
Selective Catalytic Reduction of NOx over Perovskite-Based Catalysts Using CxHy(Oz), H2 and CO as Reducing Agents—A Review of the Latest Developments

 ,
, 
 ,
, 

Abstract
:
1. Introduction
2. Perovskite-Catalyzed SCR of NOx
2.1. Perovskite Catalysts in CxHy/CxHyOz-SCR of NOx
2.2. Perovskite Catalysts in H2-SCR of NOx
2.3. Perovskite Catalysts in CO-SCR of NOx
3. General Outcomes and Future Perspectives
- The SCR of NOx behavior of perovskites (both in activity and N2-selectivity) is comparable, if not better (especially at low temperatures) to that of NM-based catalysts. Their time-on-stream stability and SO2 tolerance are also remarkable.
- Τhe partial replacement of A and/or B-sites with other suitable elements allows a significant improvement and controlled optimization of their SCR performance. For example, partially substituted with Cu perovskites were found to be significantly more active in comparison with the bare sample, due to the additional effect of the advantageous in catalysis Cu2+/Cu+ redox couple.
- Preparation methods capable of providing perovskites with a larger specific surface area are particularly advantageous due to the increased number of accessible active sites exposed to the reaction mixture. The typical specific surface of perovskites produced by traditional methods ranges between 5 and 20 m2∙g−1. Advanced or modified classical methods have been reported which can raise these values by two or even three times resulting in a significant catalytic benefit during SCR of NOx. Currently, significant efforts have been put to synthesize perovskites with surface areas as high as 100 m2∙g−1.
- Extensive characterizations of the synthesized perovskite materials that were frequently applied allowed the researchers to better understand the reaction pathways, the nature, and the role of the active sites, thus extracting relatively reliable morphology–activity correlations. However, apart from the in situ DRIFTS studies, no other operando techniques such as in situ XRD and in situ TEM were found to have been applied to the studies included herein. In light of such shortcomings, the frequent borrowing of reaction mechanisms from those proposed for analogous NM-based systems is justified. However, the use of perovskites in the SCR process is more likely to introduce new, easier reaction pathways that need to be in-depth understood in order to proceed with a coordinated optimization of perovskite composition for the SCR of NOx. At the same time, DFT calculations that are generally missing from the documents included herein can be particularly helpful in the above objectives.
- On the other hand, modern approaches in catalysis have emerged following the pioneering work of the Hamada team [50] on what is now described as “exsolution” that offers new perspectives on the use of perovskites. The creation of different kinds of alloy or metal particles at nano or even atomic sizes on the surface of perovskites may provide the chance of tailoring the local surface properties and metal–support interactions, leading to enhanced performance. The active interfaces generated by the exsolution process can also result in higher activity and stability for this type of catalyst. The method could provide effective solutions in the field of SCR of NOx as well. Due to the recency of the discovery, applications focused on the specific topic of this review have not yet been found (the work of Hamada and co-workers was implemented in TWC conditions). We could assume that the exsolution concept will be an intense research approach in the coming years on NOx abatement under lean conditions.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BET | Brunauer, Emmett and Teller |
| CBM | Coal Bed Methane |
| DFT | Density Functional Theory |
| DLS | Dynamic Light Scattering |
| DRIFTS | Diffuse Reflectance Infrared Fourier Transform Spectroscopy |
| DSC | Differential Scanning Calorimetry |
| EDS | Energy-Dispersive X-ray Spectroscopy |
| EF-TEM | Energy Filtering Transmission Electron Microscopy |
| FE-SEM | Field Emission Scanning Electron Microscopy |
| FTIR | Fourier Transform Infrared Spectroscopy |
| HCs | Hydrocarbons |
| ICP-AES | Inductively Coupled Plasma Atomic Emission Spectroscopy |
| ICP-OES | Inductively Coupled Plasma Optical Emission Spectroscopy |
| PM | Particulate Matter |
| SCR | Selective Catalytic Reduction |
| SEM | Scanning Electron Microscopy |
| TEM | Transmission Electron Microscopy |
| TPD | Temperature-Programmed Desorption |
| TPR | Temperature-Programmed Reduction |
| TWC | Three-Way Catalysts |
| WGHSV | Weight-basis Gas Hourly Space Velocity |
| XRD | X-ray Diffraction |
| XPS | X-ray Photoelectron Spectroscopy |
References
- Granger, P.; Parvulescu, V.I. Catalytic NOx abatement systems for mobile sources: From three-way to lean burn after-treatment technologies. Chem. Rev. 2011, 111, 3155–3207. [Google Scholar] [CrossRef] [PubMed]
- Yentekakis, I.V.; Konsolakis, M. Three-way Catalysis. In Perovskites and Related Mixed Oxides; Wiley-VCH, Vergal GmbH & Co. KGaA: Weinheim, Germany, 2016; pp. 559–586. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Vernoux, P. Emissions control catalysis. Catalysts 2019, 9, 912. [Google Scholar] [CrossRef] [Green Version]
- Yentekakis, I.V.; Vernoux, P.; Goula, G.; Caravaca, A. Electropositive promotion by Alkalis or Alkaline earths of Pt-group metals in emissions control catalysis: A status report. Catalysts 2019, 9, 157. [Google Scholar] [CrossRef] [Green Version]
- Yentekakis, I.V.; Dong, F. Grand Challenges for Catalytic Remediation in Environmental and Energy Applications Toward a Cleaner and Sustainable Future. Front. Environ. Chem. 2020, 1, 5. [Google Scholar] [CrossRef]
- Damma, D.; Ettireddy, P.R.; Reddy, B.M.; Smirniotis, P.G. A review of low temperature NH3-SCR for removal of NOx. Catalysts 2019, 9, 349. [Google Scholar] [CrossRef] [Green Version]
- Yentekakis, I.V.; Tellou, V.; Botzolaki, G.; Rapakousios, I.A. A comparative study of the C3H6 + NO + O2, C3H6 + O2 and NO + O2 reactions in excess oxygen over Na-modified Pt/γ-Al2O3 catalysts. Appl. Catal. B Environ. 2005, 56, 229–239. [Google Scholar] [CrossRef]
- Goula, M.A.; Charisiou, N.D.; Papageridis, K.N.; Delimitis, A.; Papista, E.; Pachatouridou, E.; Iliopoulou, E.F.; Marnellos, G.; Konsolakis, M.; Yentekakis, I.V. A comparative study of the H2-assisted selective catalytic reduction of nitric oxide by propene over noble metal (Pt, Pd, Ir)/γ-Al2O3 catalysts. J. Environ. Chem. Eng. 2016, 4, 1629–1641. [Google Scholar] [CrossRef]
- Costa, C.N.; Savva, P.G.; Andronikou, C.; Lambrou, P.S.; Polychronopoulou, K.; Belessi, V.C.; Stathopoulos, V.N.; Pomonis, P.J.; Efstathiou, A.M. An investigation of the NO/H2/O2 (Lean De-NOx) reaction on a highly active and selective Pt/La0.7Sr0.2Ce0.1FeO3 catalyst at low temperatures. J. Catal. 2002, 209, 456–471. [Google Scholar] [CrossRef]
- Polychronopoulou, K.; Efstathiou, A.M. NOx Control via H2-Selective Catalytic Reduction (H2-SCR) Technology for Stationary and Mobile Applications. Recent Patents Mater. Sci. 2012, 5, 84–107. [Google Scholar] [CrossRef]
- Machida, M.; Ikeda, S.; Kurogi, D.; Kijima, T. Low temperature catalytic NOx–H2 reactions over Pt/TiO2-ZrO2 in an excess oxygen. Appl. Catal. B Environ. 2001, 35, 107–116. [Google Scholar] [CrossRef]
- Macleod, N.; Lambert, R.M. An in situ DRIFTS study of efficient lean NOx reduction with H2 + CO over Pd/Al2O3: The key role of transient NCO formation in the subsequent generation of ammonia. Appl. Catal. B Environ. 2003, 46, 483–495. [Google Scholar] [CrossRef]
- Konsolakis, M.; Vrontaki, M.; Avgouropoulos, G.; Ioannides, T.; Yentekakis, I.V. Novel doubly-promoted catalysts for the lean NOx reduction by H2 + CO: Pd(K)/Al2O3–(TiO2). Appl. Catal. B Environ. 2006, 68, 59–67. [Google Scholar] [CrossRef]
- Pekridis, G.; Kaklidis, N.; Komvokis, V.; Athanasiou, C.; Konsolakis, M.; Yentekakis, I.V.; Marnellos, G.E. Surface and catalytic elucidation of Rh/γ-Al2O3 catalysts during NO reduction by C3H8 in the presence of excess O2, H2O, and SO2. J. Phys. Chem. A 2010, 114, 3969–3980. [Google Scholar] [CrossRef]
- Burch, R. Knowledge and know-how in emission control for mobile applications. Catal. Rev. Sci. Eng. 2004, 46, 271–334. [Google Scholar] [CrossRef]
- Macleod, N.; Isaac, J.; Lambert, R.M. Sodium Promotion of the NO+C3H6 Reaction over Rh/γ-Al2O3 Catalysts. J. Catal. 2000, 193, 115–122. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Konsolakis, M.; Rapakousios, I.A.; Matsouka, V. Novel electropositively promoted monometallic (Pt-only) catalytic converters for automotive pollution control. Top. Catal. 2007, 42, 393–397. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Lambert, R.M.; Konsolakis, M.; Kiousis, V. The effect of sodium on the Pd-catalyzed reduction of NO by methane. Appl. Catal. B Environ. 1998, 18, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Konsolakis, M.; Yentekakis, I.V. Strong promotional effects of Li, K, Rb and Cs on the Pt-catalysed reduction of NO by propene. Appl. Catal. B Environ. 2001, 29, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Konsolakis, M.; Yentekakis, I.V. The reduction of NO by propene over Ba-promoted Pt/γ-Al2O3 catalysts. J. Catal. 2001, 198, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Tanikawa, K.; Egawa, C. Effect of barium addition over palladium catalyst for CO-NO-O2 reaction. J. Mol. Catal. A Chem. 2011, 349, 94–99. [Google Scholar] [CrossRef]
- Palermo, A.; Lambert, R.M.; Harkness, I.R.; Yentekakis, I.V.; Marina, O.; Vayenas, C.G. Electrochemical promotion by Na of the platinum-catalyzed reaction between CO and NO. J. Catal. 1996, 161, 471–479. [Google Scholar] [CrossRef] [Green Version]
- Papadakis, V.G.; Pliangos, C.A.; Yentekakis, I.V.; Verykios, X.E.; Vayenas, C.G. Development of high performance, Pd-based, three-way catalysts. Catal. Today 1996, 29, 71–75. [Google Scholar] [CrossRef]
- Palermo, A.; Tikhov, M.S.; Filkin, N.C.; Lambert, R.M.; Yentekakis, I.V.; Vayenas, C.G. Electrochemical promotion of NO reduction by CO and by propene. Stud. Surf. Sci. Catal. 1996, 101, 513–522. [Google Scholar] [CrossRef]
- Matsouka, V.; Konsolakis, M.; Yentekakis, I.V.; Papavasiliou, A.; Tsetsekou, A.; Boukos, N. Thermal aging behavior of Pt-only TWC converters under simulated exhaust conditions: Effect of rare earths (CeO2, La2O3) and alkali (Na) modifiers. Top. Catal. 2011, 54, 1124–1134. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Goula, G.; Panagiotopoulou, P.; Kampouri, S.; Taylor, M.J.; Kyriakou, G.; Lambert, R.M. Stabilization of catalyst particles against sintering on oxide supports with high oxygen ion lability exemplified by Ir-catalyzed decomposition of N2O. Appl. Catal. B Environ. 2016, 192, 357–364. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Goula, G.; Kampouri, S.; Betsi-Argyropoulou, I.; Panagiotopoulou, P.; Taylor, M.J.; Kyriakou, G.; Lambert, R.M. Ir-Catalysed Nitrous oxide (N2O) Decomposition: Effect of Ir Particle Size and Metal–Support Inter-actions. Catal. Letters 2018, 148, 341–347. [Google Scholar] [CrossRef] [Green Version]
- Yentekakis, I.V.; Goula, G.; Panagiotopoulou, P.; Katsoni, A.; Diamadopoulos, E.; Mantzavinos, D.; Delimitis, A. Dry reforming of methane: Catalytic performance and stability of Ir catalysts supported on γ-Al2O3, Zr0.92Y0.08O2-δ (YSZ) or Ce0.9Gd0.1O2-δ (GDC) supports. Top. Catal. 2015, 58, 1228–1241. [Google Scholar] [CrossRef]
- Goula, G.; Botzolaki, G.; Osatiashtiani, A.; Parlett, C.M.A.; Kyriakou, G.; Lambert, R.M.; Yentekakis, I.V. Oxidative thermal sintering and redispersion of Rh nanoparticles on supports with high oxygen ion lability. Catalysts 2019, 9, 541. [Google Scholar] [CrossRef] [Green Version]
- Nikolaraki, E.; Goula, G.; Panagiotopoulou, P.; Taylor, M.J.; Kousi, K.; Kyriakou, G.; Kondarides, D.I.; Lambert, R.M.; Yentekakis, I.V. Support induced effects on the Ir nanoparticles activity, selectivity and stability performance under CO2 reforming of methane. Nanomaterials 2021, 11, 2880. [Google Scholar] [CrossRef]
- Parvulescu, V.I.; Kaliaguine, S.; Prellier, W. (Eds.) Perovskites and Related Mixed Oxides; Wiley-VCH, Vergal GmbH & Co. KGaA: Weinheim, Germany, 2016; Volumes 1 and 2, ISBN 978-3-527-33763-7. [Google Scholar]
- Zhu, J.; Li, H.; Zhong, L.; Xiao, P.; Xu, X.; Yang, X.; Zhao, Z.; Li, J. Perovskite oxides: Preparation, characterizations, and applications in heterogeneous catalysis. ACS Catal. 2014, 4, 2917–2940. [Google Scholar] [CrossRef]
- Royer, S.; Duprez, D.; Can, F.; Courtois, X.; Batiot-Dupeyrat, C.; Laassiri, S.; Alamdari, H. Perovskites as substitutes of noble metals for heterogeneous catalysis: Dream or reality. Chem. Rev. 2014, 114, 10292–10368. [Google Scholar] [CrossRef]
- Hwang, J.; Rao, R.R.; Giordano, L.; Katayama, Y.; Yu, Y.; Shao-Horn, Y. Perovskites in catalysis and electrocatalysis. Science 2017, 358, 751–756. [Google Scholar] [CrossRef] [Green Version]
- Tarjomannejad, A.; Niaei, A.; Gómez, M.J.I.; Farzi, A.; Salari, D.; Albaladejo-Fuentes, V. NO + CO reaction over LaCu0.7B0.3O3 (B = Mn, Fe, Co) and La0.8A0.2Cu0.7Mn0.3O3 (A = Rb, Sr, Cs, Ba) perovskite-type catalysts. J. Therm. Anal. Calorim. 2017, 129, 671–680. [Google Scholar] [CrossRef]
- Bhattar, S.; Abedin, M.A.; Kanitkar, S.; Spivey, J.J. A review on dry reforming of methane over perovskite derived catalysts. Catal. Today 2021, 365, 2–23. [Google Scholar] [CrossRef]
- Sim, Y.; Kwon, D.; An, S.; Ha, J.M.; Oh, T.S.; Jung, J.C. Catalytic behavior of ABO3 perovskites in the oxidative coupling of methane. Mol. Catal. 2020, 489, 110925. [Google Scholar] [CrossRef]
- Yang, E.-h.; Noh, Y.S.; Hong, G.H.; Moon, D.J. Combined steam and CO2 reforming of methane over La1-xSrxNiO3 perovskite oxides. Catal. Today 2018, 299, 242–250. [Google Scholar] [CrossRef]
- Lima, S.M.; Assaf, J.M.; Peña, M.A.; Fierro, J.L.G. Structural features of La1-xCexNiO3 mixed oxides and performance for the dry reforming of methane. Appl. Catal. A Gen. 2006, 311, 94–104. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, T.; Dong, X.; Li, M.; Wang, H. Effects of Ce substitution at the A-site of LaNi0.5Fe0.5O3 perovskite on the enhanced catalytic activity for dry reforming of methane. Appl. Catal. B Environ. 2018, 224, 214–221. [Google Scholar] [CrossRef]
- Peña, M.A.; Fierro, J.L.G. Chemical structures and performance of perovskite oxides. Chem. Rev. 2001, 101, 1981–2017. [Google Scholar] [CrossRef]
- Shen, M.; Zhao, Z.; Chen, J.; Su, Y.; Wang, J.; Wang, X. Effects of calcium substitute in LaMnO3 perovskites for NO catalytic oxidation. J. Rare Earths 2013, 31, 119–123. [Google Scholar] [CrossRef]
- Zhang, R.; Villanueva, A.; Alamdari, H.; Kaliaguine, S. Reduction of NO by CO over nanoscale LaCo1-xCuxO3 and LaMn1-xCuxO3 perovskites. J. Mol. Catal. A Chem. 2006, 258, 22–34. [Google Scholar] [CrossRef]
- Misono, M. A view on the future of mixed oxide catalysts: The case of heteropolyacids (polyoxometalates) and perovskites. Catal. Today 2005, 100, 95–100. [Google Scholar] [CrossRef]
- Yentekakis, I.V. Open- and closed-circuit study of an intermediate temperature SOFC directly fueled with simulated biogas mixtures. J. Power Sources 2006, 29, 422–425. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Papadam, T.; Goula, G. Electricity production from wastewater treatment via a novel biogas-SOFC aided process. Solid State Ionics 2008, 179, 1521–1525. [Google Scholar] [CrossRef]
- Buciuman, F.-C.; Joubert, E.; Menezo, J.-C.; Barbier, J. Catalytic properties of La0.8A0.2MnO3 (A = Sr, Ba, K, Cs) and LaMn0.8B0.2O3 (B = Ni, Zn, Cu) perovskites. Appl. Catal. B Environ. 2001, 35, 149–156. [Google Scholar] [CrossRef]
- Wu, X.; Xu, L.; Weng, D. The NO selective reduction on the La1-xSrxMnO3 catalysts. Catal. Today 2004, 90, 199–206. [Google Scholar] [CrossRef]
- Kousi, K.; Tang, C.; Metcalfe, I.S.; Neagu, D. Emergence and future of exsolved materials. Small 2021, 17, 2006479. [Google Scholar] [CrossRef]
- Nishihata, Y.; Mizuki, J.; Akao, T.; Tanaka, H.; Uenishi, M.; Kimura, M.; Okamoto, T.; Hamada, N. Self-regeneration of a Pd-perovskite catalyst for automotive emissions control. Nature 2002, 418, 164–167. [Google Scholar] [CrossRef]
- Kwon, O.; Joo, S.; Choi, S.; Sengodan, S.; Kim, G. Review on exsolution and its driving forces in perovskites. J. Phys Energy 2020, 2, 032001. [Google Scholar] [CrossRef]
- He, H.; Dai, H.X.; Au, C.T. An investigation on the utilization of perovskite-type oxides La1-xSrxMO3 (M = Co0.77Bi0.20Pd0.03) as three-way catalysts. Appl. Catal. B Environ. 2001, 33, 65–80. [Google Scholar] [CrossRef]
- Zhu, J.; Zhao, Z.; Xiao, D.; Li, J.; Yang, X.; Wu, Y. Study of La2-xSrxCuO4 (x = 0.0, 0.5, 1.0) catalysts for NO + CO reaction from the measurements of O2-TPD, H2-TPR and cyclic voltammetry. J. Mol. Catal. A Chem. 2005, 238, 35–40. [Google Scholar] [CrossRef]
- Fino, D.; Fino, P.; Saracco, G.; Specchia, V. Studies on kinetics and reactions mechanism of La2-xKxCu1-yVyO4 layered perovskites for the combined removal of diesel particulate and NOx. Appl. Catal. B Environ. 2003, 43, 243–259. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. Nature of active species in copper-based catalysts and their chemistry of transformation of nitrogen oxides. Appl. Catal. A Gen. 1995, 132, 179–259. [Google Scholar] [CrossRef]
- Yahiro, H.; Iwamoto, M. Copper ion-exchanged zeolite catalysts in deNOx reaction. Appl. Catal. A Gen. 2001, 222, 163–181. [Google Scholar] [CrossRef]
- Zhang, R.; Villanueva, A.; Alamdari, H.; Kaliaguine, S. Catalytic reduction of NO by propene over LaCo1-xCuxO3 perovskites synthesized by reactive grinding. Appl. Catal. B Environ. 2006, 64, 220–233. [Google Scholar] [CrossRef]
- Zhang, R.; Villanueva, A.; Alamdari, H.; Kaliaguine, S. SCR of NO by propene over nanoscale LaMn1-xCuxO3 perovskites. Appl. Catal. A Gen. 2006, 307, 85–97. [Google Scholar] [CrossRef]
- Glisenti, A.; Pacella, M.; Guiotto, M.; Natile, M.M.; Canu, P. Largely Cu-doped LaCo1-xCuxO3 perovskites for TWC: Toward new PGM-free catalysts. Appl. Catal. B Environ. 2016, 180, 94–105. [Google Scholar] [CrossRef]
- Levasseur, B.; Kaliaguine, S. Effects of iron and cerium in La1-yCeyCo1-xFexO3 perovskites as catalysts for VOC oxidation. Appl. Catal. B Environ. 2009, 88, 305–314. [Google Scholar] [CrossRef]
- Deng, C.; Huang, Q.; Zhu, X.; Hu, Q.; Su, W.; Qian, J.; Dong, L.; Li, B.; Fan, M.; Liang, C. The influence of Mn-doped CeO2 on the activity of CuO/CeO2 in CO oxidation and NO + CO model reaction. Appl. Surf. Sci. 2016, 389, 1033–1049. [Google Scholar] [CrossRef]
- Deng, C.; Qian, J.; Yu, C.; Yi, Y.; Zhang, P.; Li, W.; Dong, L.; Li, B.; Fan, M. Influences of doping and thermal stability on the catalytic performance of CuO/Ce20M1Ox (M = Zr, Cr, Mn, Fe, Co, Sn) catalysts for NO reduction by CO. RSC Adv. 2016, 6, 113630–113647. [Google Scholar] [CrossRef]
- Ma, J.; Jin, G.; Gao, J.; Li, Y.; Dong, L.; Huang, M.; Huang, Q.; Li, B. Catalytic effect of two-phase intergrowth and coexistence CuO-CeO2. J. Mater. Chem. A 2015, 3, 24358–24370. [Google Scholar] [CrossRef]
- Giannakas, A.E.; Leontiou, A.A.; Ladavos, A.K.; Pomonis, P.J. Characterization and catalytic investigation of NO + CO reaction on perovskites of the general formula LaxM1-xFeO3 (M = Sr and/or Ce) prepared via a reverse micelles microemulsion route. Appl. Catal. A Gen. 2006, 309, 254–262. [Google Scholar] [CrossRef]
- Costa, C.N.; Efstathiou, A.M. Low-temperature H2-SCR of NO on a novel Pt/MgO-CeO2 catalyst. Appl. Catal. B Environ. 2007, 72, 240–252. [Google Scholar] [CrossRef]
- Engelmann-Pirez, M.; Granger, P.; Leclercq, G. Investigation of the catalytic performances of supported noble metal based catalysts in the NO + H2 reaction under lean conditions. Catal. Today 2005, 107, 315–322. [Google Scholar] [CrossRef]
- Mondragón Rodríguez, G.C.; Saruhan, B. Effect of Fe/Co-ratio on the phase composition of Pd-integrated perovskites and its H2-SCR of NOx performance. Appl. Catal. B Environ. 2010, 93, 304–313. [Google Scholar] [CrossRef]
- Sato, S.; Yu-u, Y.; Yahiro, H.; Mizuno, N.; Iwamoto, M. Cu-ZSM-5 zeolite as highly active catalyst for removal of nitrogen monoxide from emission of diesel engines. Appl. Catal. 1991, 70, 3–7. [Google Scholar] [CrossRef]
- Vasala, S.; Karppinen, M. A2B′B″O6 perovskites: A review. Prog. Solid State Chem. 2015, 43, 1–36. [Google Scholar] [CrossRef]
- Li, X.; Chen, C.; Liu, C.; Xian, H.; Guo, L.; Lv, J.; Jiang, Z. Pd-Doped Perovskite: An Effective Catalyst for Removal of NO. ACS Catal. 2013, 3, 1071–1075. [Google Scholar] [CrossRef]
- Kucherov, A.V.; Gerlock, J.L.; Jen, H.W.; Shelef, M. In Situ ESR Monitoring of CuH-ZSM-5 Up to 500 °C in Flowing Dry Mixtures of NO(NO2), C3H6(C2H5OH), and Excess O2. J. Catal. 1995, 152, 63–69. [Google Scholar] [CrossRef]
- Ukisu, Y.; Miyadera, T.; Abe, A.; Yoshida, K. Infrared study of catalytic reduction of lean NOx with alcohols over alumina-supported silver catalyst. Catal. Letters 1996, 39, 265–267. [Google Scholar] [CrossRef]
- Wu, Q.; He, H.; Yu, Y. In situ DRIFTS study of the selective reduction of NOx with alcohols over Ag/Al2O3 catalyst: Role of surface enolic species. Appl. Catal. B Environ. 2005, 61, 107–113. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, R.; Li, P.; Royer, S.; Dacquin, J.P. Mechanistic insight into the methanol selective catalytic reduction of NO reaction over Cu-containing perovskites. J. Catal. 2019, 377, 480–493. [Google Scholar] [CrossRef]
- Teng, Z.; Huang, S.; Zhang, H.; Yu, H.; Li, N.; Zhou, Q. A system including enriching coal bed methane by solar energy and selective catalytic reduction. Appl. Therm. Eng. 2018, 130, 822–829. [Google Scholar] [CrossRef]
- Teng, Z.; Zhang, H.; Huang, S.; Li, N.; Zhou, Q. Experimental study on reduction of NO by CH4 over La0.8Sr0.2MnO3/α-Al2O3 in excess of O2. J. Taiwan Inst. Chem. Eng. 2018, 87, 204–210. [Google Scholar] [CrossRef]
- Giroir-Fendler, A.; Gil, S.; Baylet, A. (La0.8A0.2)MnO3 (A = Sr, K) perovskite catalysts for NO and C10H22 oxidation and selective reduction of NO by C10H22. Cuihua Xuebao Chin. J. Catal. 2014, 35, 1299–1304. [Google Scholar] [CrossRef]
- Tabata, K.; Hirano, Y.; Suzuki, E. XPS studies on the oxygen species of LaMn1-xCuxO3+λ. Appl. Catal. A Gen. 1998, 170, 245–254. [Google Scholar] [CrossRef]
- Baylet, A.; Royer, S.; Labrugère, C.; Valencia, H.; Marécot, P.; Tatibouët, J.M.; Duprez, D. Effect of palladium on the reducibility of Mn based materials: Correlation with methane oxidation activity. Phys. Chem. Chem. Phys. 2008, 10, 5983–5992. [Google Scholar] [CrossRef]
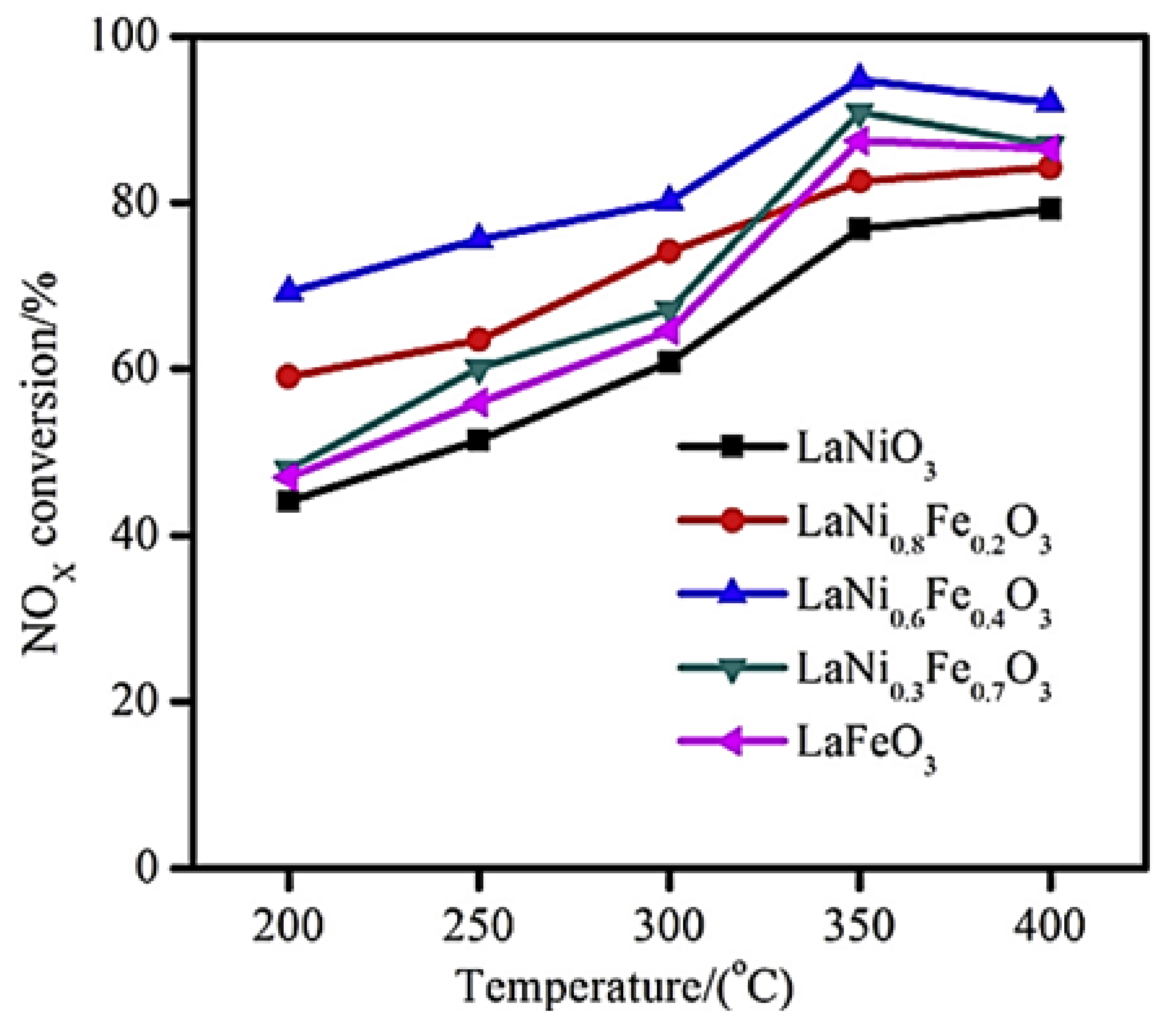
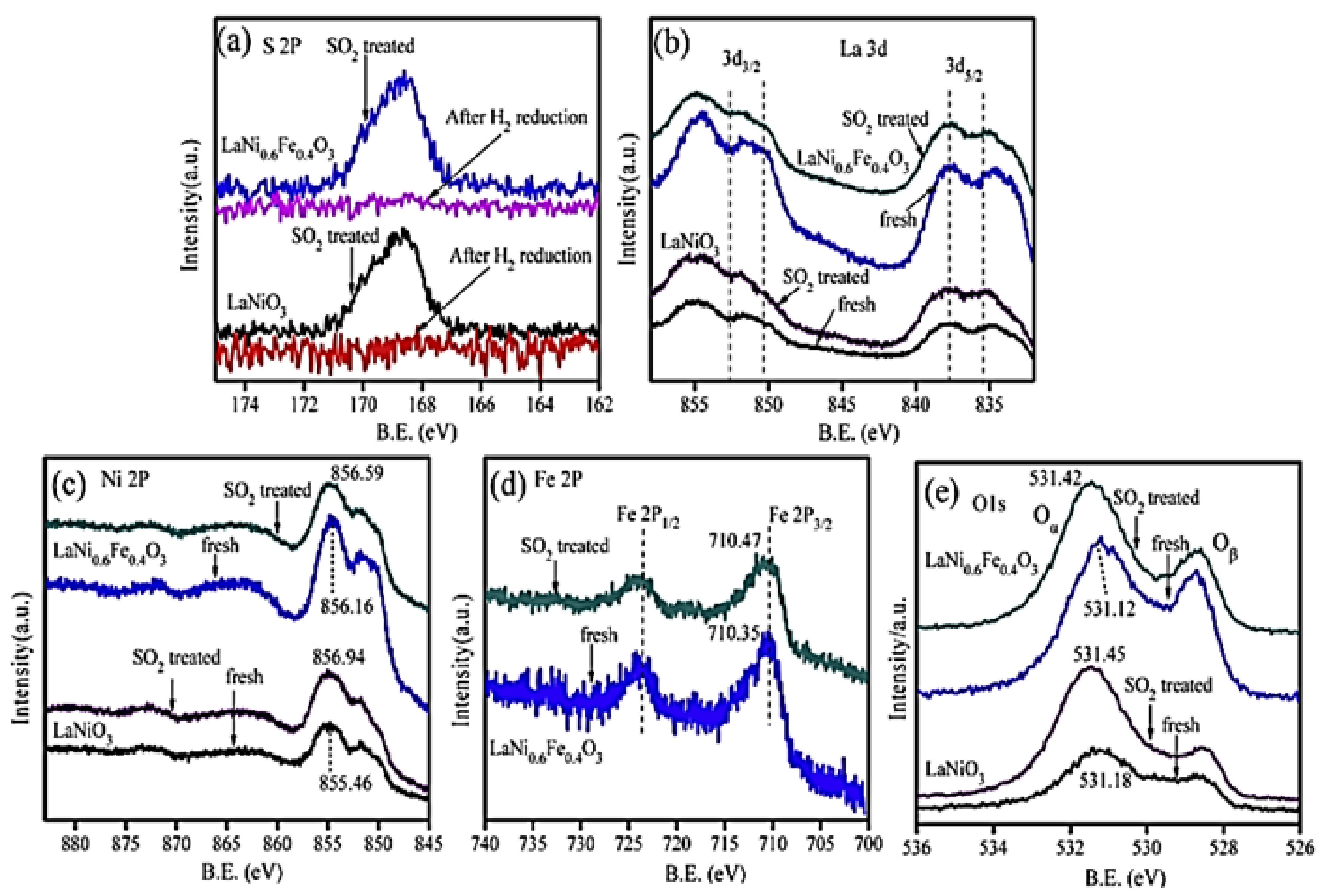
- Luo, Y.; Wang, X.; Qian, Q.; Chen, Q. Studies on B sites in Fe-doped LaNiO3 perovskite for SCR of NOx with H2. Int. J. Hydrogen Energy 2014, 39, 15836–15843. [Google Scholar] [CrossRef]
- Furfori, S.; Russo, N.; Fino, D.; Saracco, G.; Specchia, V. NO SCR reduction by hydrogen generated in line on perovskite-type catalysts for automotive diesel exhaust gas treatment. Chem. Eng. Sci. 2010, 65, 120–127. [Google Scholar] [CrossRef]
- Burch, R.; Coleman, M.D. An investigation of the NO/H2/O2 reaction on noble-metal catalysts at low temperatures under lean-burn conditions. Appl. Catal. B Environ. 1999, 23, 115–121. [Google Scholar] [CrossRef]
- Dhainaut, F.; Pietrzyk, S.; Granger, P. Kinetic investigation of the NO reduction by H2 over noble metal based catalysts. Catal. Today 2007, 119, 94–99. [Google Scholar] [CrossRef]
- Barrera, A.; Viniegra, M.; Bosch, P.; Lara, V.H.; Fuentes, S. Pd/Al2O3-La2O3 catalysts prepared by sol-gel: Characterization and catalytic activity in the NO reduction by H2. Appl. Catal. B Environ. 2001, 34, 97–111. [Google Scholar] [CrossRef]
- Mondragon Rodriguez, G.C.; Kelm, K.; Saruhan, B. H2-selective catalytic reduction of NOx activity and microstructural analysis of new BaTi0.95Pd0.05O3 catalyst. Appl. Catal. A Gen. 2010, 387, 173–184. [Google Scholar] [CrossRef]
- Costa, C.N.; Efstathiou, A.M.; Stathopoulos, V.N.; Belessi, V.C. An investigation of the NO/H2/O2 (Lean-deNOx) reaction on a highly active and selective Pt/La0.5Ce0.5MnO3 catalyst. J. Catal. 2001, 197, 350–364. [Google Scholar] [CrossRef]
- Macleod, N.; Lambert, R.M. Lean NOx reduction with CO + H2 mixtures over Pt/Al2O3 and Pd/Al2O3 catalysts. Appl. Catal. B Environ. 2002, 35, 269–279. [Google Scholar] [CrossRef]
- Dhainaut, F.; Pietrzyk, S.; Granger, P. NO + H2 reaction on Pd/Al2O3 under lean conditions: Kinetic study. Top. Catal. 2007, 42, 135–141. [Google Scholar] [CrossRef]
- Matsouka, V.; Konsolakis, M.; Lambert, R.M.; Yentekakis, I.V. In situ DRIFTS study of the effect of structure (CeO2–La2O3) and surface (Na) modifiers on the catalytic and surface behaviour of Pt/γ-Al2O3 catalyst under simulated exhaust conditions. Appl. Catal. B Environ. 2008, 84, 715–722. [Google Scholar] [CrossRef]
- Qin, Y.; Sun, L.; Zhang, D.; Huang, L. Role of ceria in the improvement of SO2 resistance of LaxCe1-XFeO3 catalysts for catalytic reduction of NO with CO. Catal. Commun. 2016, 79, 53–57. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, H.; Li, G.; Jin, L.; Li, X.; Ou, X.; Dong, L.; Jin, G.; Li, B. Tuning composition on B sites of LaM0.5Mn0.5O3 (M = Cu, Co, Fe, Ni, Cr) perovskite catalysts in NOx efficient reduction. Appl. Surf. Sci. 2020, 508, 145158. [Google Scholar] [CrossRef]
- Wu, Y.; Chu, B.; Zhang, M.; Yi, Y.; Dong, L.; Fan, M.; Jin, G.; Zhang, L.; Li, B. Influence of calcination temperature on the catalytic properties of LaCu0.25Co0.75O3 catalysts in NOx reduction. Appl. Surf. Sci. 2019, 481, 1277–1286. [Google Scholar] [CrossRef]
- Wu, Y.; Li, G.; Chu, B.; Dong, L.; Tong, Z.; He, H.; Zhang, L.; Fan, M.; Li, B.; Dong, L. NO Reduction by CO over Highly Active and Stable Perovskite Oxide Catalysts La0.8Ce0.2M0.25Co0.75O3 (M = Cu, Mn, Fe): Effect of the Role in B Site. Ind. Eng. Chem. Res. 2018, 57, 15670–15682. [Google Scholar] [CrossRef]
- Yi, Y.; Liu, H.; Chu, B.; Qin, Z.; Dong, L.; He, H.; Tang, C.; Fan, M.; Bin, L. Catalytic removal NO by CO over La-Ni0.5M0.5O3 (M = Co, Mn, Cu) perovskite oxide catalysts: Tune surface chemical composition to improve N2 selectivity. Chem. Eng. J. 2019, 369, 511–521. [Google Scholar] [CrossRef]
- Tarjomannejad, A.; Farzi, A.; Gómez, M.J.I.; Niaei, A.; Salari, D.; Albaladejo-Fuentes, V. Catalytic Reduction of NO by CO over LaMn1−xFexO3 and La0.8A0.2Mn0.3Fe0.7O3 (A = Sr, Cs, Ba, Ce) Perovskite Catalysts. Catal. Letters 2016, 146, 2330–2340. [Google Scholar] [CrossRef]
- Tarjomannejad, A.; Farzi, A.; Niaei, A.; Salari, D. NO reduction by CO over LaB0.5B′0.5O3 (B = Fe, Mn, B′ = Fe, Mn, Co, Cu) perovskite catalysts, an experimental and kinetic study. J. Taiwan Inst. Chem. Eng. 2017, 78, 200–211. [Google Scholar] [CrossRef]
- Lorimer, D.; Bell, A.T. Reduction of NO by CO over a silica-supported platinum catalyst: Infrared and kinetic studies. J. Catal. 1979, 59, 223–238. [Google Scholar] [CrossRef]
- Zhdanov, V.P.; Kasemo, B. Mechanism and kinetics of the NO-CO reaction on Rh. Surf. Sci. Rep. 1997, 29, 31–33. [Google Scholar] [CrossRef]
- De Lima, R.K.C.; Batista, M.S.; Wallau, M.; Sanches, E.A.; Mascarenhas, Y.P.; Urquieta-González, E.A. High specific surface area LaFeCo perovskites-Synthesis by nanocasting and catalytic behavior in the reduction of NO with CO. Appl. Catal. B Environ. 2009, 90, 441–450. [Google Scholar] [CrossRef]
- Giannakas, A.E.; Ladavos, A.K.; Pomonis, P.J. Preparation, characterization and investigation of catalytic activity for NO + CO reaction of LaMnO3 and LaFeO3 perovskites prepared via microemulsion method. Appl. Catal. B Environ. 2004, 49, 147–158. [Google Scholar] [CrossRef]
- Leontiou, A.A.; Ladavos, A.K.; Armatas, G.S.; Trikalitis, P.N.; Pomonis, P.J. Kinetics investigation of NO + CO reaction on La-Sr-Mn-O perovskite-type mixed oxides. Appl. Catal. A Gen. 2004, 263, 227–239. [Google Scholar] [CrossRef]
- Konsolakis, M.; Yentekakis, I.V.; Palermo, A.; Lambert, R.M. Optimal promotion by rubidium of the CO + NO reaction over Pt/γ-Al2O3 catalysts. Appl. Catal. B Environ. 2001, 33, 293–302. [Google Scholar] [CrossRef]







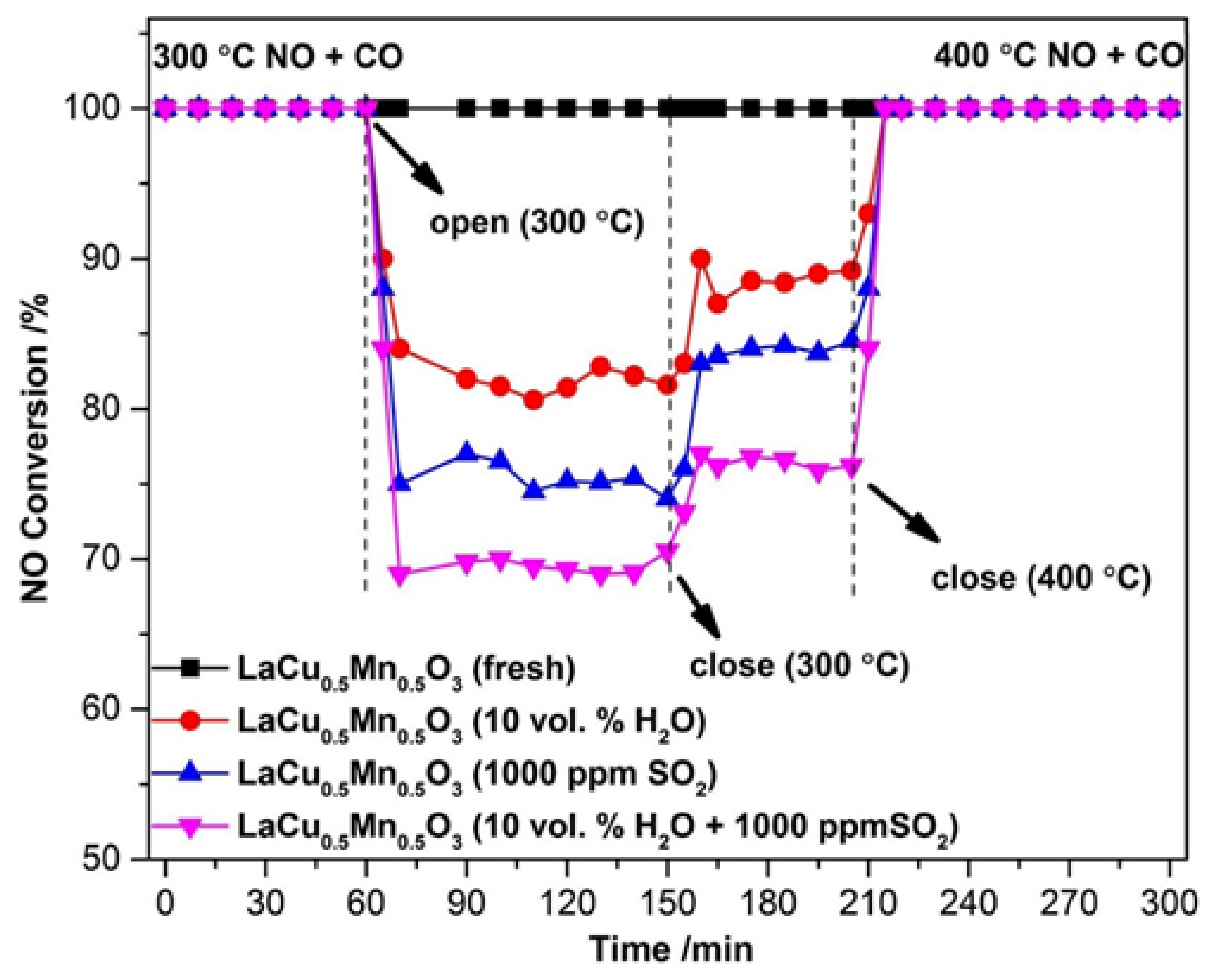
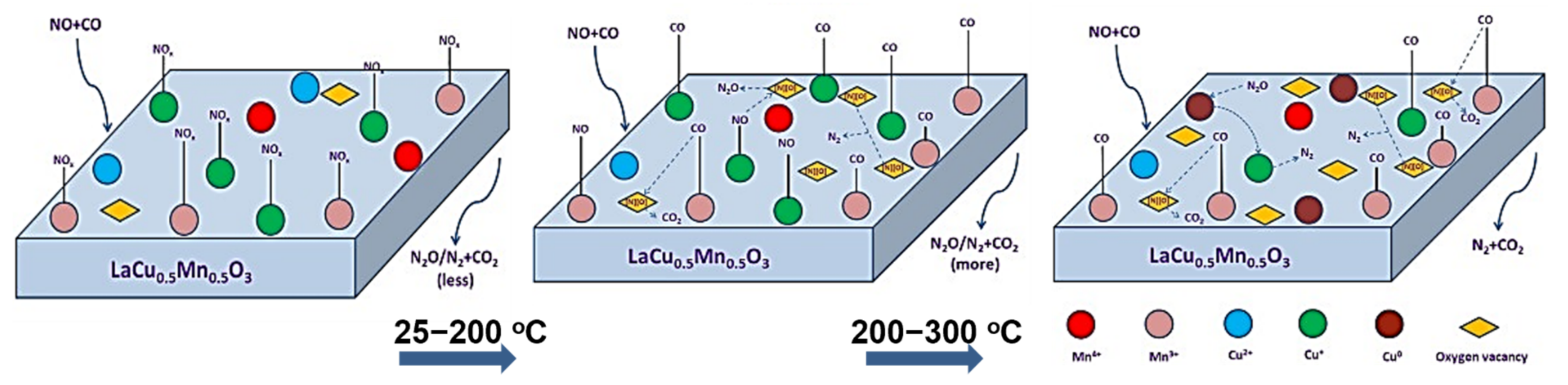
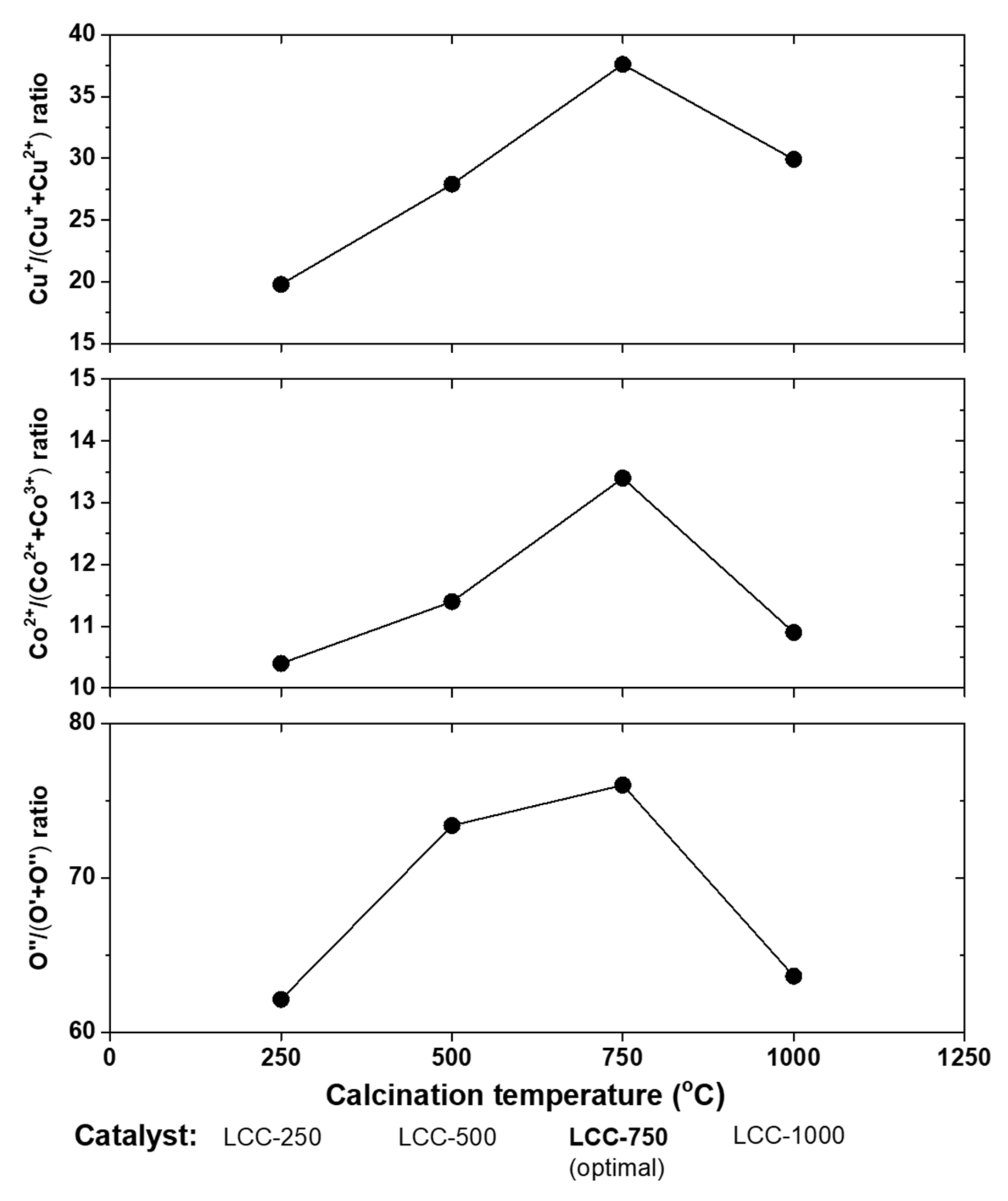
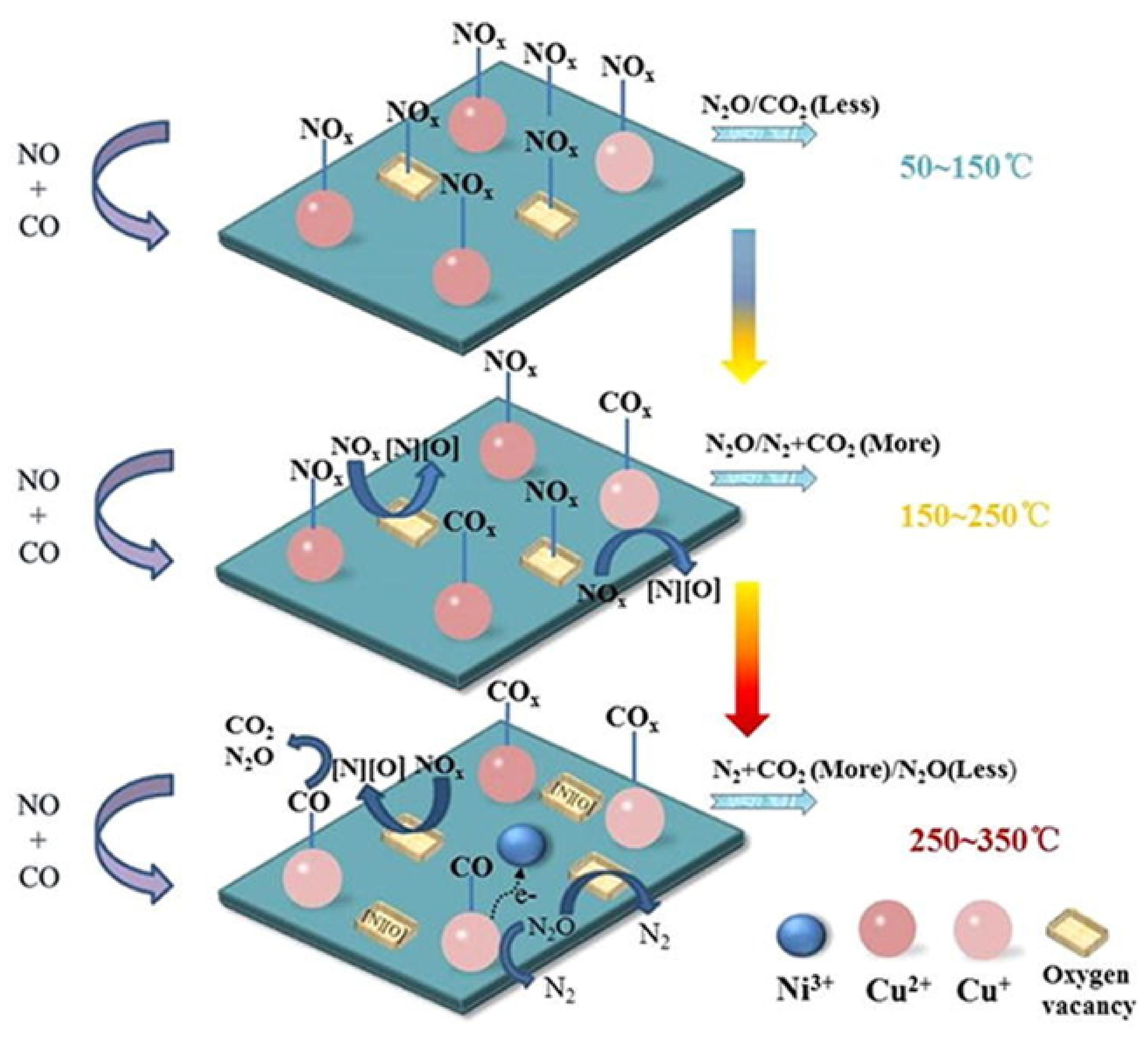

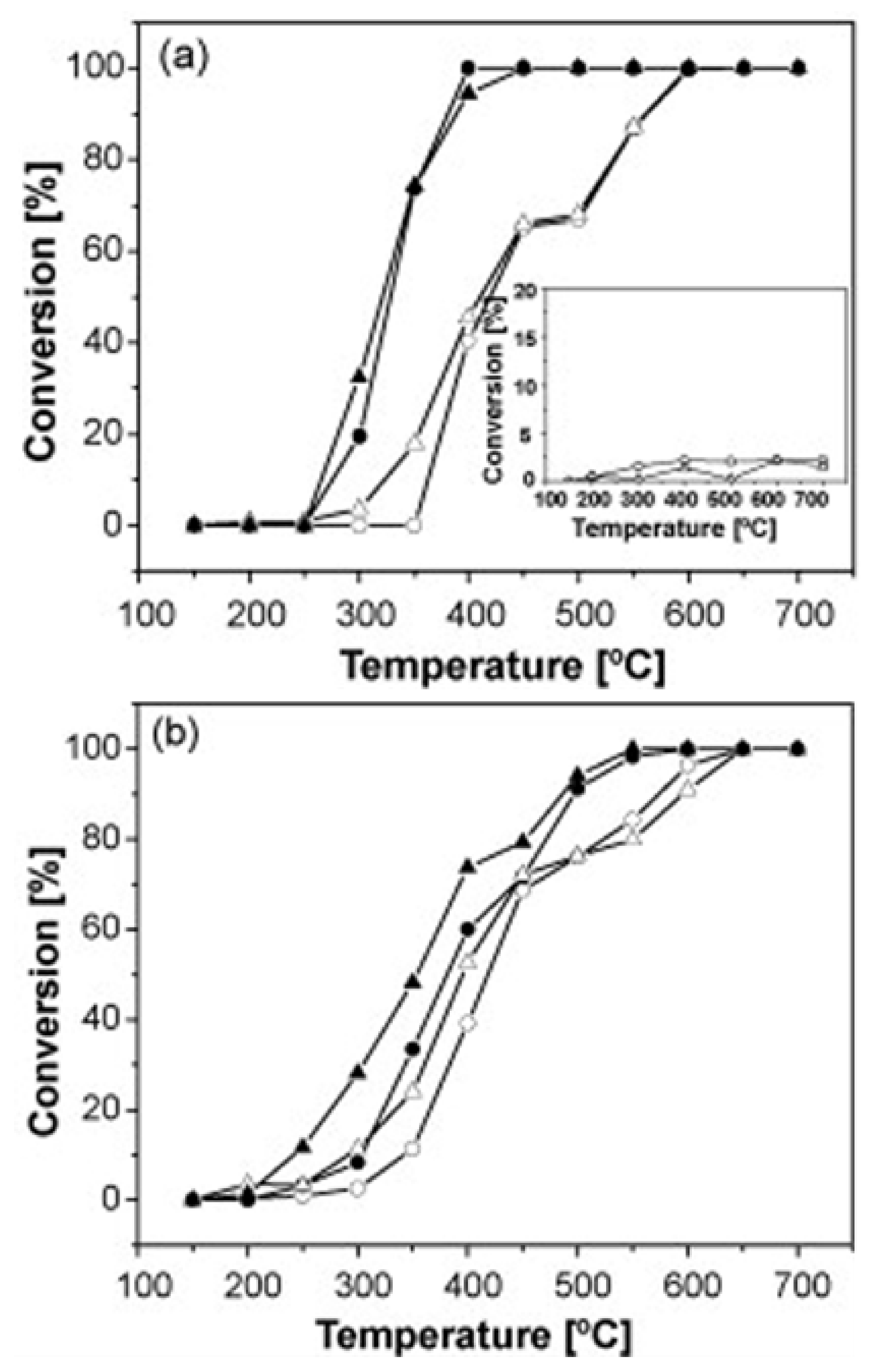
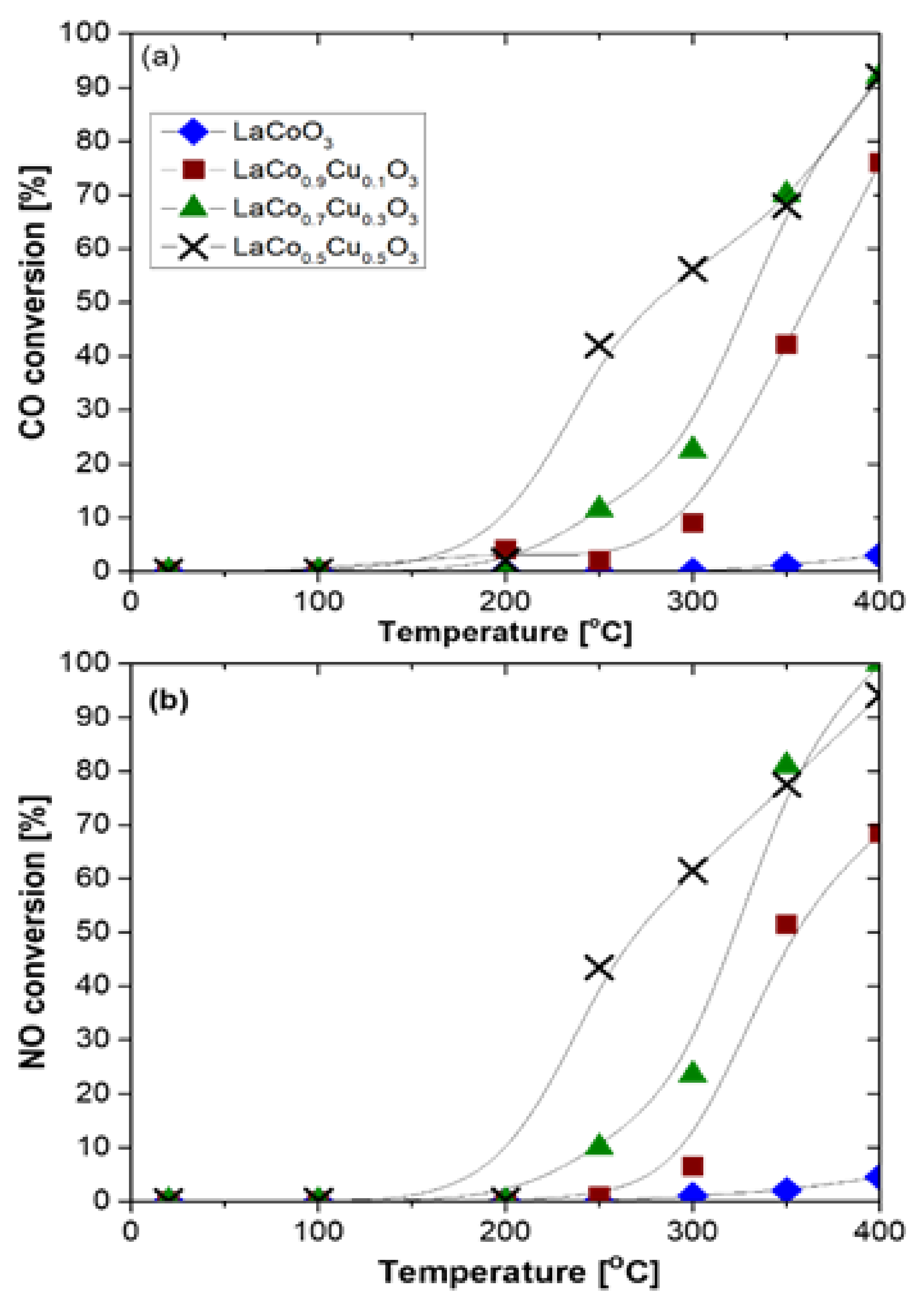

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Reaction Feed Conditions | Achievements | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|
| NO (%) | HC (%) | O2 (%) | Other (%) | WGHSV (mL∙g−1∙h−1) | XNO (%) | at T (°C) | SN2 (%) | ||
| Perovskite and NM/perovskite catalysts | |||||||||
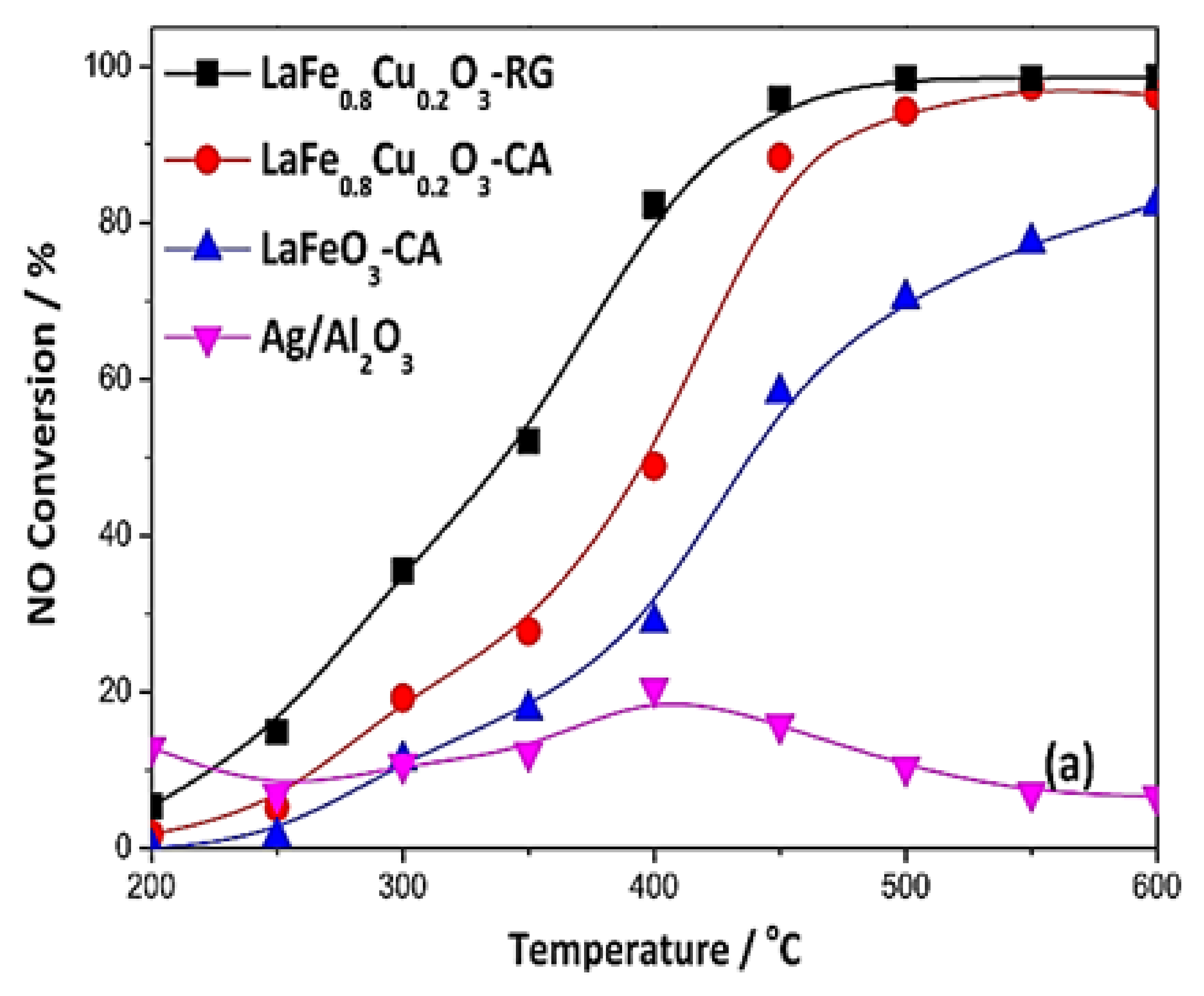
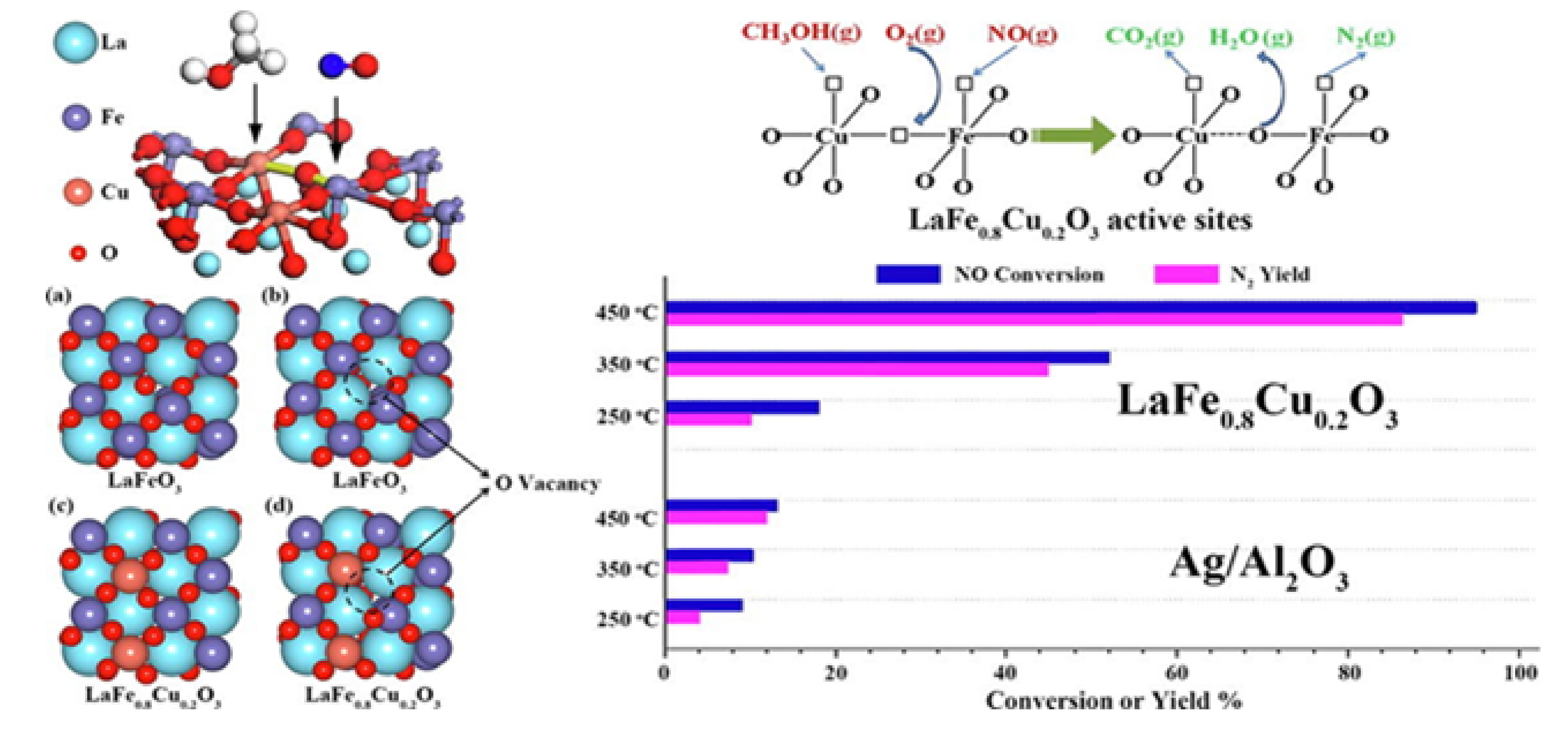
| LaFe0.8Cu0.2O3-RG LaFe0.8Cu0.2O3-CA LaFeO3-CA | 0.1 0.1 0.1 | 0.3 (CH3OH) 0.3 (CH3OH) 0.3 (CH3OH) | 8 8 8 | - - - | 30,000 30,000 30,000 | >90 >90 >80 | >430 >475 >575 | n/a n/a n/a | [74] [74] [74] |
| La0.8Sr0.2MnO3/α-Al2O3 La0.8Sr0.2MnO3/α-Al2O3 | 0.1 0.1 | 0.12 (CH4) 0.12 (CH4) | 0 5 | - - | 1636 h−1 (GHSV) 1636 h−1 (GHSV) | >90 96 | >875 800 | n/a n/a | [76] [76] |
| La0.8Sr0.2MnO3 | 0.4 | 0.24 (C10H22) | 9 | 1.5 (H2O) | 36,000 | 20–65 | 200–275 | 13 (max at 210 °C) | [77] |
| Conventional, supported on oxide supports NM catalysts | |||||||||
| 2wt%Pt/SiO2 | 0.4 | 0.24 (C10H22) | 9 | 1.5 (H2O) | 36,000 | >90 | 200–250 | 18 (max at 200 oC) | [77] |
| 0.5wt%Pt/γ-Al2O3 0.5wt%Pt(1.6wt%Na)/γ-Al2O3 | 0.1 0.1 | 0.1 (C3H6) 0.1 (C3H6) | 5 5 | - - | 180,000 * 53,485 * | >50 >50 | 300–400 225–375 | 40 (at 300 °C) 75 (at 225 °C) | [7] [7] |
| Catalyst | Reaction Feed Conditions | Achievements | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|
| NO (%) | H2 (%) | O2 (%) | Other (%) | WGHSV (mL∙g−1∙h−1) | XNO (%) | at T (°C) | max. SN2 (%) | ||
| Perovskite and NM/perovskite catalysts | |||||||||
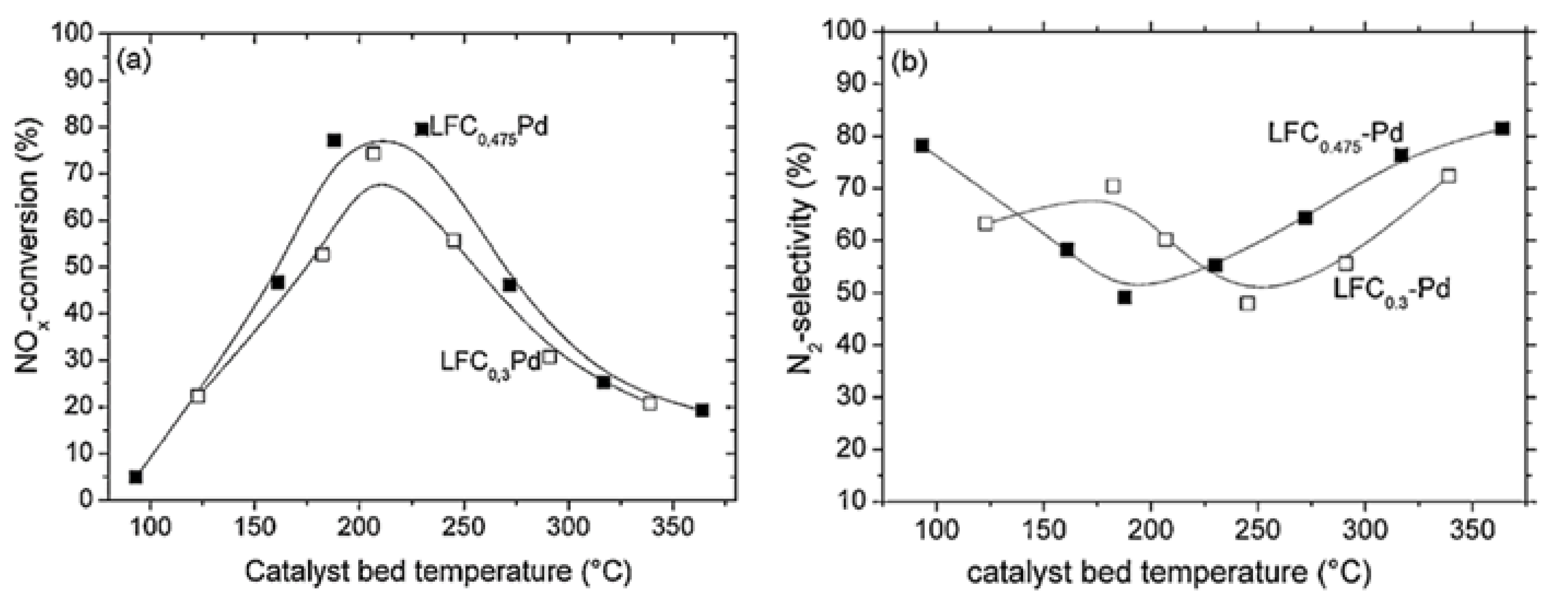
| LaFe0.65Co0.3Pd0.05O3 | 0.072 | 1 | 5 | 7.2 (H2O) + 7.2 (CO2) | 55,400 | >50 (max. 57) | 200–250 | 75 (at 200 °C) | [67] |
| LaFe0.475Co0.475Pd0.05O3 | 0.072 | 1 | 5 | 7.2 (H2O) + 7.2 (CO2) | 55,400 | >50 (max. 85) | 175–300 | 76 (at 250 °C) | [67] |
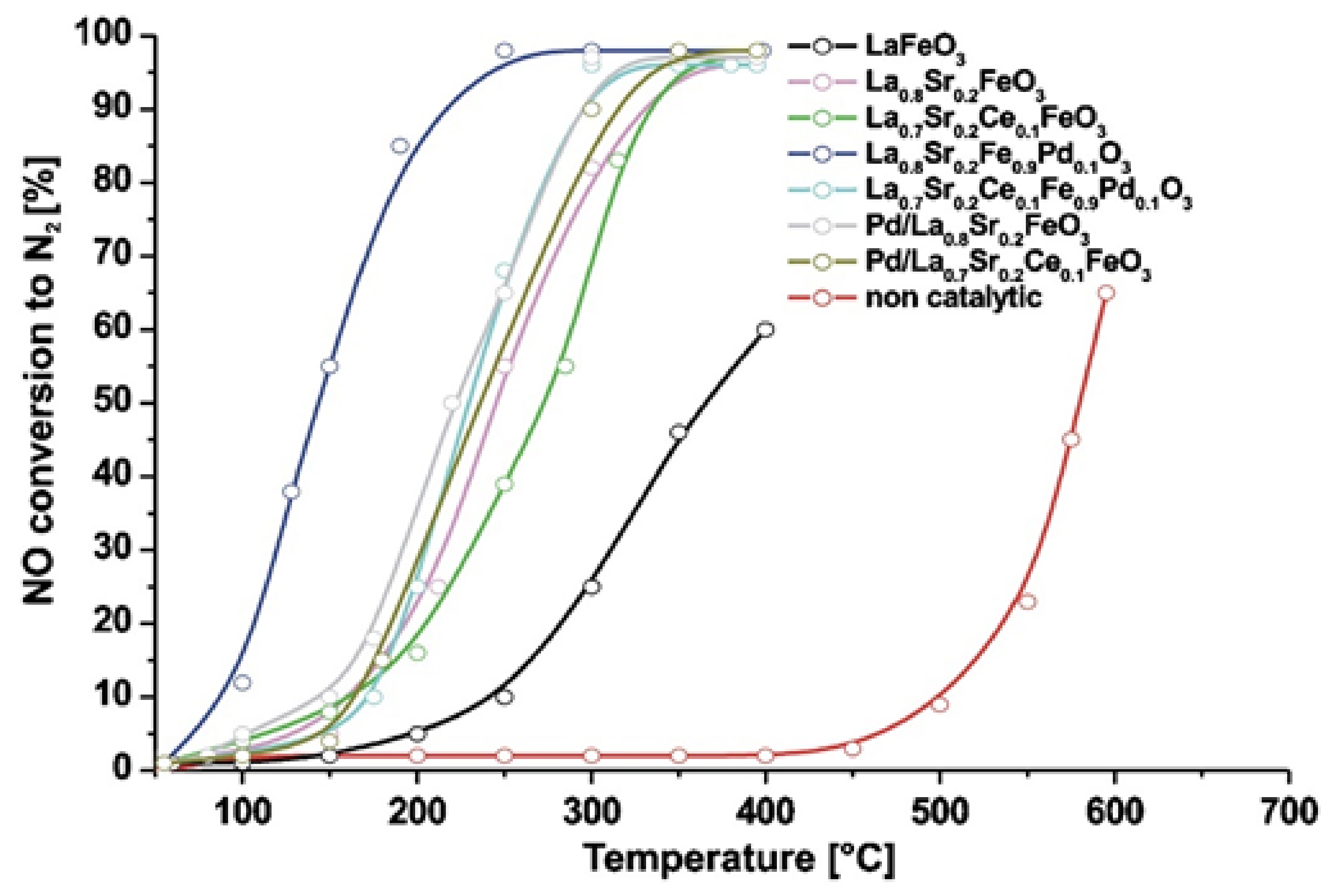
| La0.8Sr0.2Fe0.9Pd0.1O3 | 0.1 | 1 | 5 | - | 180,000 | >50 (max. 96) | 120–210 | 67 (at 160 °C) | [81] |
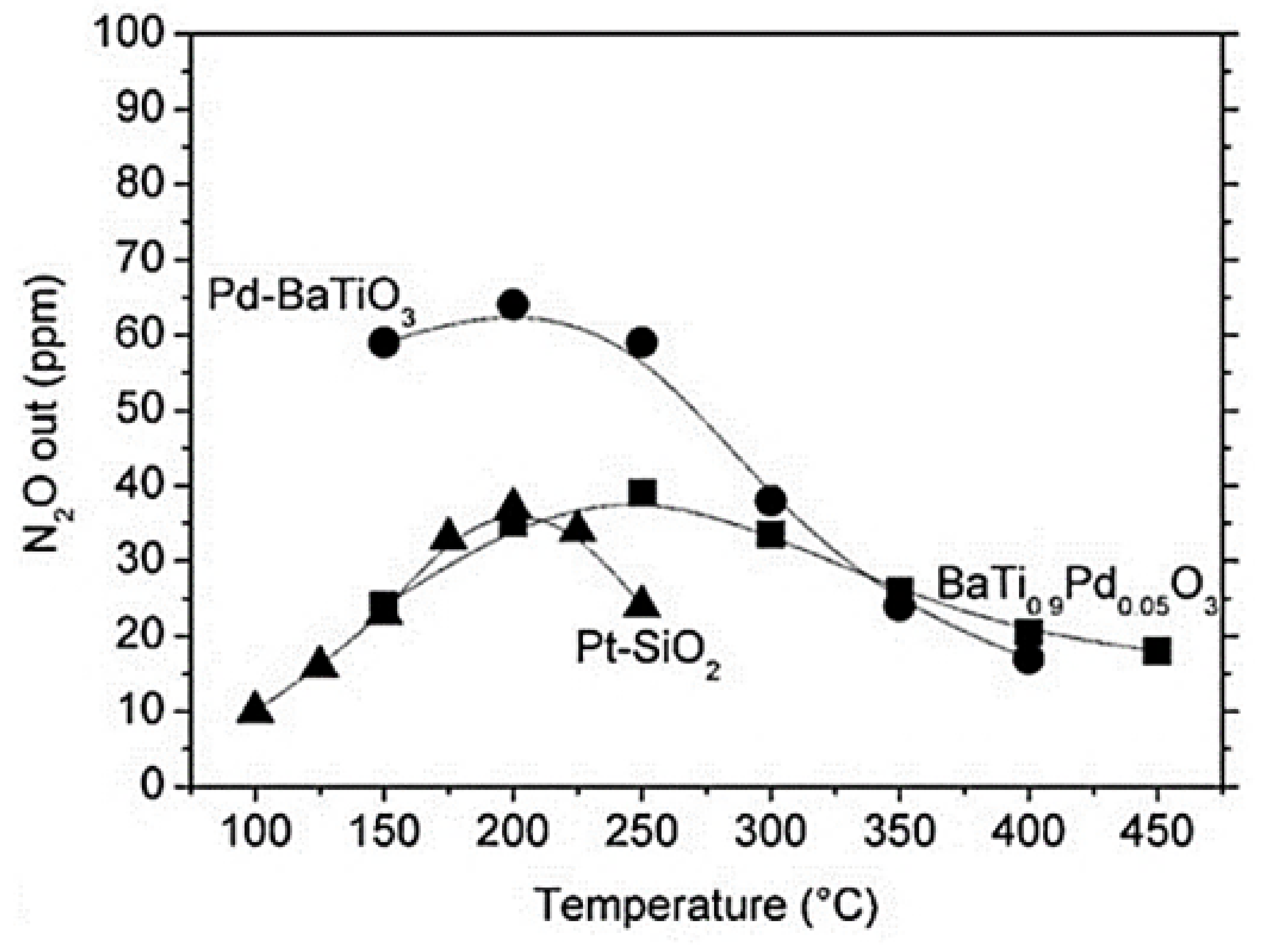
| BaTi0.95Pd0.05O3 | 0.072 | 1 | 5 | 7.2 (H2O) + 7.2 (CO2) | 55,400 | >50 (max. 92) | 150–300 | 72 (at 200 °C) | [85] |
| BaTi0.95Pd0.05O3 | 0.045 | 0.8 | 5 | - | 1.61 × 106 | >50 (max. 55) | 200–300 | 68 (at 250 °C) | [85] |
| Pd/BaTiO3 | 0.045 | 0.8 | 5 | - | 1.61 × 106 | >50 (max. 70) | 125–250 | 60 (at 150 °C) | [85] |
| 0.3%Pt/La0.7Sr0.2Ce0.1FeO3 | 0.25 | 1 | 5 | - | 40,000 | >50 (max. 83) | 125–225 | 93 (at 170 °C) | [9] |
| 0.1%Pt/La0.5Ce0.5MnO3 | 0.25 | 1 | 5 | 5(H2O) | 40,000 | >50 (max. 88) | 125–175 | 78 (at 150 °C) | [86] |
| Conventional, supported on oxide supports, NM catalysts | |||||||||
| 1%Pt/SiO2 | 0.072 | 1 | 5 | 7.2 (H2O) + 7.2 (CO2) | 55,400 | >50 (max. 80) | 100–175 | 51 (at 125 °C) | [67] |
| 1%Pt/Al2O3 | 0.05 | 0.2 | 6 | - | 120,000 | 50 | 150 | 30 | [82] |
| 1%Pt/SiO2 | 0.05 | 0.2 | 6 | - | 120,000 | >50 (max. 76) | 85–110 | 20 | [82] |
| 0.5%Pt/Al2O3 | 0.05 | 0.4 | 5 | - | 120,000 | >50 (max. 80) | 100–225 | 60 (at 175 °C) | [87] |
| 0.5%Pd/Al2O3 | 0.05 | 0.4 | 5 | - | 120,000 | 9 | 275 | 72 | [87] |
| 1%Pd/Al2O3 | 1 | 1 | 1 | 100,000 | 30 | 160 | 23 | [88] | |
| 0.1%Pt/MgO-CeO2 | 0.25 | 1 | 5 | 5(H2O) | 40,000 | >50 (max. 95) | 90–250 | 78 (at 150 °C) | [65] |
| 0.5%Pd/Al2O3 | 0.1 | 0.75 | 6 | 0.25(CO) | 240,000 | 30 | 210 | 70 | [89] |
| 0.5%Pd/Al2O3-(10%TiO2) | 0.1 | 0.75 | 6 | 0.25(CO) | 240,000 | >50 (max. 92) | 160–450 | 70 (at 265 °C) | [89] |
| Catalyst | Reaction Conditions | Achievements | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|
| NO (%) | CO (%) | O2 (%) | Other (%) | WGHSV (mL/g∙h) | XNO (%) | at T (°C) | max. SN2 (%) | ||
| Perovskite catalysts | |||||||||
| La0.6Ce0.4FeO3 (a) | 0.04 | 0.05 | - | - | 24,000 h−1 (GHSV) | >50 (max. 88) | 350–500 | 90 (at 350) | [90] |
| La0.6Ce0.4FeO3 (a) | 0.04 | 0.05 | - | 0.01 (SO2) | 24,000 h−1 (GHSV) | 76 | 500 | n/a | [90] |
| La0.6Ce0.4FeO3 (a) | 0.04 | 0.05 | 3 | 0.01 (SO2) + 3 (H2O) | 24,000 h−1 (GHSV) | >50 (max. 74) | 350–500 | n/a | [90] |
| LaCu0.5Mn0.5O3 (b) | 5 | 10 | - | - | 60,000 | 100 | 300–600 | 100 | [91] |
| LaCu0.25Co0.75O3-750 (c) | 5 | 10 | - | - | 60,000 | 100 | 350–600 | 100 | [92] |
| La0.8Ce0.2Cu0.25Co0.75O3 (d) | 5 | 10 | - | - | 60,000 | 100 | 290–600 | 100 | [93] |
| LaNi0.5Cu0.5O3 (e) | 5 | 10 | - | - | 36,000 | 100 | 375–500 | 100 (at 450 °C) | [94] |
| LaMn0.3Fe0.7O3 (f) | 0.3 | 0.3 | - | - | 12,000 h−1 (GHSV) | 90–100 | 390–450 | 90–100 | [95] |
| La0.8Ce0.2Fe0.7Mn0.3O3 (g) | 0.3 | 0.3 | - | - | 12,000 h−1 (GHSV) | 90–100 | 340–450 | 92–96 | [95] |
| LaFe0.5Mn0.5O3 (h) | 0.3 | 0.3 | - | - | 12,000 h−1 (GHSV) | 90–100 | 420–450 | 92–96 | [96] |
| LaMn0.5Cu0.5O3 (i) | 0.3 | 0.3 | - | - | 12,000 h−1 (GHSV) | 90–100 | 400–450 | 90–98 | [96] |
| LaFeO3-nanocast | 0.5 | 0.5 | - | - | 30,000 | 100 | 375–700 | 100 | [99] |
| LaFeO3-uncast | 0.5 | 0.5 | - | - | 30,000 | 100 | 600–700 | 100 | [99] |
| LaFe0.6Co0.4O3-nanocast | 0.5 | 0.5 | - | - | 30,000 | 100 | 550–700 | 100 | [99] |
| LaFe0.6Co0.4O3-uncast | 0.5 | 0.5 | - | - | 30,000 | 100 | 650–700 | 100 | [99] |
| LaCo0.5Cu0.5O3 (j) | 4 | 4 | - | - | 150,000 | 95 | 400 | n/a | [59] |
| LaCu0.7Mn0.3O3 (k) | 0.3 | 0.3 | - | - | 12,000 h−1 (GHSV) | >90 | 360–450 | n/a | [35] |
| La0.8Sr0.2Cu0.7Mn0.3O3 (l) | 0.3 | 0.3 | - | - | 12,000 h−1 (GHSV) | >90 | 320–450 | n/a | [35] |
| La0.8Ce0.2FeO3 (m) | 2 | 2 | - | - | 30,000 | >90 | 330–500 | n/a | [64] |
| LaMnO3 (r); LaFeO3 (r) * | 2 | 2 | - | - | 30,000 | >90 | 420–500 | n/a | [100] |
| LaMnO3 (n) | 2 | 2 | - | - | 30,000 | >90 | 510–570 | 100 | [101] |
| Conventional, supported on oxide supports, NM catalysts | |||||||||
| 0.5wt%Pt/γ-Al2O3 | 0.05 | 0.4 | 5 | - | 120,000 | 26 | 250 | 80 | [87] |
| 0.5wt%Pd/γ-Al2O3 | 0.05 | 0.4 | 5 | - | 120,000 | 13 | 180 | 62 | [87] |
| 0.5wt%Pt/γ-Al2O3 | 0.1 | 0.1 | - | - | 600,000 | 60 | 480 | 60 | [102] |
| 0.5wt%Pt(9.7%Rb)/γ-Al2O3 | 0.1 | 0.1 | - | - | 600,000 | >90 | 320–500 | 100 (at 350 °C) | [102] |
| 0.5wt%Rh/γ-Al2O3 | 0.1 | 0.1 | - | - | 600,000 | >90 | 250–500 | 100 (at 300 °C) | [102] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yentekakis, I.V.; Georgiadis, A.G.; Drosou, C.; Charisiou, N.D.; Goula, M.A. Selective Catalytic Reduction of NOx over Perovskite-Based Catalysts Using CxHy(Oz), H2 and CO as Reducing Agents—A Review of the Latest Developments. Nanomaterials 2022, 12, 1042. https://doi.org/10.3390/nano12071042
Yentekakis IV, Georgiadis AG, Drosou C, Charisiou ND, Goula MA. Selective Catalytic Reduction of NOx over Perovskite-Based Catalysts Using CxHy(Oz), H2 and CO as Reducing Agents—A Review of the Latest Developments. Nanomaterials. 2022; 12(7):1042. https://doi.org/10.3390/nano12071042
Chicago/Turabian StyleYentekakis, Ioannis V., Amvrosios G. Georgiadis, Catherine Drosou, Nikolaos D. Charisiou, and Maria A. Goula. 2022. "Selective Catalytic Reduction of NOx over Perovskite-Based Catalysts Using CxHy(Oz), H2 and CO as Reducing Agents—A Review of the Latest Developments" Nanomaterials 12, no. 7: 1042. https://doi.org/10.3390/nano12071042
APA StyleYentekakis, I. V., Georgiadis, A. G., Drosou, C., Charisiou, N. D., & Goula, M. A. (2022). Selective Catalytic Reduction of NOx over Perovskite-Based Catalysts Using CxHy(Oz), H2 and CO as Reducing Agents—A Review of the Latest Developments. Nanomaterials, 12(7), 1042. https://doi.org/10.3390/nano12071042