective Hydrogenation of Acetic Acid to Acetaldehyde. J. Catal. 1997, 168, 255–264. [Google Scholar] [CrossRef]
- He, Z.; Wang, X. Hydrodeoxygenation of model compounds and catalytic systems for pyrolysis bio-oils upgrading. Catal. Sustain. Energy 2013, 1, 28–52. [Google Scholar] [CrossRef]
- Whiffen, V.M.L.; Smith, K.J. Hydrodeoxygenation of 4-Methylphenol over Unsupported MoP, MoS2, and MoOx Catalysts. Energy Fuels 2010, 24, 4728–4737. [Google Scholar] [CrossRef]
- Kanda, Y.; Chiba, T.; Aranai, R.; Yasuzawa, T.; Ueno, R.; Toyao, T.; Kato, K.; Obora, Y.; Shimizu, K.; Uemichi, Y. Catalytic Activity of Rhodium Phosphide for Selective Hydrodeoxygenation of Phenol. Chem. Lett. 2019, 48, 471–474. [Google Scholar] [CrossRef] [Green Version]
- Feitosa, L.F.; Berhault, G.; Laurenti, D.; da Silva, V.T. Effect of the nature of the carbon support on the guaiacol hydrodeoxygenation performance of nickel phosphide: Comparison between carbon nanotubes and a mesoporous carbon support. Ind. Eng. Chem. Res. 2019, 58, 16164–16181. [Google Scholar] [CrossRef]
- Yu, Z.; Wang, A.; Liu, S.; Yao, Y.; Sun, Z.; Li, X.; Wang, Y.; Camaioni, D.M.; Lercher, J.A. Hydrodeoxygenation of phenolic compounds to cycloalkanes over supported nickel phosphides. Catal. Today 2019, 319, 48–56. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, K.; Liu, H.; Qiao, Z.; Yang, Y.; Ren, K. Hydrodeoxygenation of p-cresol on unsupported Ni–P catalysts prepared by thermal decomposition method. Catal. Commun. 2013, 41, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Jing, Y.; Wang, Y. Catalytic Hydrodeoxygenation of Lignin-Derived Feedstock into Arenes and Phenolics. Front. Chem. Eng. 2020, 10, 10. [Google Scholar] [CrossRef]
- Zhao, H.Y.; Li, D.; Bui, P.; Oyamaa, S.T. Hydrodeoxygenation of guaiacol as model compound for pyrolysis oil on transition metal phosphide hydroprocessing catalysts. Appl. Catal. A Gen. 2011, 391, 305–310. [Google Scholar] [CrossRef]
- Hurff, S.J.; Klein, M.T. Reaction pathway analysis of thermal and catalytic lignin fragmentation by use of model compounds. Ind. Eng. Chem. Fundam. 1983, 22, 426–430. [Google Scholar] [CrossRef]
- Bredenberg, J.S.; Huuska, M.; Toropainen, P. Hydrogenolysis of differently substituted methoxyphenols. J. Catal. 1989, 120, 401–408. [Google Scholar] [CrossRef]
- Huuska, M.; Rintala, J. Effect of catalyst acidity on the hydrogenolysis of anisole. J. Catal. 1985, 94, 230–238. [Google Scholar] [CrossRef]
- Ferrari, M.; Maggi, R.; Delmon, B.; Grange, P. Influences of the hydrogen sulfide partial pressure and of a nitrogen compound on the hydrodeoxygenation activity of a CoMo/carbon catalyst. J. Catal. 2001, 198, 47–55. [Google Scholar] [CrossRef]
- Gonçalves, V.O.; de Souza, P.M.; Cabioc’h, T.; da Silva, V.T.; Noronha, F.B.; Richard, F. Hydrodeoxygenation of m-cresol over nickel and nickel phosphide based catalysts. Influence of the nature of the active phase and the support. Appl. Catal. B Environ. 2017, 219, 619–628. [Google Scholar] [CrossRef]
- Shi, H.; Chen, J.X.; Yang, Y.; Tian, S.S. Catalytic deoxygenation of methyl laurate as a model compound to hydrocarbons on nickel phosphide catalysts: Remarkable support effect. Fuel Process. Technol. 2014, 118, 161–170. [Google Scholar] [CrossRef]
- Peroni, M.; Lee, I.; Huang, X.; Baráth, E.; Gutiérrez, O.Y.; Lercher, J.A. Deoxygenation of palmitic acid on unsupported transition metal phosphides. ACS Catal. 2017, 7, 6331–6341. [Google Scholar] [CrossRef]
- Lee, S.P.; Chen, Y.W. Selective hydrogenation of furfural on Ni− P, Ni− B, and Ni− P− B ultrafine materials. Ind. Eng. Chem. Res. 1999, 38, 2548–2556. [Google Scholar] [CrossRef]
- Zhang, J.; Matsubara, K.; Yun, G.N.; Zheng, H.; Takagaki, A.; Kikuchi, R.; Oyama, S.T. Comparison of phosphide catalysts prepared by temperature-programmed reduction and liquid-phase methods in the hydrodeoxygenation of 2-methylfuran. Appl. Catal. A Gen. 2017, 548, 39–46. [Google Scholar] [CrossRef]
- Yu, Z.; Meng, F.; Wang, Y.; Sun, Z.; Liu, Y.; Shi, C.; Wang, W.; Wang, A. Catalytic transfer hydrogenation of levulinic acid to γ-valerolactone over Ni3P-CePO4 catalysts. Ind. Eng. Chem. Res. 2020, 59, 7416–7425. [Google Scholar] [CrossRef]
- Wilson, C.L. Reactions of furan compounds. Part V. Formation of furan from furfuraldehyde by the action of nickel or cobalt catalysts: Importance of added hydrogen. J. Chem. Soc. 1945, 61–63. [Google Scholar] [CrossRef]
- Kijeński, J.K.; Winiarek, P.; Paryjczak, T.; Lewicki, A.; Mikołajska, A. Platinum deposited on monolayer supports in selective hydrogenation of furfural to furfuryl alcohol. Appl. Catal. A 2002, 233, 171–182. [Google Scholar] [CrossRef]
- Sitthisa, S.; Pham, T.; Prasomsri, T.; Sooknoi, T.; Mallinson, R.G.; Resasco, D.E. Conversion of furfural and 2-methylpentanal on Pd/SiO2 and Pd–Cu/SiO2 catalysts. J. Catal. 2011, 280, 17–27. [Google Scholar] [CrossRef]
- Movick, W.J.; Yun, G.; Vargheese, V.; Bando, K.K.; Kikuchi, R.; Oyama, S.T. Hydrodeoxygenation of benzofuran on novel CoPdP catalysts supported on potassium ion exchanged ultra-stable Y-zeolites. J. Catal. 2021, 403, 160–172. [Google Scholar] [CrossRef]
- Cecilia, J.A.; Infantes-Molina, A.; Rodríguez-Castellón, E.; Jiménez-López, A.; Oyama, S.T. Oxygen-removal of dibenzofuran as a model compound in biomass derived bio-oil on nickel phosphide catalysts: Role of phosphorus. Appl. Catal. B Environ. 2013, 136–137, 140–149. [Google Scholar] [CrossRef]
- Liu, T.; Li, A.; Wang, C.; Zhou, W.; Liu, S.; Guo, L. Interfacial Electron Transfer of Ni2P–NiP2 Polymorphs Inducing Enhanced Electrochemical Properties. Adv. Mater. 2018, 30, 1803590. [Google Scholar] [CrossRef]
- Bonita, Y.; Hicks, J.C. Periodic Trends from Metal Substitution in Bimetallic Mo-Based Phosphides for Hydrodeoxygenation and Hydrogenation Reactions. J. Phys. Chem. C 2018, 122, 13322–13332. [Google Scholar] [CrossRef]
- Cho, A.; Shin, J.; Takagaki, A.; Kikuchi, R.; Oyama, S.T. Ligand and ensemble effects in bimetallic NiFe phosphide catalysts for the hydrodeoxygenation of 2-methyltetrahydrofuran. Top. Catal. 2012, 55, 969–980. [Google Scholar] [CrossRef]
- Kang, Q.; Li, M.; Shi, J.; Lu, Q.; Gao, F. A Universal Strategy for Carbon-Supported Transition Metal Phosphides as High-Performance Bifunctional Electrocatalysts towards Efficient Overall Water Splitting. ACS Appl. Mater. Interfaces 2020, 12, 19447–19456. [Google Scholar] [CrossRef]
- Weigold, H. Behaviour of Co-Mo-Al2O3 catalysts in the hydrodeoxygenation of phenols. Fuel 1982, 61, 1021–1026. [Google Scholar] [CrossRef]
- Liu, P.; Rodriguez, J.A.; Takahashi, Y.; Nakamura, K. Water-gas-shift reaction on a Ni2P(001) catalyst: Formation of oxy- phosphides and highly active reaction sites. J. Catal. 2009, 262, 294–303. [Google Scholar] [CrossRef]
- Li, K.; Wang, R.; Chen, J. Hydrodeoxygenation of Anisole over Silica-Supported Ni2P, MoP, and NiMoP Catalysts. Energy Fuels 2011, 25, 854–863. [Google Scholar] [CrossRef]
- Wongnongwa, Y.; Jungsuttiwong, S.; Pimsuta, M.; Khemthong, P.; Kunaseth, M. Mechanistic and thermodynamic insights into the deoxygenation of palm oils using Ni2P catalyst: A combined experimental and theoretical study. Chem. Eng. J. 2020, 399, 125586. [Google Scholar] [CrossRef]
- Moon, J.S.; Kim, E.G.; Lee, Y.K. Active sites of Ni2P/SiO2 catalyst for hydrodeoxygenation of guaiacol: A joint XAFS and DFT study. J. Catal. 2014, 311, 144–152. [Google Scholar] [CrossRef]
- Jain, V.; Bonita, Y.; Brown, A.; Taconi, A.; Hicks, J.C.; Rai, N. Mechanistic insights into hydrodeoxygenation of phenol on bimetallic phosphide catalysts. Catal. Sci. Technol. 2018, 8, 4083–4096. [Google Scholar] [CrossRef]
- He, Y.; Laursen, S. The surface and catalytic chemistry of the first row transition metal phosphides in deoxygenation. Catal. Sci. Technol. 2018, 8, 5302–5314. [Google Scholar] [CrossRef]
- He, Y.; Laursen, S. Trends in the Surface and Catalytic Chemistry of Transition-Metal Ceramics in the Deoxygenation of a Woody Biomass Pyrolysis Model Compound. ACS Catal. 2017, 7, 3169–3180. [Google Scholar] [CrossRef]
- Jia, Z.; Ji, N.; Diao, X.; Li, X.; Zhao, Y.; Lu, X.; Liu, Q.; Liu, C.; Chen, G.; Ma, L.; et al. Highly Selective Hydrodeoxygenation of Lignin to Naphthenes over Three-Dimensional Flower-like Ni2P Derived from Hydrotalcite. ACS Catal. 2022, 12, 1338–1356. [Google Scholar] [CrossRef]
- Wang, S.; Xu, D.; Chen, Y.; Zhou, S.; Zhu, D.; Wen, X.; Yang, Y.; Li, Y. Hydrodeoxygenation of anisole to benzene over an Fe2P catalyst by a direct deoxygenation pathway. Catal. Sci. Technol. 2020, 10, 3015–3023. [Google Scholar] [CrossRef]
- Witzke, M.E.; Almithn, A.; Conrad, C.L.; Triezenberg, M.D.; Hibbitts, D.D.; Flaherty, D.W. In Situ Methods for Identifying Reactive Surface Intermediates during Hydrogenolysis Reactions: C-O Bond Cleavage on Nanoparticles of Nickel and Nickel Phosphides. J. Am. Chem. Soc. 2019, 141, 16671–16684. [Google Scholar] [CrossRef] [PubMed]
- Witzke, M.E.; Almithn, A.; Conrad, C.L.; Hibbitts, D.D.; Flaherty, D.W. Mechanisms and Active Sites for C-O Bond Rupture within 2-Methyltetrahydrofuran over Ni, Ni12P5, and Ni2P Catalysts. ACS Catal. 2018, 8, 7141–7157. [Google Scholar] [CrossRef]
- Lan, X.; Hensen, E.J.M.; Weber, T. Hydrodeoxygenation of Guaiacol over Ni2P/SiO2–Reaction Mechanism and Catalyst Deactivation. Appl. Catal. A Gen. 2018, 550, 57–66. [Google Scholar] [CrossRef]
- Li, D.; Baydoun, H.; Verani, C.N.; Brock, S.L. Efficient Water Oxidation Using CoMnP Nanoparticles. J. Am. Chem. Soc. 2016, 138, 4006–4009. [Google Scholar] [CrossRef]
- Rensel, D.J.; Kim, J.; Jain, V.; Bonita, Y.; Rai, N.; Hicks, J.C. Composition-directed FeXMo2-XP bimetallic catalysts for hydrodeoxygenation reactions. Catal. Sci. Technol. 2017, 7, 1857–1867. [Google Scholar] [CrossRef]
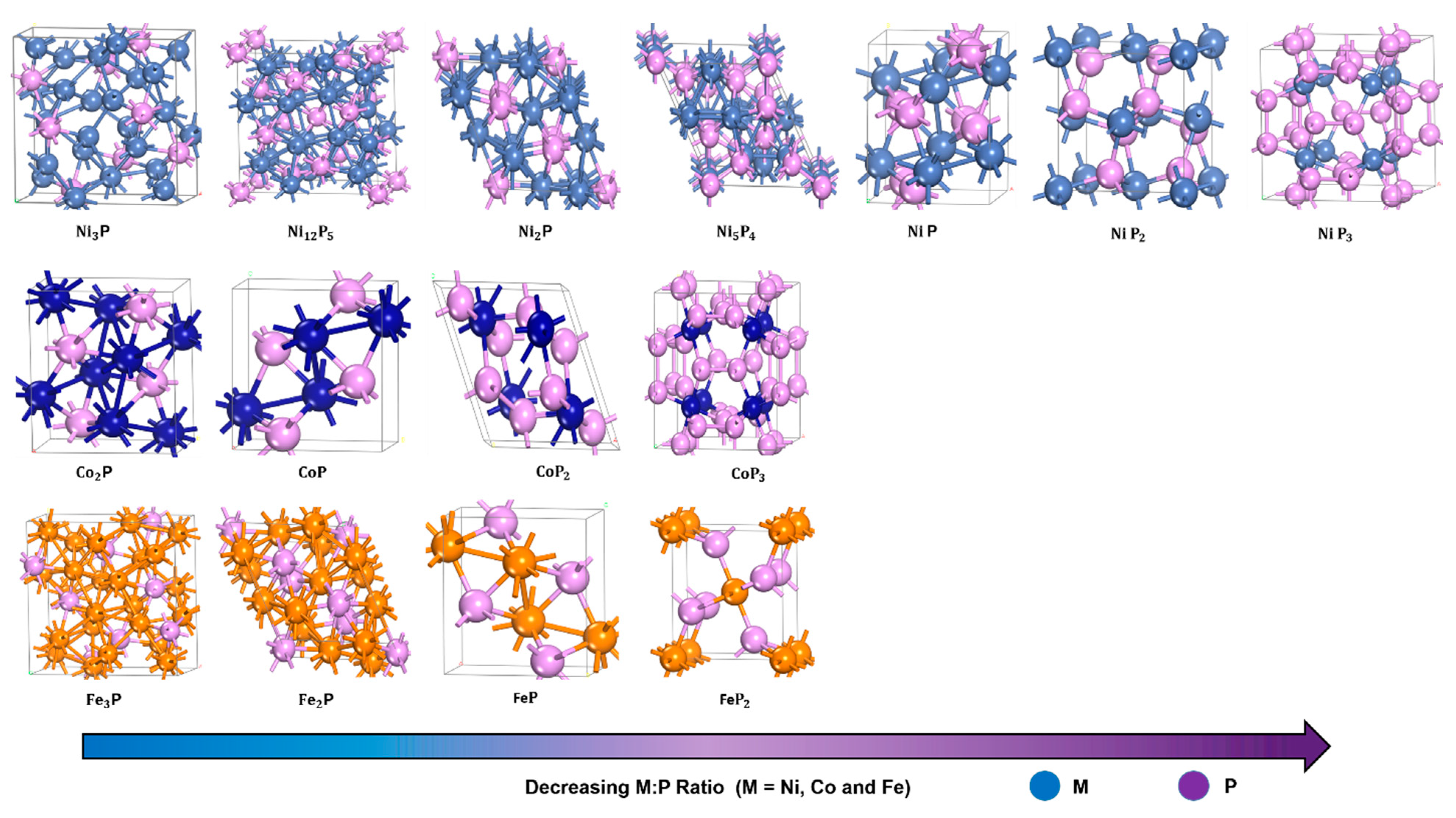

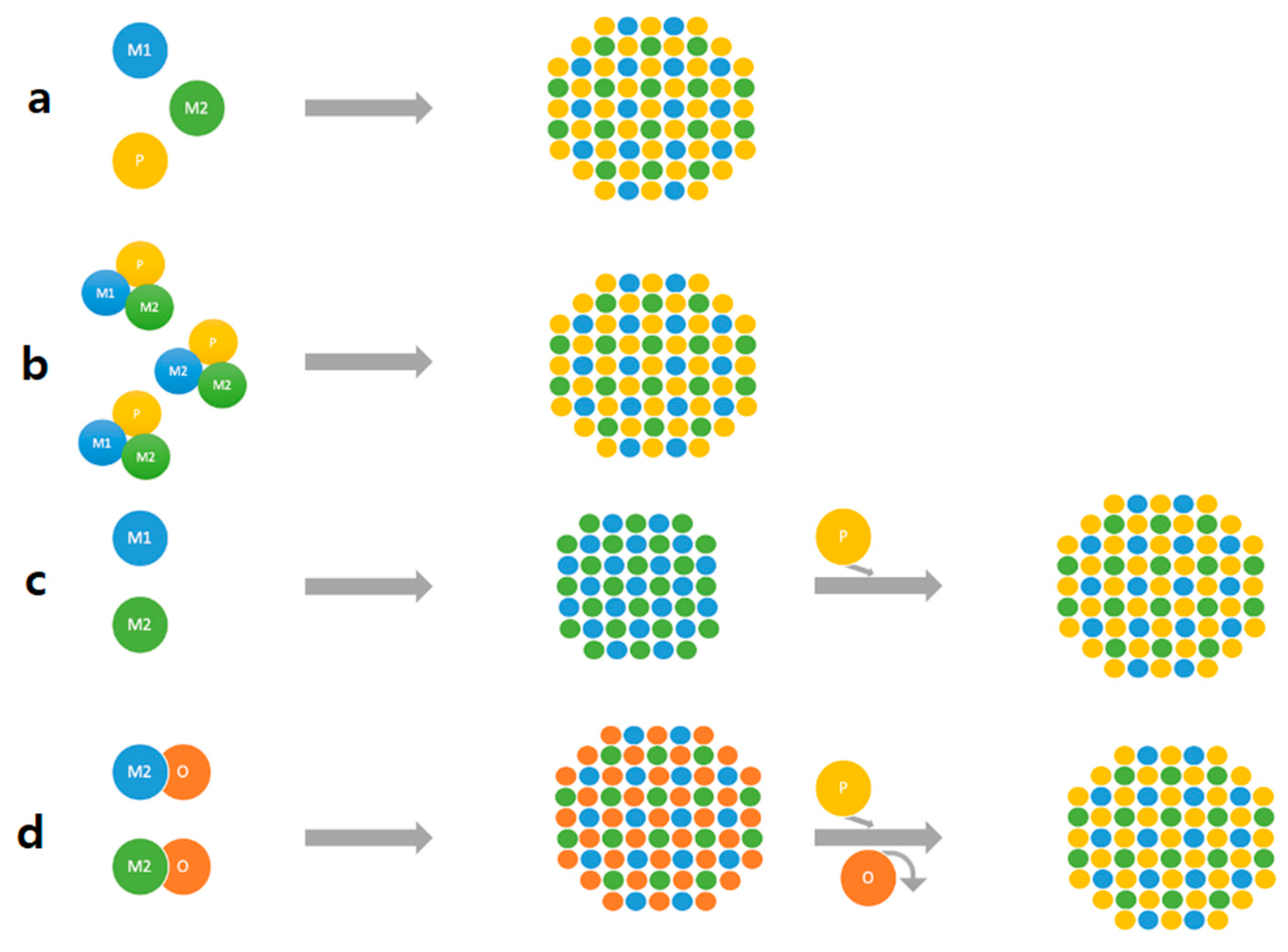
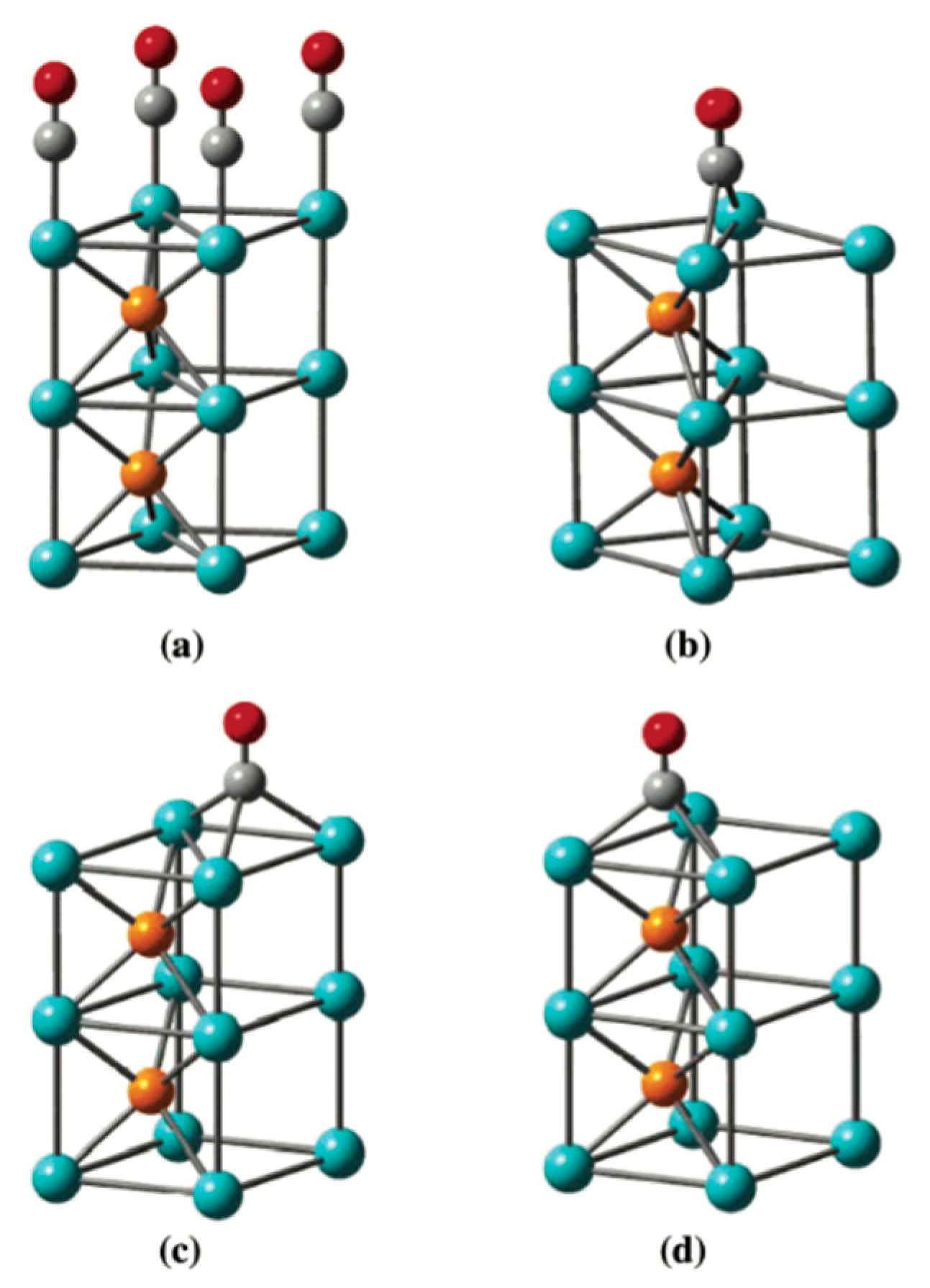
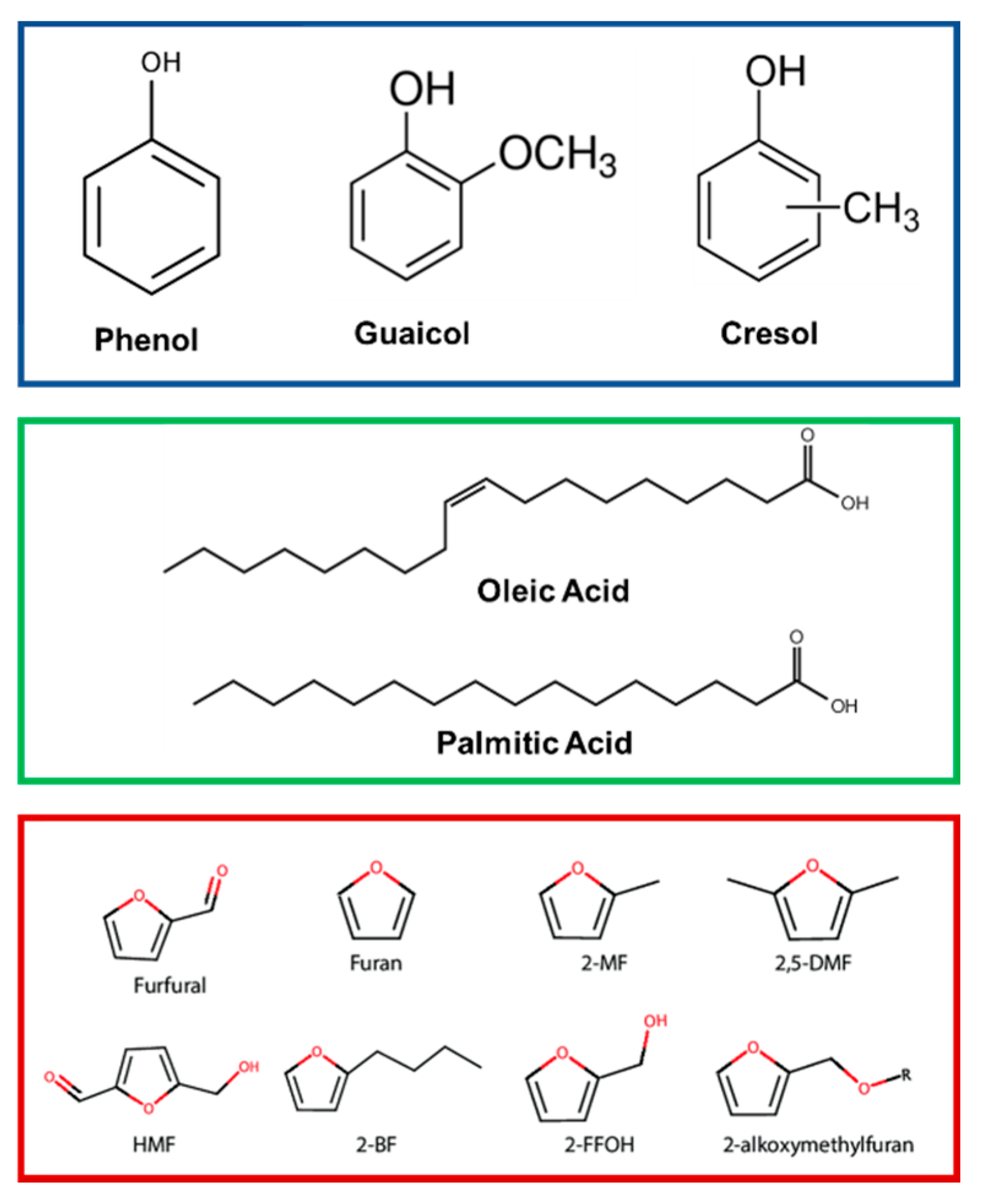
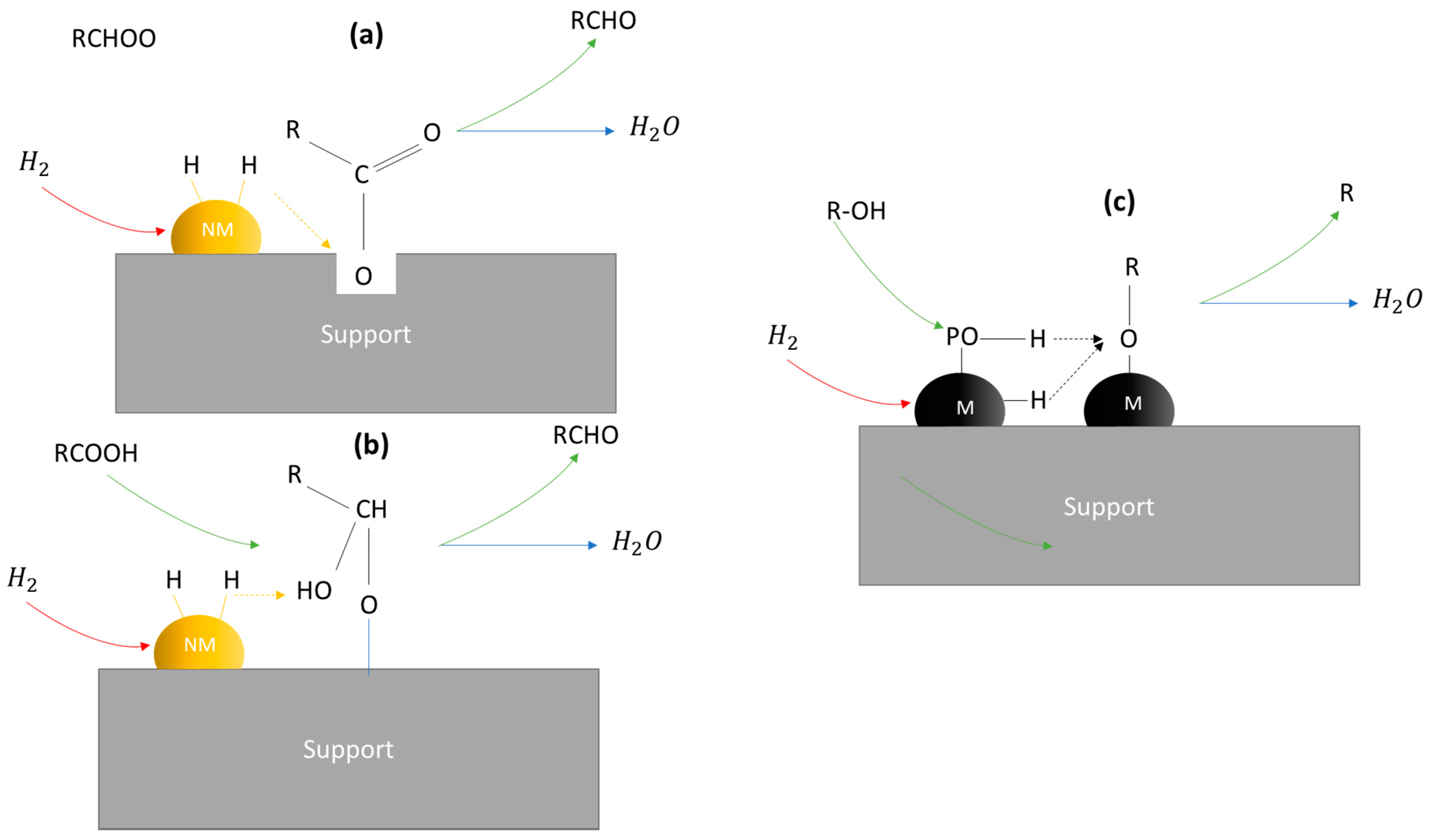
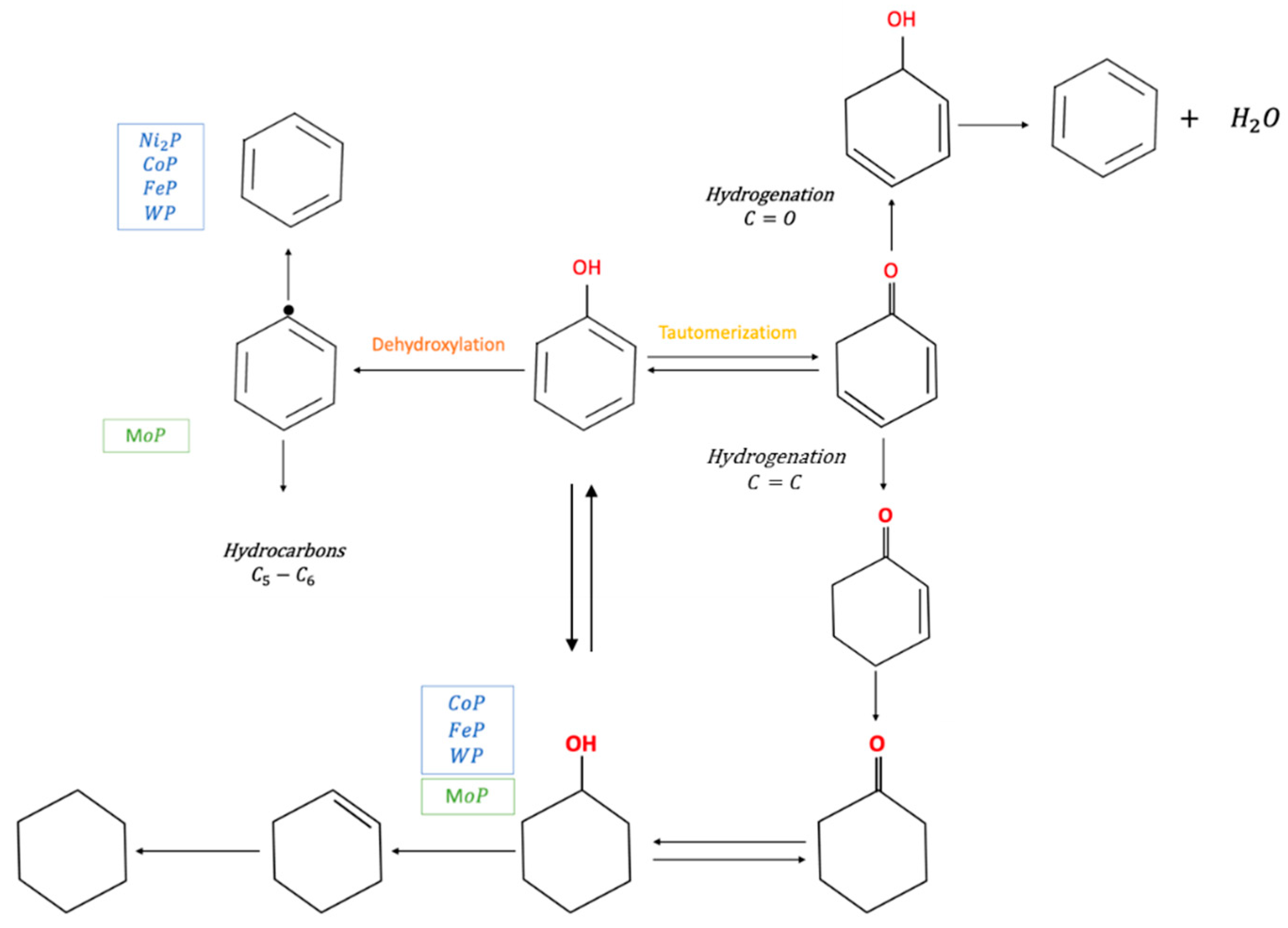
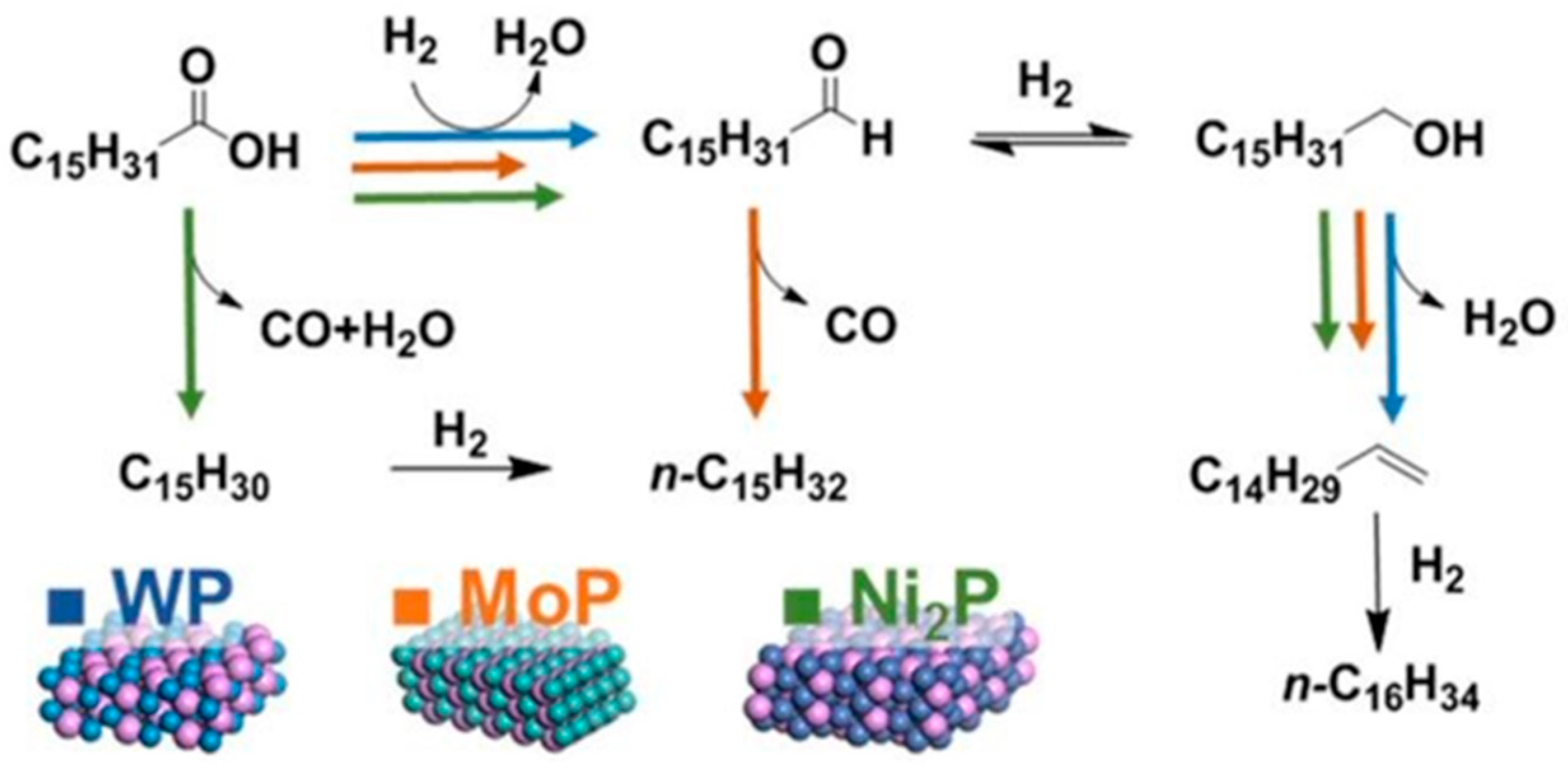
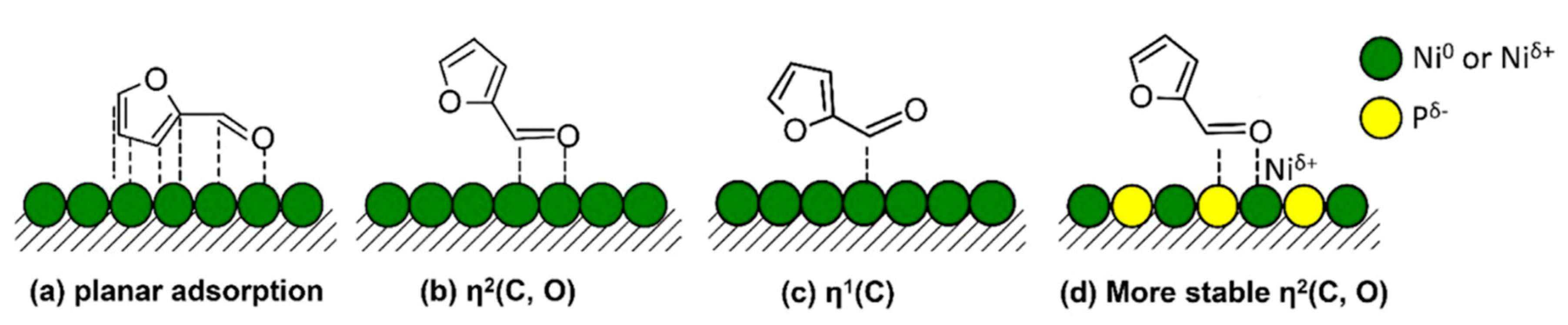
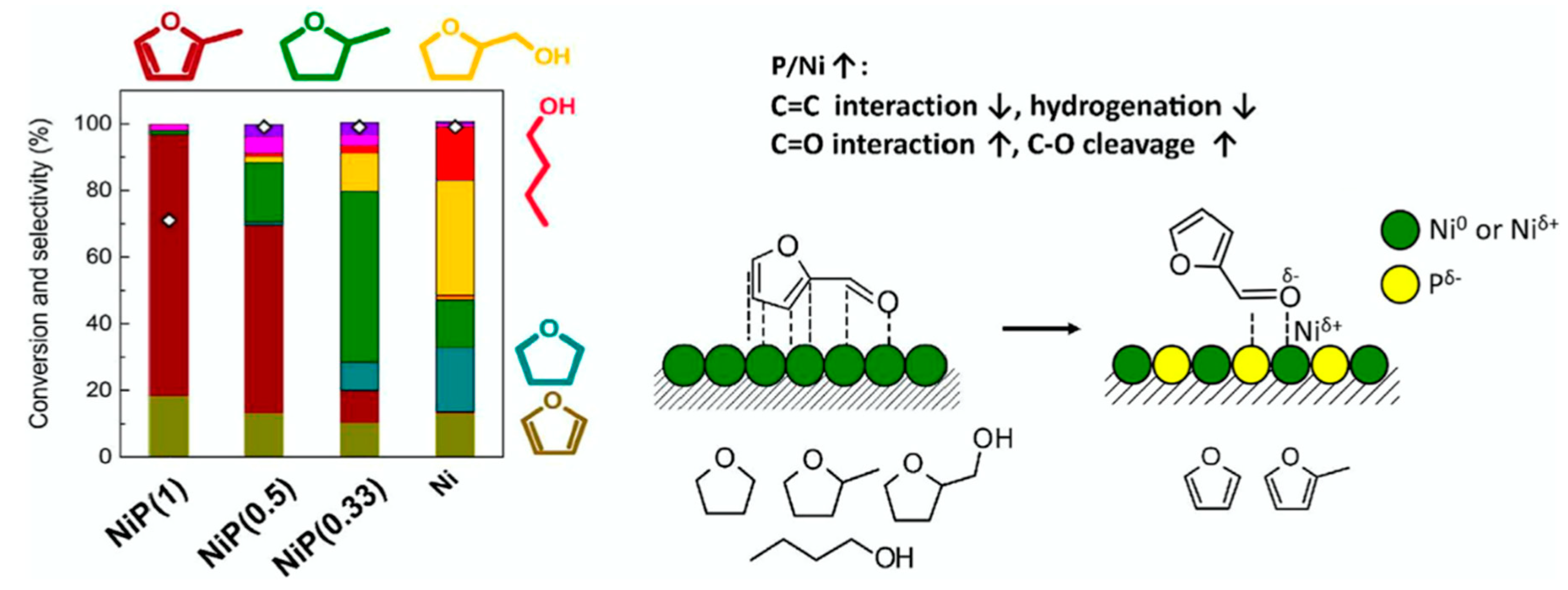

| Metal Phosphide Classes | Examples of Metal Phosphides |
|---|---|
| Metal-rich phosphides | |
| Monophosphides | |
| Phosphorous-rich phosphides | |
| Ionic phosphides |
| Ceramic Properties | Metallic Properties | ||||
|---|---|---|---|---|---|
| Melting point | 830–1530 | Electrical resistivity | 900–25,000 | ||
| Microhardness | 600–1100 | Magnetic susceptibility | 110–620 | ||
| Heat of formation | 30–180 | Heat capacity | 20–50 | ||
| Phosphide Phase | Crystal System, Space Group | Number of Formula Units | References |
|---|---|---|---|
| 3 | [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40] | ||
| 2 | [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40] | ||
| 8 | [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40] | ||
| 4 | [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40] | ||
| 4 | JCPDS No.29-0497 | ||
| 4 | JCPDS No.89-3030 | ||
| 1 | [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40] | ||
| 8 | [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40] | ||
| 4 | JCPDS No.29-1364 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Ali, L.I.; Elmutasim, O.; Al Ali, K.; Singh, N.; Polychronopoulou, K. Transition Metal Phosphides (TMP) as a Versatile Class of Catalysts for the Hydrodeoxygenation Reaction (HDO) of Oil-Derived Compounds. Nanomaterials 2022, 12, 1435. https://doi.org/10.3390/nano12091435
Al-Ali LI, Elmutasim O, Al Ali K, Singh N, Polychronopoulou K. Transition Metal Phosphides (TMP) as a Versatile Class of Catalysts for the Hydrodeoxygenation Reaction (HDO) of Oil-Derived Compounds. Nanomaterials. 2022; 12(9):1435. https://doi.org/10.3390/nano12091435
Chicago/Turabian StyleAl-Ali, Latifa Ibrahim, Omer Elmutasim, Khalid Al Ali, Nirpendra Singh, and Kyriaki Polychronopoulou. 2022. "Transition Metal Phosphides (TMP) as a Versatile Class of Catalysts for the Hydrodeoxygenation Reaction (HDO) of Oil-Derived Compounds" Nanomaterials 12, no. 9: 1435. https://doi.org/10.3390/nano12091435
APA StyleAl-Ali, L. I., Elmutasim, O., Al Ali, K., Singh, N., & Polychronopoulou, K. (2022). Transition Metal Phosphides (TMP) as a Versatile Class of Catalysts for the Hydrodeoxygenation Reaction (HDO) of Oil-Derived Compounds. Nanomaterials, 12(9), 1435. https://doi.org/10.3390/nano12091435