
Atomic Simulations of (8,0)CNT-Graphene by SCC-DFTB Algorithm

Abstract
:1. Introduction
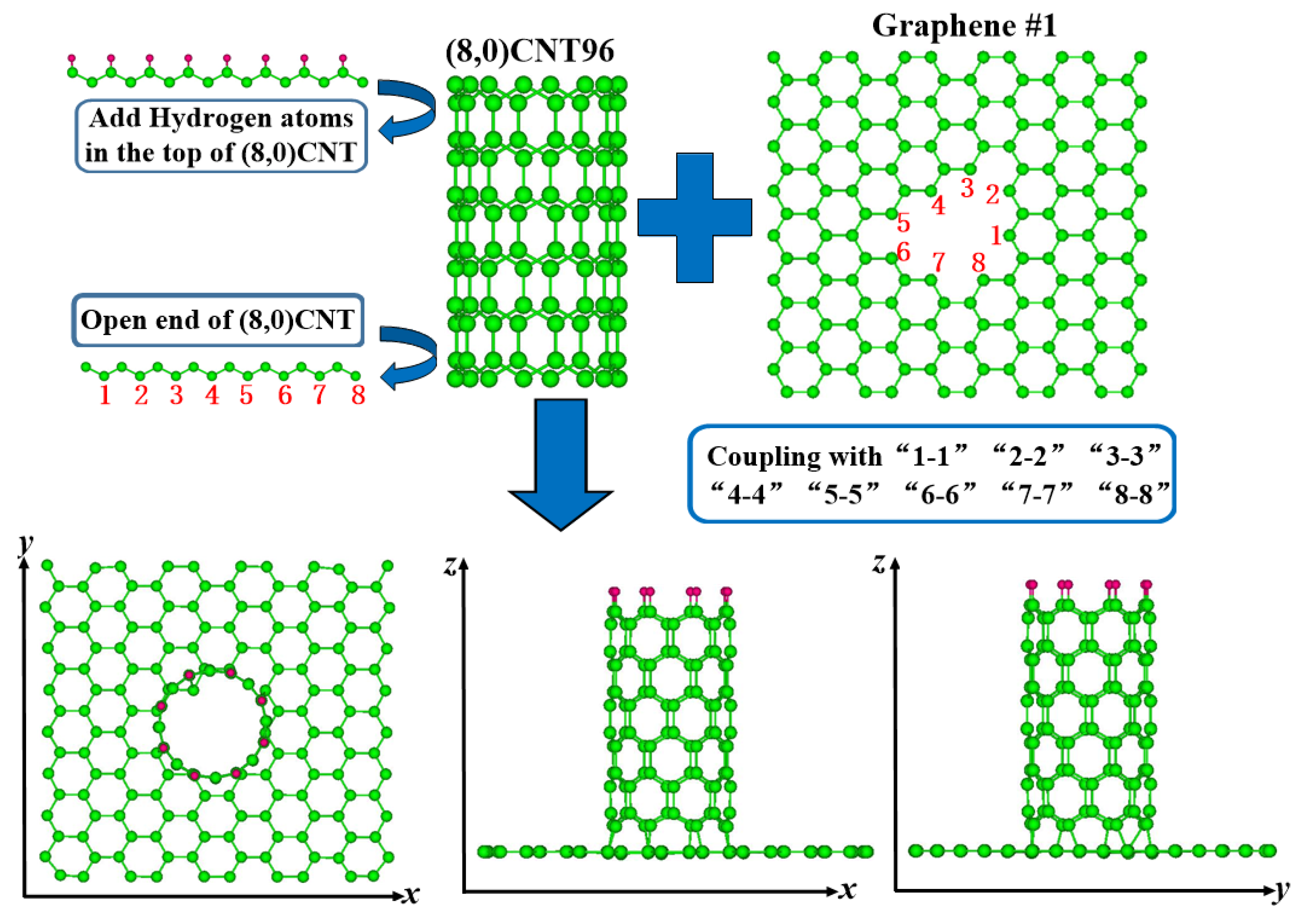
2. Simulation Methodology
3. Results and Discussion
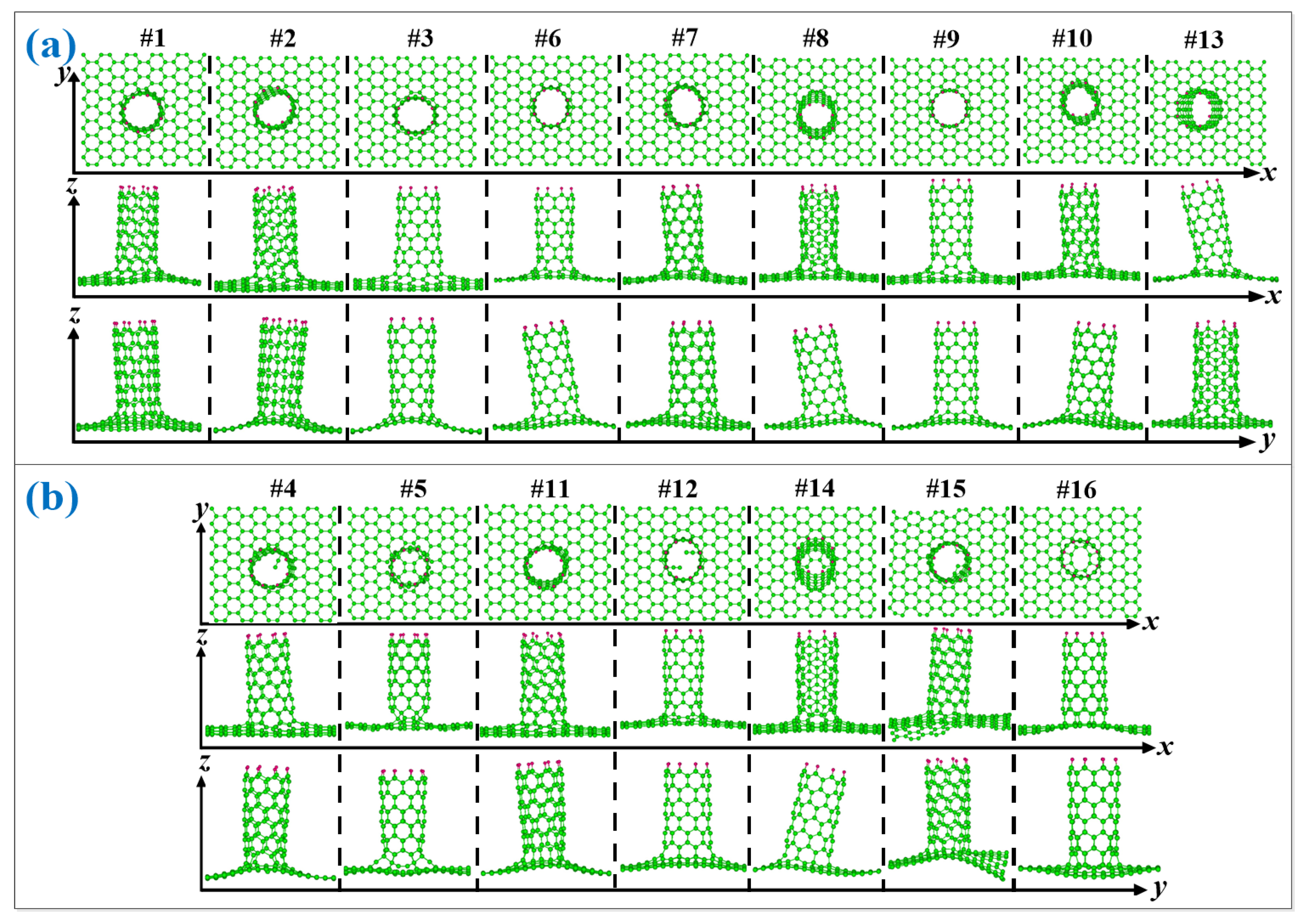
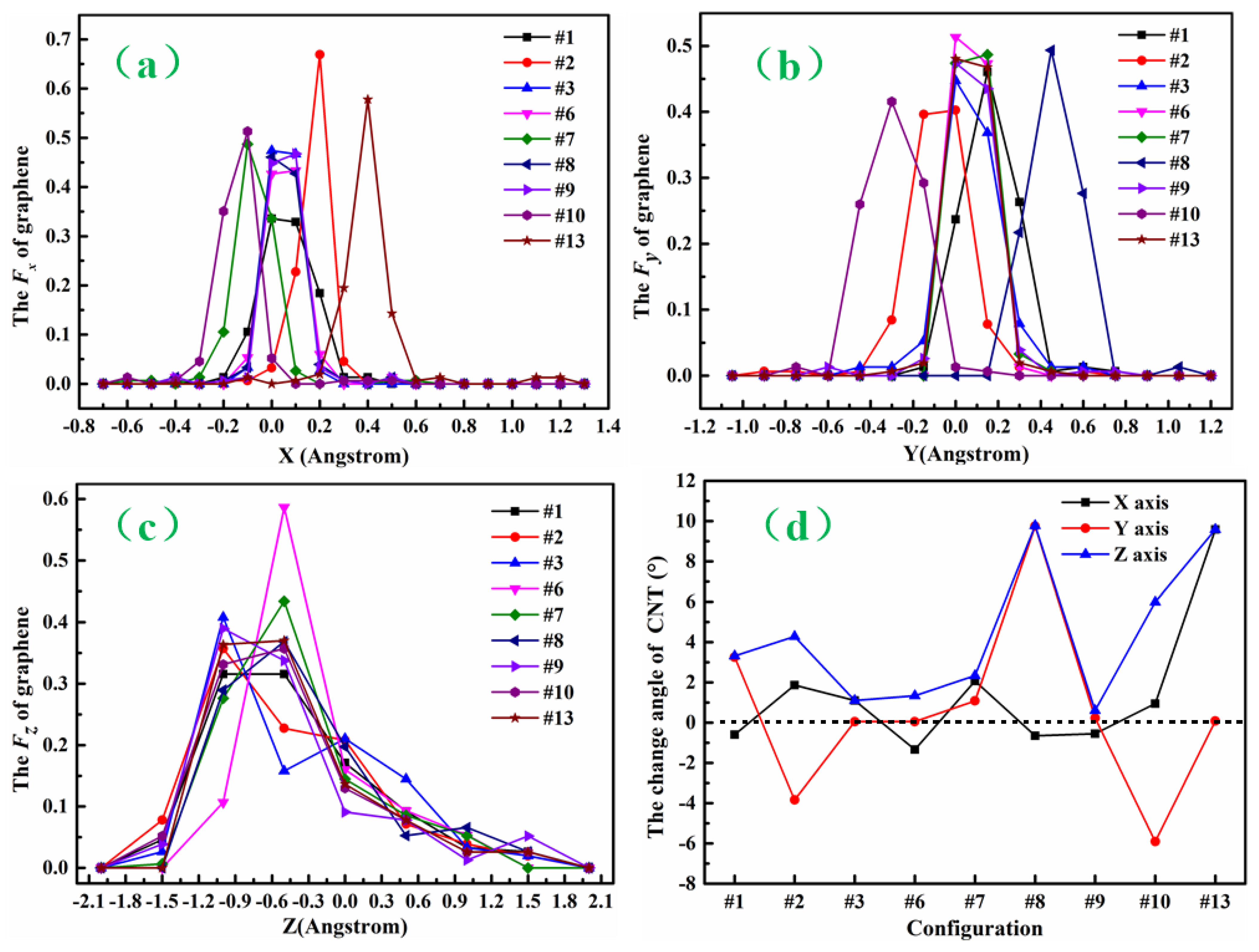
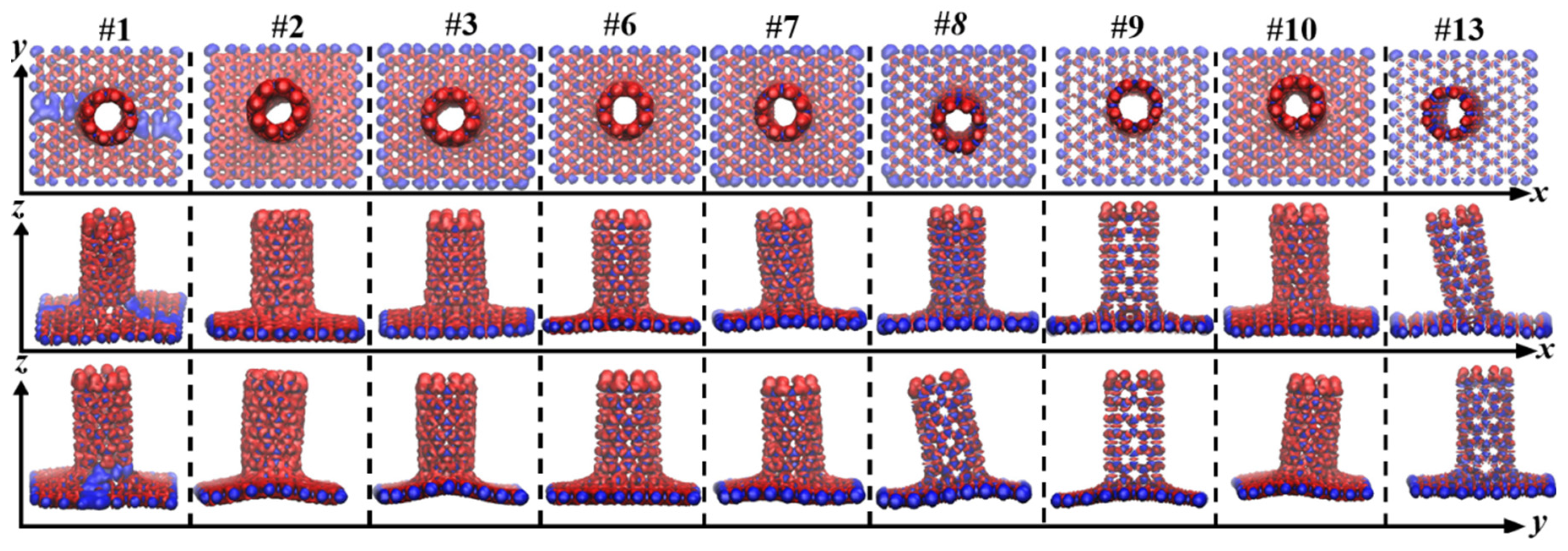
3.1. Structure and Energy Analysis
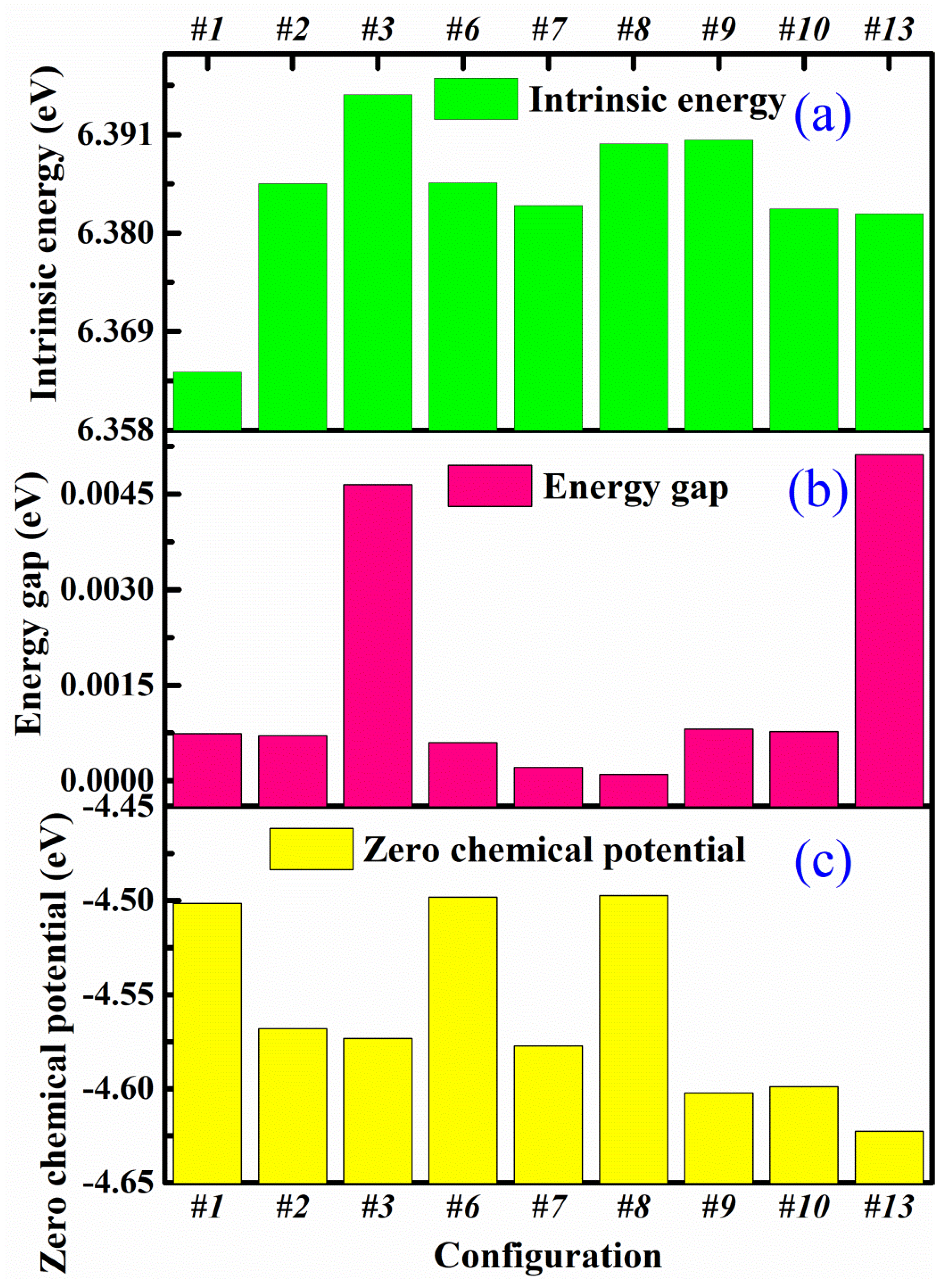
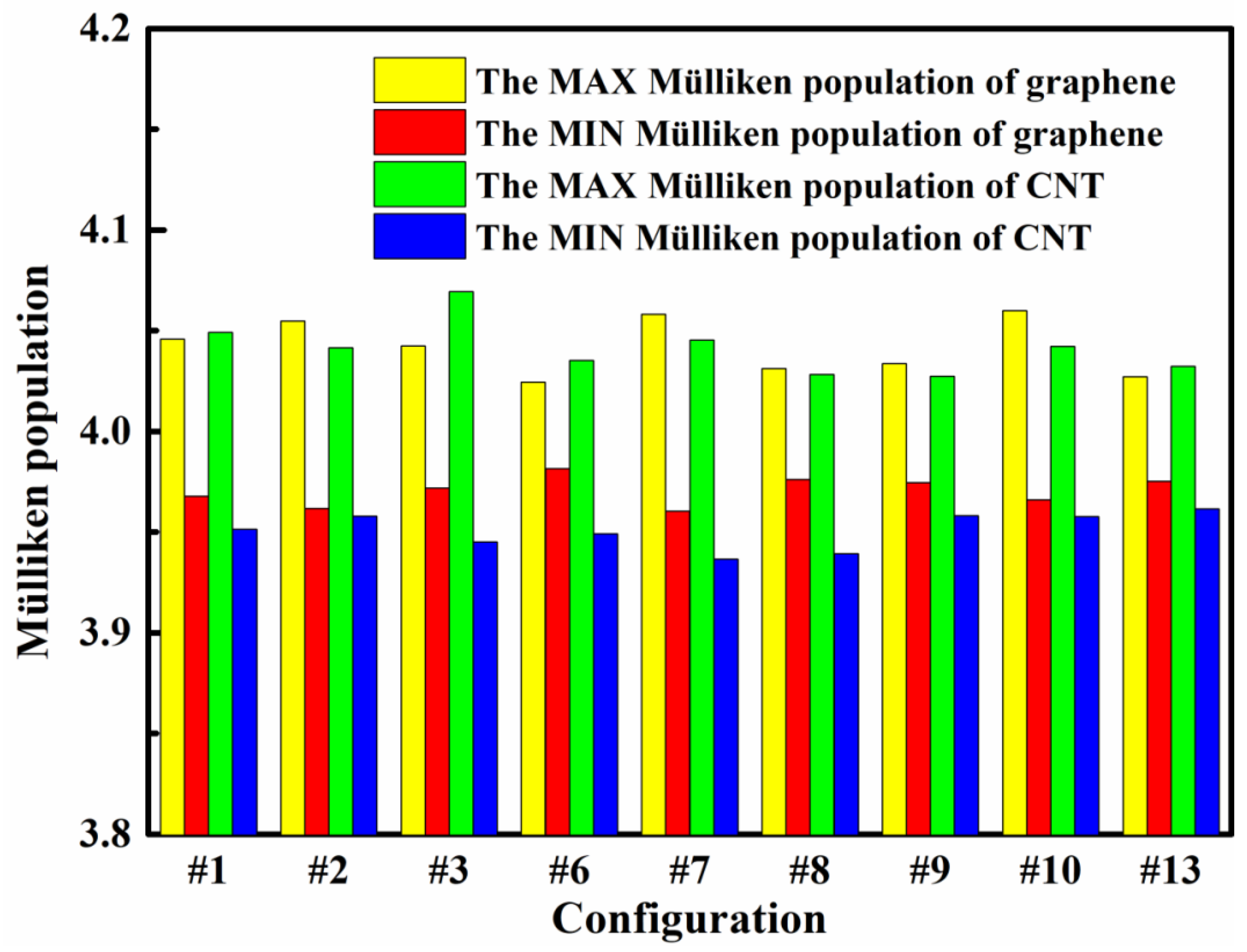
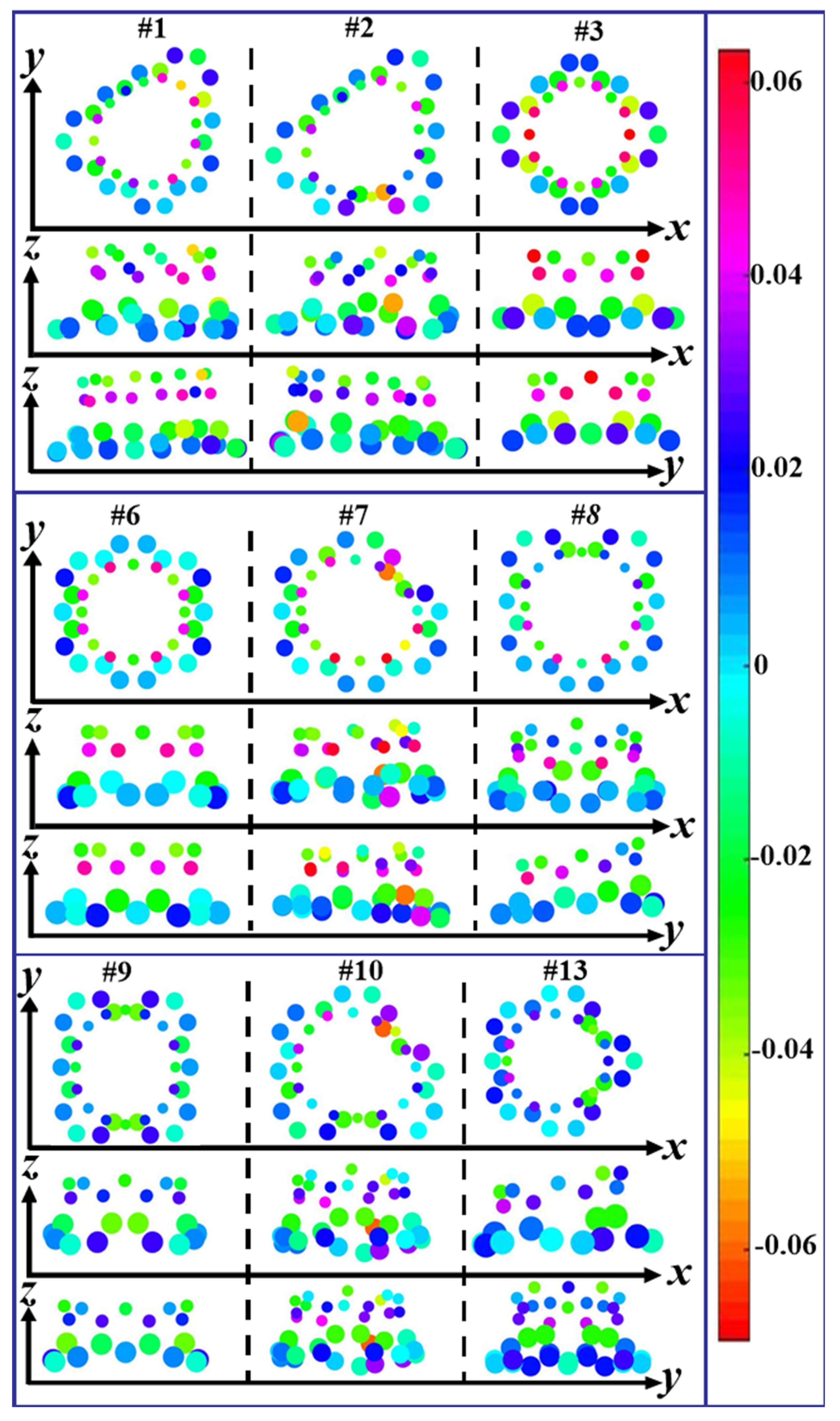
3.2. Differential Charge Density and Mülliken Population
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Qu, Y.; Cheng, C.; Ma, R.; Fan, R. Spark plasma sintered GR-CNT/CaCu3Ti4O12 ceramic nanocomposites with tunable epsilon-negative and epsilon-near-zero property. Ceram. Int. 2021, 47, 17345–17352. [Google Scholar] [CrossRef]
- Zhao, B.; Li, Y.; Ji, H.; Bai, P.; Wang, S.; Fan, B.; Guo, X.; Zhang, R. Lightweight graphene aerogels by decoration of 1D CoNi chains and CNTs to achieve ultra-wide microwave absorption. Carbon 2021, 176, 411–420. [Google Scholar] [CrossRef]
- Fengab, L.; Zuoa, Y.; Hea, X.; Houa, X.; Fub, Q.; Lib, H.; Songb, Q. Development of light cellular carbon nanotube@graphene/carbon nanocomposites with effective mechanical and EMI shielding performance. Carbon 2020, 168, 719–731. [Google Scholar] [CrossRef]
- Yousefi, K. Graphene-Carbon Nanotube Hybrids: Synthesis and Application. J. Environ. Treat. Tech. 2020, 9, 224–232. [Google Scholar] [CrossRef]
- He, Y.; Wu, D.; Zhou, M.; Zheng, Y.; Wang, T.; Lu, C.; Zhang, L.; Liu, H.; Liu, C. Wearable Strain Sensors Based on a Porous Polydimethylsiloxane Hybrid with Carbon Nanotubes and Graphene. ACS Appl. Mater. Interfaces 2021, 13, 15572–15583. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Y.; Wang, G.; Shi, X.; Qiao, Y.; Liu, J.; Liu, H.; Ganesh, A.; Li, L. Direct Graphene-Carbon Nanotube Composite Ink Writing All-Solid-State Flexible Microsupercapacitors with High Areal Energy Density. Adv. Funct. Mater. 2020, 30, 1907284. [Google Scholar] [CrossRef]
- Han, K.; An, F.; Wan, Q.; Xing, L.; Wang, L.; Liu, Q.; Wang, W.; Liu, Y.; Li, P.; Qu, X. Confining Pyrrhotite Fe 7 S 8 in Carbon Nanotubes Covalently Bonded onto 3D Few-Layer Graphene Boosts Potassium-Ion Storage and Full-Cell Applications. Small 2021, 17, 2006719. [Google Scholar] [CrossRef]
- Wang, G.; Sun, X.; Lu, F.; Sun, H.; Yu, M.; Jiang, W.; Liu, C.; Lian, J. Flexible Pillared Graphene-Paper Electrodes for High-Performance Electrochemical Supercapacitors. Small 2012, 8, 452–459. [Google Scholar] [CrossRef]
- Paul, R.K.; Ghazinejad, M.; Penchev, M.; Lin, J.; Ozkan, M.; Ozkan, C.S. Synthesis of a Pillared Graphene Nanostructure: A Counterpart of Three-Dimensional Carbon Architectures. Small 2010, 6, 2309–2313. [Google Scholar] [CrossRef]
- Lin, J.; Zhong, J.; Bao, D.; Reiber-Kyle, J.; Wang, W.; Vullev, V.; Ozkan, M.; Ozkan, C.S. Supercapacitors Based on Pillared Graphene Nanostructures. J. Nanosci. Nanotechnol. 2012, 12, 1770–1775. [Google Scholar] [CrossRef]
- You, B.; Wang, L.; Yao, L.; Yang, J. Three dimensional N-doped graphene–CNT networks for supercapacitor. Chem. Commun. 2013, 49, 5016–5018. [Google Scholar] [CrossRef] [PubMed]
- Georgios, D.K.; Emmanuel, T.; George, F.E. Pillared Graphene: A New 3-D Innovative Network Nanostructure Augments Hydrogen Storage. AIP Conf. Proc. 2009, 1148, 388. [Google Scholar]
- Wesołowski, R.P.; Terzyk, A.P. Pillared graphene as a gas separation membrane. Phys. Chem. Chem. Phys. 2011, 13, 17027–17029. [Google Scholar] [CrossRef] [PubMed]
- Pedrielli, A.; Taioli, S.; Garberoglio, G.; Pugno, N.M. Gas adsorption and dynamics in Pillared Graphene Frameworks. Microporous Mesoporous Mater. 2018, 257, 222–231. [Google Scholar] [CrossRef] [Green Version]
- Ghazinejad, M.; Guo, S.; Paul, R.K.; George, A.S.; Penchev, M.; Ozkan, M.; Ozkan, C.S. Synthesis of Graphene-CNT Hybrid Nanostructures. MRS Proc. 2011, 1344. [Google Scholar] [CrossRef]
- Li, X.; Tong, T.; Wu, Q.; Guo, S.; Song, Q.; Han, J.; Huang, Z. Unique Seamlessly Bonded CNT@Graphene Hybrid Nanostructure Introduced in an Interlayer for Efficient and Stable Perovskite Solar Cells. Adv. Funct. Mater. 2018, 28, 1800475. [Google Scholar] [CrossRef]
- Maarouf, A.; Kasry, A.; Chandra, B.; Martyna, G.J. A graphene–carbon nanotube hybrid material for photovoltaic applications. Carbon 2016, 102, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Ali, F.; Irfan, M.; Muhammad, F.; Glowacz, A.; Antonino-Daviu, J.A.; Caesarendra, W.; Qamar, S. Mechanical Pressure Characterization of CNT-Graphene Composite Material. Micromachines 2020, 11, 1000. [Google Scholar] [CrossRef]
- Peng, M.; Shi, D.; Sun, Y.; Cheng, J.; Zhao, B.; Xie, Y.; Zhang, J.; Guo, W.; Jia, Z.; Liang, Z.; et al. 3D Printed Mechanically Robust Graphene/CNT Electrodes for Highly Efficient Overall Water Splitting. Adv. Mater. 2020, 32, e1908201. [Google Scholar] [CrossRef]
- Park, M.; Lee, D.-M.; Park, S.; Kim, T.-W.; Lee, S.H.; Lee, S.-K.; Jeong, H.S.; Hong, B.H.; Bae, S. Performance enhancement of graphene assisted CNT/Cu composites for lightweight electrical cables. Carbon 2021, 179, 53–59. [Google Scholar] [CrossRef]
- Baowan, D.; Cox, B.J.; Hill, J.M. Two least squares analyses of bond lengths and bond angles for the joining of carbon nanotubes to graphenes. Carbon 2007, 45, 2972–2980. [Google Scholar] [CrossRef]
- Novaes, F.D.; Rurali, R.; Ordejón, P. Electronic Transport between Graphene Layers Covalently Connected by Carbon Nanotubes. ACS Nano 2010, 4, 7596–7602. [Google Scholar] [CrossRef] [PubMed]
- Taheri, S.; Lakmehsari, M.S.; Ganji, M.D.; Azar, Y.T. Configuration-tuning of vacancy induced magnetism in 3-D pillared graphene: An ab initio study. Diam. Relat. Mater. 2018, 82, 1–6. [Google Scholar] [CrossRef]
- Karimzadeh, S.; Safaei, B.; Jen, T.-C. Predicting phonon scattering and tunable thermal conductivity of 3D pillared graphene and boron nitride heterostructure. Int. J. Heat Mass Transf. 2021, 172, 121145. [Google Scholar] [CrossRef]
- Varshney, V.; Patnaik, S.S.; Roy, A.K.; Froudakis, G.; Farmer, B.L. Modeling of Thermal Transport in Pillared-Graphene Architectures. ACS Nano 2010, 4, 1153–1161. [Google Scholar] [CrossRef]
- Lee, J.; Varshney, V.; Brown, J.S.; Roy, A.K.; Farmer, B.L. Single mode phonon scattering at carbon nanotube-graphene junction in pillared graphene structure. Appl. Phys. Lett. 2012, 100, 183111. [Google Scholar] [CrossRef]
- Rutter, G.M.; Crain, J.N.; Guisinger, N.P.; Li, T.; First, P.N.; Stroscio, J.A. Scattering and Interference in Epitaxial Graphene. Science 2007, 317, 219–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, A.; Suenaga, K.; Gloter, A.; Urita, K.; Iijima, S. Direct evidence for atomic defects in graphene layers. Nature 2004, 430, 870–873. [Google Scholar] [CrossRef]
- Nguyen, V.; Etz, B.D.; Pylypenko, S.; Vyas, S. Periodic Trends behind the Stability of Metal Catalysts Supported on Graphene with Graphitic Nitrogen Defects. ACS Omega 2021, 6, 28215–28228. [Google Scholar] [CrossRef]
- Kattel, S.; Atanassov, P.; Kiefer, B. Stability, Electronic and Magnetic Properties of In-Plane Defects in Graphene: A First-Principles Study. J. Phys. Chem. C 2012, 116, 8161–8166. [Google Scholar] [CrossRef]
- Jeong, B.W.; Ihm, J.; Lee, G.-D. Stability of dislocation defect with two pentagon-heptagon pairs in graphene. Phys. Rev. B 2008, 78, 165403. [Google Scholar] [CrossRef]
- Zhang, L. Studying Stability of Atom Packing for Ti Nanoparticles on Heating by Molecular Dynamics Simulations. Adv. Eng. Mater. 2019, 21, 21. [Google Scholar] [CrossRef]
- Lina, W.; Dandan, Z.; Lijing; Junjun, L.; Lin, Z. Atomic simulations of bamboo-like N-doped CNTs with spaced nitrogen and carbon atoms by DFTB algorithm. Diam. Relat. Mater. 2021, 116, 108383. [Google Scholar] [CrossRef]
- Wu, L.; Han, Y.; Zhao, Q.; Zhang, L. Effects of chiral indices on the atomic arrangements and electronic properties of Si double-walled nanotubes (6,min)@(9,mout) (min = 0 to 6, mout = 0 to 9) by SCC-DFTB calculations. Mater. Sci. Semicond. Process. 2021, 129, 105775. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, L.; Qi, Y. Study of Atomic Arrangements and Charge Distribution on the Surfaces of a Si Ultra-Thin-Substrate by Using DFTB Simulations. Sci. Adv. Mater. 2017, 9, 1775–1779. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, L.; Shen, L. Study of atomic arrangements and charge distribution on Si(0 0 1) surfaces with the adsorption of one Ge atom by DFTB calculations. Appl. Surf. Sci. 2018, 447, 22–30. [Google Scholar] [CrossRef]
- Wu, L.; Xu, X.; Zhang, L.; Qi, Y. Atomic packing characteristics and electronic structures of Si nanowires from density functional tight binding calculations. Superlattices Microstruct. 2019, 135, 106261. [Google Scholar] [CrossRef]
- Kim, J.; Bae, S.; Kotal, M.; Stalbaum, T.; Oh, I. Polymer Actuators: Soft but Powerful Artificial Muscles Based on 3D Graphene-CNT-Ni Heteronanostructures (Small 31/2017). Small 2017, 13, 1701314. [Google Scholar] [CrossRef]
- Hourahine, B.; Aradi, B.; Blum, V.; Bonafé, F.; Buccheri, A.; Camacho, C.; Cevallos, C.; Deshaye, M.Y.; Dumitrică, T.; Dominguez, A.; et al. DFTB+, a software package for efficient approximate density functional theory based atomistic simulations. J. Chem. Phys. 2020, 152, 124101. [Google Scholar] [CrossRef]
- Elstner, M.; Porezag, D.; Jungnickel, G.; Elsner, J.; Haugk, M.; Frauenheim, T.; Suhai, S.; Seifert, G. Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Phys. Rev. B 1998, 58, 7260–7268. [Google Scholar] [CrossRef]
- Witek, H.; Irle, S.; Morokuma, K. Analytical second-order geometrical derivatives of energy for the self-consistent-charge density-functional tight-binding method. J. Chem. Phys. 2004, 121, 5163–5170. [Google Scholar] [CrossRef] [PubMed]
- Mulliken, R.S. Electronic Population Analysis on LCAO–MO Molecular Wave Functions. II. Overlap Populations, Bond Orders, and Covalent Bond Energies. J. Chem. Phys. 1955, 23, 1841–1846. [Google Scholar] [CrossRef]
- Roothaan, C.C.J. New Developments in Molecular Orbital Theory. Rev. Mod. Phys. 1951, 23, 69–89. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic population analysis on LCAO-MO molecular wave functions, I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Defects | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| graphene | Y | Y | Y | Y | N | Y | Y | Y | Y | Y | N | Y | Y | N | N | N |
| (8,0)CNT-graphene | Y | Y | Y | N | N | Y | Y | Y | Y | Y | N | N | Y | N | N | N |
| Configuration | C-C Bond Length/Å | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Connection | 1-1 | 2-2 | 3-3 | 4-4 | 5-5 | 6-6 | 7-7 | 8-8 | Average |
| #1 | 1.403 | 1.405 | 1.444 | 1.389 | 1.441 | 1.411 | 1.413 | 1.431 | 1.417 |
| #2 | 1.408 | 1.418 | 1.439 | 1.389 | 1.437 | 1.434 | 1.435 | 1.473 | 1.429 |
| #3 | 1.417 | 1.417 | 1.414 | 1.413 | 1.415 | 1.415 | 1.413 | 1.414 | 1.415 |
| #6 | 1.414 | 1.414 | 1.432 | 1.432 | 1.414 | 1.414 | 1.432 | 1.432 | 1.423 |
| #7 | 1.409 | 1.439 | 1.476 | 1.408 | 1.406 | 1.407 | 1.425 | 1.412 | 1.423 |
| #8 | 1.410 | 1.407 | 1.472 | 1.472 | 1.407 | 1.410 | 1.416 | 1.416 | 1.426 |
| #9 | 1.410 | 1.410 | 1.468 | 1.468 | 1.410 | 1.410 | 1.468 | 1.468 | 1.439 |
| #10 | 1.401 | 1.443 | 1.471 | 1.415 | 1.410 | 1.405 | 1.468 | 1.474 | 1.436 |
| #13 | 1.455 | 1.455 | 1.462 | 1.382 | 1.397 | 1.396 | 1.383 | 1.462 | 1.424 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, L.; Zhang, L. Atomic Simulations of (8,0)CNT-Graphene by SCC-DFTB Algorithm. Nanomaterials 2022, 12, 1361. https://doi.org/10.3390/nano12081361
Wei L, Zhang L. Atomic Simulations of (8,0)CNT-Graphene by SCC-DFTB Algorithm. Nanomaterials. 2022; 12(8):1361. https://doi.org/10.3390/nano12081361
Chicago/Turabian StyleWei, Lina, and Lin Zhang. 2022. "Atomic Simulations of (8,0)CNT-Graphene by SCC-DFTB Algorithm" Nanomaterials 12, no. 8: 1361. https://doi.org/10.3390/nano12081361
APA StyleWei, L., & Zhang, L. (2022). Atomic Simulations of (8,0)CNT-Graphene by SCC-DFTB Algorithm. Nanomaterials, 12(8), 1361. https://doi.org/10.3390/nano12081361