
X-ray Absorption Near-Edge Structure (XANES) at the O K-Edge of Bulk Co3O4: Experimental and Theoretical Studies


 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
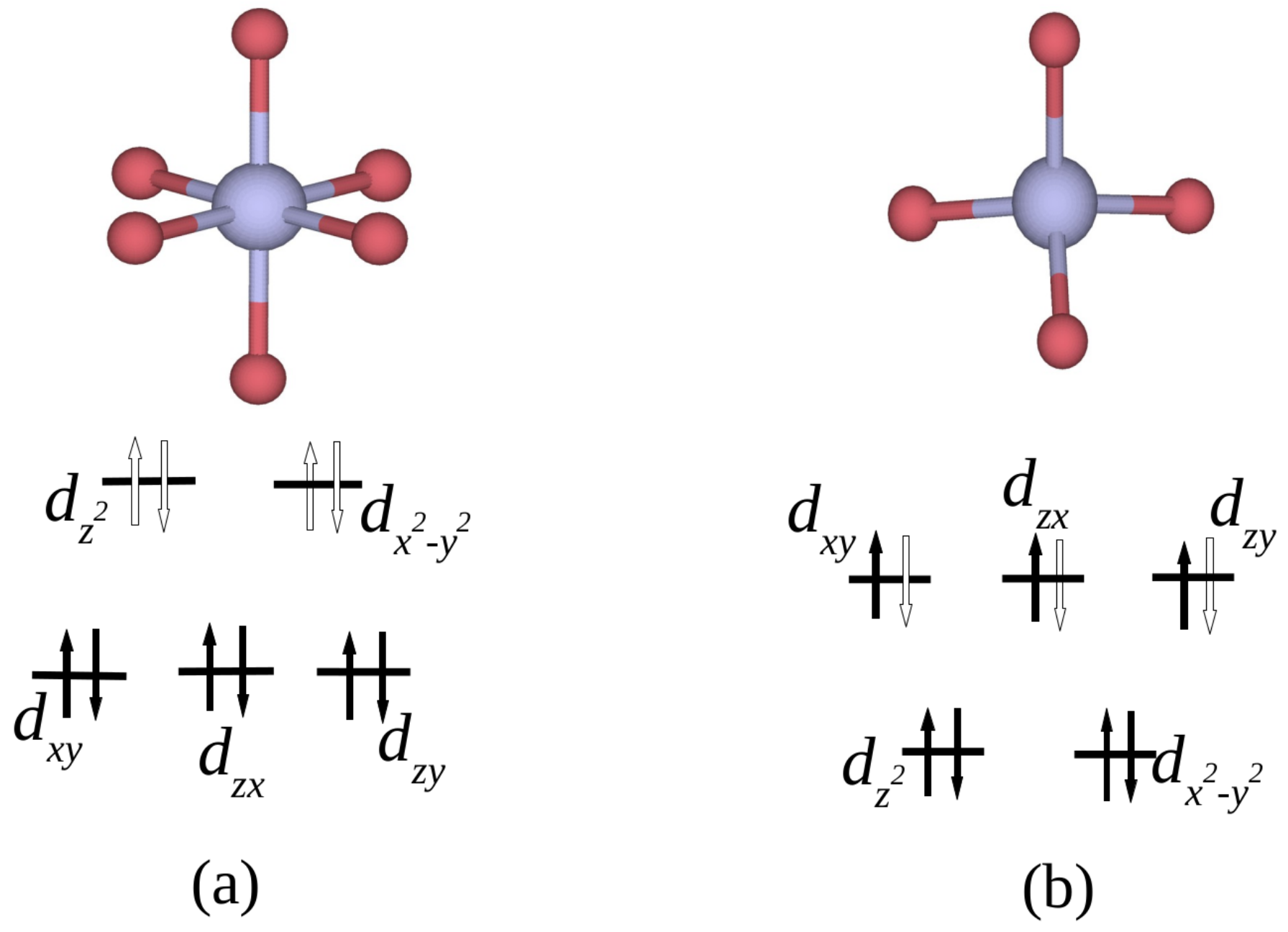
:1. Introduction
2. Experimental and Computational Details
2.1. Experimental Details
2.2. Computational Details
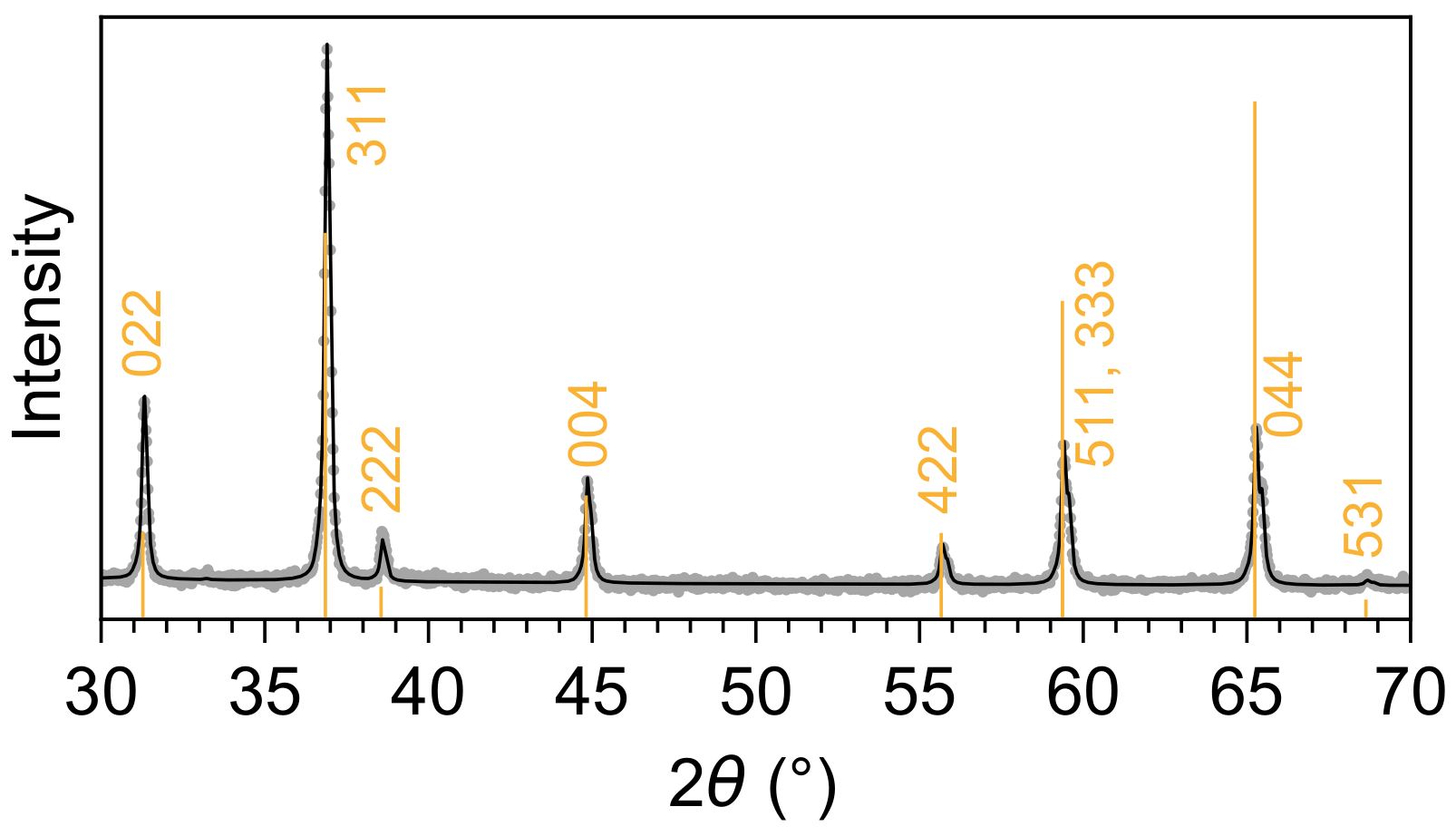
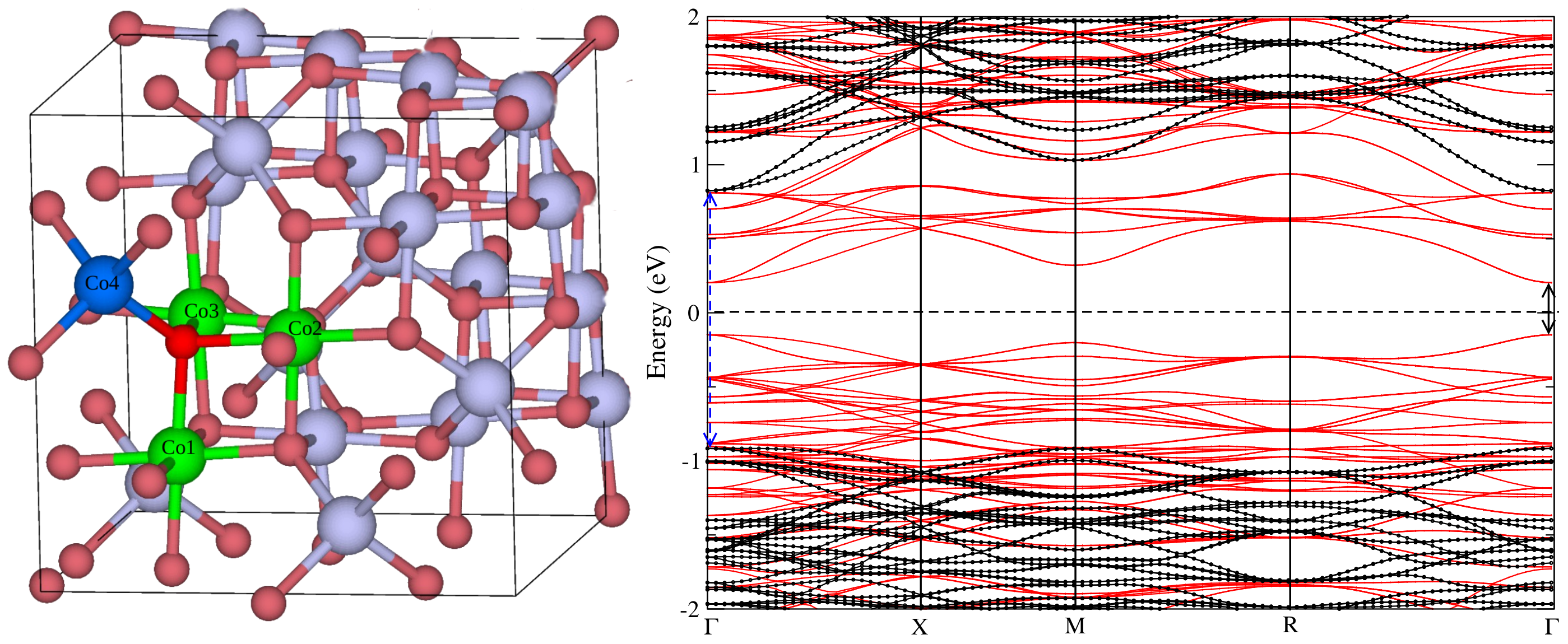
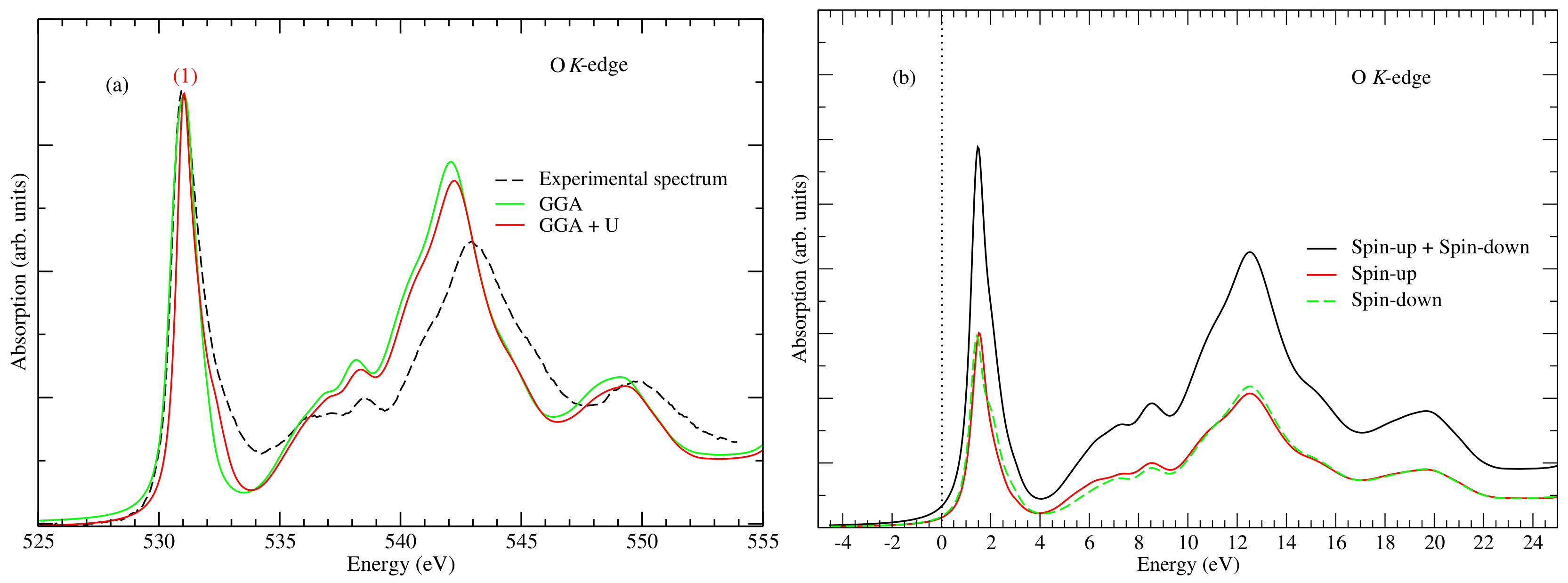
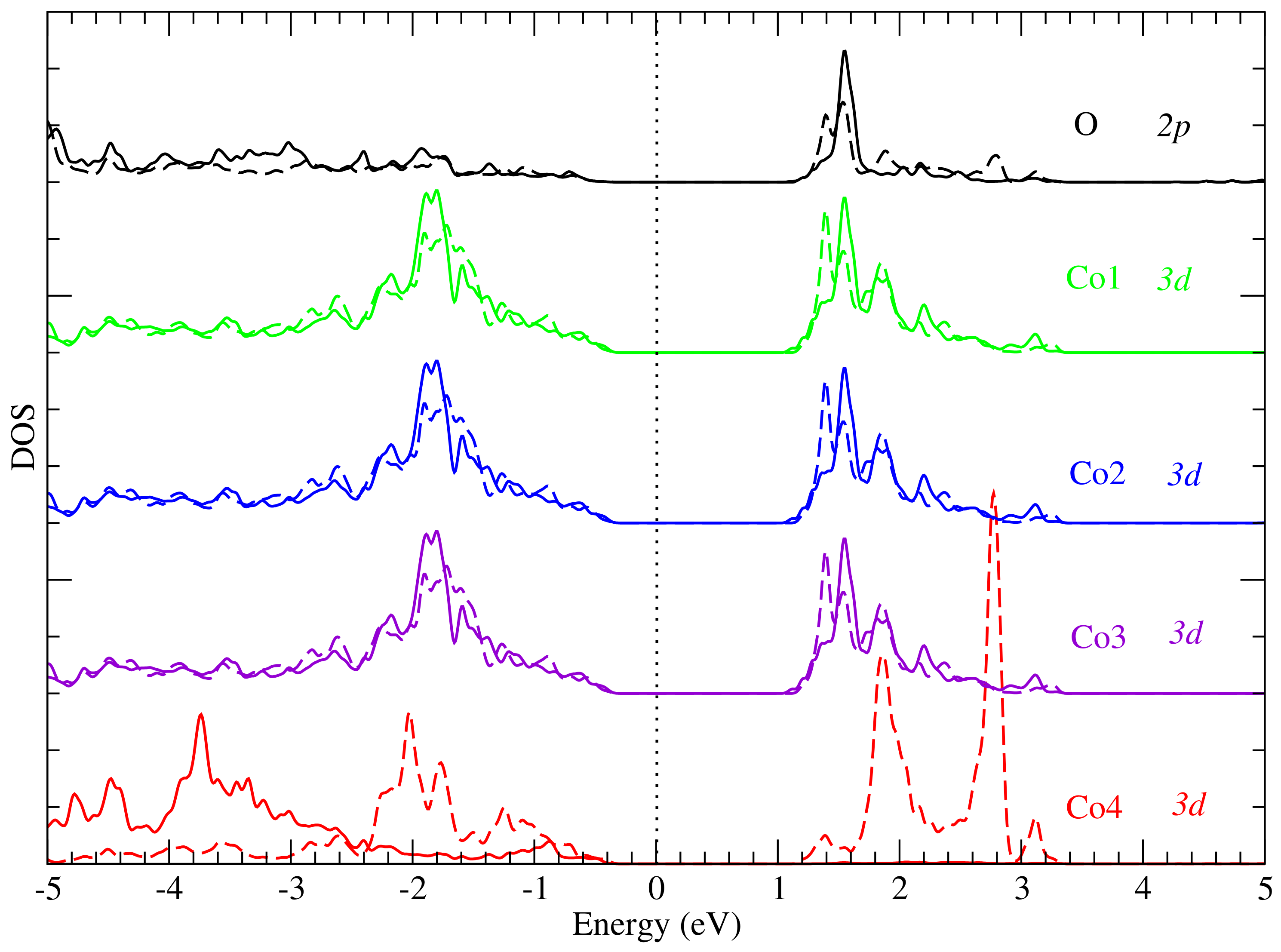
3. Results
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Deng, X.; Tüysüz, H. Cobalt-Oxide-Based Materials as Water Oxidation Catalyst: Recent Progress and Challenges. ACS Catal. 2014, 4, 3701–3714. [Google Scholar] [CrossRef]
- Cai, Y.; Xu, J.; Guo, Y.; Liu, J. Ultrathin, Polycrystalline, Two-Dimensional Co3O4 for Low-Temperature CO Oxidation. ACS Catal. 2019, 9, 2558–2567. [Google Scholar] [CrossRef]
- Anke, S.; Bendt, G.; Sinev, I.; Hajiyani, H.; Antoni, H.; Zegkinoglou, I.; Jeon, H.; Pentcheva, R.; Roldan Cuenya, B.; Schulz, S.; et al. Selective 2-Propanol Oxidation over Unsupported Co3O4 Spinel Nanoparticles: Mechanistic Insights into Aerobic Oxidation of Alcohols. ACS Catal. 2019, 9, 5974–5985. [Google Scholar] [CrossRef]
- Llorca, J.; Ramírez de la Piscina, P.; Dalmon, J.A.; Homs, N. Transformation of Co3O4 during Ethanol Steam-Re-forming. Activation Process for Hydrogen Production. Chem. Mater. 2004, 16, 3573–3578. [Google Scholar] [CrossRef]
- Kubin, M.; Guo, M.; Kroll, T.; Löchel, H.; Källman, E.; Baker, M.L.; Mitzner, R.; Gul, S.; Kern, J.; Föhlisch, A.; et al. Probing the oxidation state of transition metal complexes: A case study on how charge and spin densities determine Mn L-edge X-ray absorption energies. Chem. Sci. 2018, 9, 6813–6829. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, B.; Frazer, B.H.; Belz, A.; Conrad, P.G.; Nealson, K.H.; Haskel, D.; Lang, J.C.; Srajer, G.; De Stasio, G. Multiple Scattering Calculations of Bonding and X-ray Absorption Spectroscopy of Manganese Oxides. J. Phys. Chem. A 2003, 107, 2839–2847. [Google Scholar] [CrossRef]
- Kowalska, J.K.; Nayyar, B.; Rees, J.A.; Schiewer, C.E.; Lee, S.C.; Kovacs, J.A.; Meyer, F.; Weyhermüller, T.; Otero, E.; DeBeer, S. Iron L2,3-Edge X-ray Absorption and X-ray Magnetic Circular Dichroism Studies of Molecular Iron Complexes with Relevance to the FeMoco and FeVco Active Sites of Nitrogenase. Inorg. Chem. 2017, 56, 8147–8158. [Google Scholar] [CrossRef] [Green Version]
- De Groot, F. Multiplet effects in X-ray spectroscopy. Coord. Chem. Rev. 2005, 249, 31–63. [Google Scholar] [CrossRef]
- Douma, D.H.; Ciprian, R.; Lamperti, A.; Lupo, P.; Cianci, E.; Sangalli, D.; Casoli, F.; Nasi, L.; Albertini, F.; Torelli, P.; et al. Experimental versus abinitio X-ray absorption of iron-doped zirconia: Trends in O K-edge spectra as a function of iron doping. Phys. Rev. B 2014, 90, 205201. [Google Scholar] [CrossRef]
- Kox, T.; Spohr, E.; Kenmoe, S. Impact of Solvation on the Structure and Reactivity of the Co3O4 (001)/H2O Interface: Insights from Molecular Dynamics Simulations. Front. Energy Res. 2020, 8, 312. [Google Scholar] [CrossRef]
- Hendel, S.; Schäfers, F.; Hävecker, M.; Reichardt, G.; Scheer, M.; Bahrdt, J.; Lips, K. The EMIL Project at BESSY II: Beamline Design and Performance. In Proceedings of the 12th International Conference on Synchrotron Radiation Instrumentation—SRI2015, New York, NY, USA, 6–10 July 2015; p. 030038. [Google Scholar] [CrossRef]
- Picard, J.; Baud, G.; Besse, J.; Chevalier, R. Croissance cristalline et étude structurale de Co3O4. J. Less Common Met. 1980, 75, 99–104. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.; Hobson, A. The structure of cobalt oxide, Co3O4. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1973, 29, 362–363. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Quantum-Espresso. Original QE PP Library. Available online: http://pseudopotentials.quantum-espresso.org/legacy_tables/original-qe-pp-library (accessed on 31 January 2022).
- Wang, L.; Maxisch, T.; Ceder, G. Oxidation energies of transition metal oxides within the GGA+ U framework. Phys. Rev. B 2006, 73, 195107. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Gougoussis, C.; Calandra, M.; Seitsonen, A.P.; Mauri, F. First-principles calculations of X-ray absorption in a scheme based on ultrasoft pseudopotentials: From α-quartz to high-Tc compounds. Phys. Rev. B 2009, 80, 075102. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [Green Version]
- Brouder, C. Angular dependence of X-ray absorption spectra. J. Phys. Condens. Matter 1990, 2, 701. [Google Scholar] [CrossRef]
- Taillefumier, M.; Cabaret, D.; Flank, A.M.; Mauri, F. X-ray absorption near-edge structure calculations with the pseudopotentials: Application to the K edge in diamond and α-quartz. Phys. Rev. B 2002, 66, 195107. [Google Scholar] [CrossRef] [Green Version]
- Messiah, A. Quantum mechanics, vol. II. Engl. Ed. Holl. Amster 1962, 2, 992. [Google Scholar]
- Kim, K.J.; Park, Y.R. Optical investigation of charge-transfer transitions in spinel Co3O4. Solid State Commun. 2003, 127, 25–28. [Google Scholar] [CrossRef]
- Shinde, V.; Mahadik, S.; Gujar, T.; Lokhande, C. Supercapacitive cobalt oxide (Co3O4) thin films by spray pyrolysis. Appl. Surf. Sci. 2006, 252, 7487–7492. [Google Scholar] [CrossRef]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5048. [Google Scholar] [CrossRef] [Green Version]
- Rinke, P.; Janotti, A.; Scheffler, M.; Van de Walle, C.G. Defect Formation Energies without the Band-Gap Problem: Combining Density-Functional Theory and the G W Approach for the Silicon Self-Interstitial. Phys. Rev. Lett. 2009, 102, 026402. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; Jia, R.; Xin, B.; Peng, B.; Zhang, Y. Effects of oxygen vacancies on the structural and optical properties of β-Ga2O3. Sci. Rep. 2017, 7, 40160. [Google Scholar] [CrossRef]
- Van Elp, J.; Wieland, J.L.; Eskes, H.; Kuiper, P.; Sawatzky, G.A.; de Groot, F.M.F.; Turner, T.S. Electronic structure of CoO, Li-doped CoO, and LiCoO2. Phys. Rev. B 1991, 44, 6090–6103. [Google Scholar] [CrossRef]
- Bethe, H. Termaufspaltung in kristallen. Ann. Phys. 1929, 395, 133–208. [Google Scholar] [CrossRef]
- Griffith, J.; Orgel, L. Ligand-field theory. Q. Rev. Chem. Soc. 1957, 11, 381–393. [Google Scholar] [CrossRef]
- Ballhausen, C.J. Introduction to Ligand Field Theory; Technical Report; McGraw-Hill: New York, NY, USA, 1962. [Google Scholar]
- Figgis, B.N. Ligand field theory. Compr. Coord. Chem. 1987, 1, 213–279. [Google Scholar]
- Chen, J.; Wu, X.; Selloni, A. Electronic structure and bonding properties of cobalt oxide in the spinel structure. Phys. Rev. B 2011, 83, 245204. [Google Scholar] [CrossRef] [Green Version]
- Roth, W. The magnetic structure of Co3O4. J. Phys. Chem. Solids 1964, 25, 1–10. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kenmoe, S.; Douma, D.H.; Raji, A.T.; M’Passi-Mabiala, B.; Götsch, T.; Girgsdies, F.; Knop-Gericke, A.; Schlögl, R.; Spohr, E. X-ray Absorption Near-Edge Structure (XANES) at the O K-Edge of Bulk Co3O4: Experimental and Theoretical Studies. Nanomaterials 2022, 12, 921. https://doi.org/10.3390/nano12060921
Kenmoe S, Douma DH, Raji AT, M’Passi-Mabiala B, Götsch T, Girgsdies F, Knop-Gericke A, Schlögl R, Spohr E. X-ray Absorption Near-Edge Structure (XANES) at the O K-Edge of Bulk Co3O4: Experimental and Theoretical Studies. Nanomaterials. 2022; 12(6):921. https://doi.org/10.3390/nano12060921
Chicago/Turabian StyleKenmoe, Stephane, Dick Hartmann Douma, Abdulrafiu Tunde Raji, Bernard M’Passi-Mabiala, Thomas Götsch, Frank Girgsdies, Axel Knop-Gericke, Robert Schlögl, and Eckhard Spohr. 2022. "X-ray Absorption Near-Edge Structure (XANES) at the O K-Edge of Bulk Co3O4: Experimental and Theoretical Studies" Nanomaterials 12, no. 6: 921. https://doi.org/10.3390/nano12060921
APA StyleKenmoe, S., Douma, D. H., Raji, A. T., M’Passi-Mabiala, B., Götsch, T., Girgsdies, F., Knop-Gericke, A., Schlögl, R., & Spohr, E. (2022). X-ray Absorption Near-Edge Structure (XANES) at the O K-Edge of Bulk Co3O4: Experimental and Theoretical Studies. Nanomaterials, 12(6), 921. https://doi.org/10.3390/nano12060921