
Borophene and Pristine Graphene 2D Sheets as Potential Surfaces for the Adsorption of Electron-Rich and Electron-Deficient π-Systems: A Comparative DFT Study

 ,
,  , , , ,
, , , ,  ,
, 

Abstract
: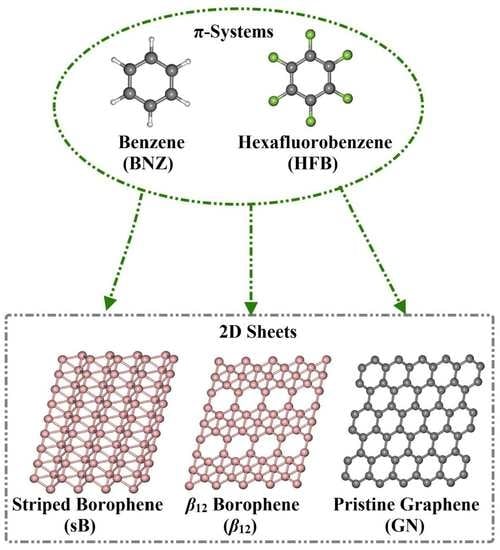
1. Introduction
2. Computational Methods
3. Results and Discussion
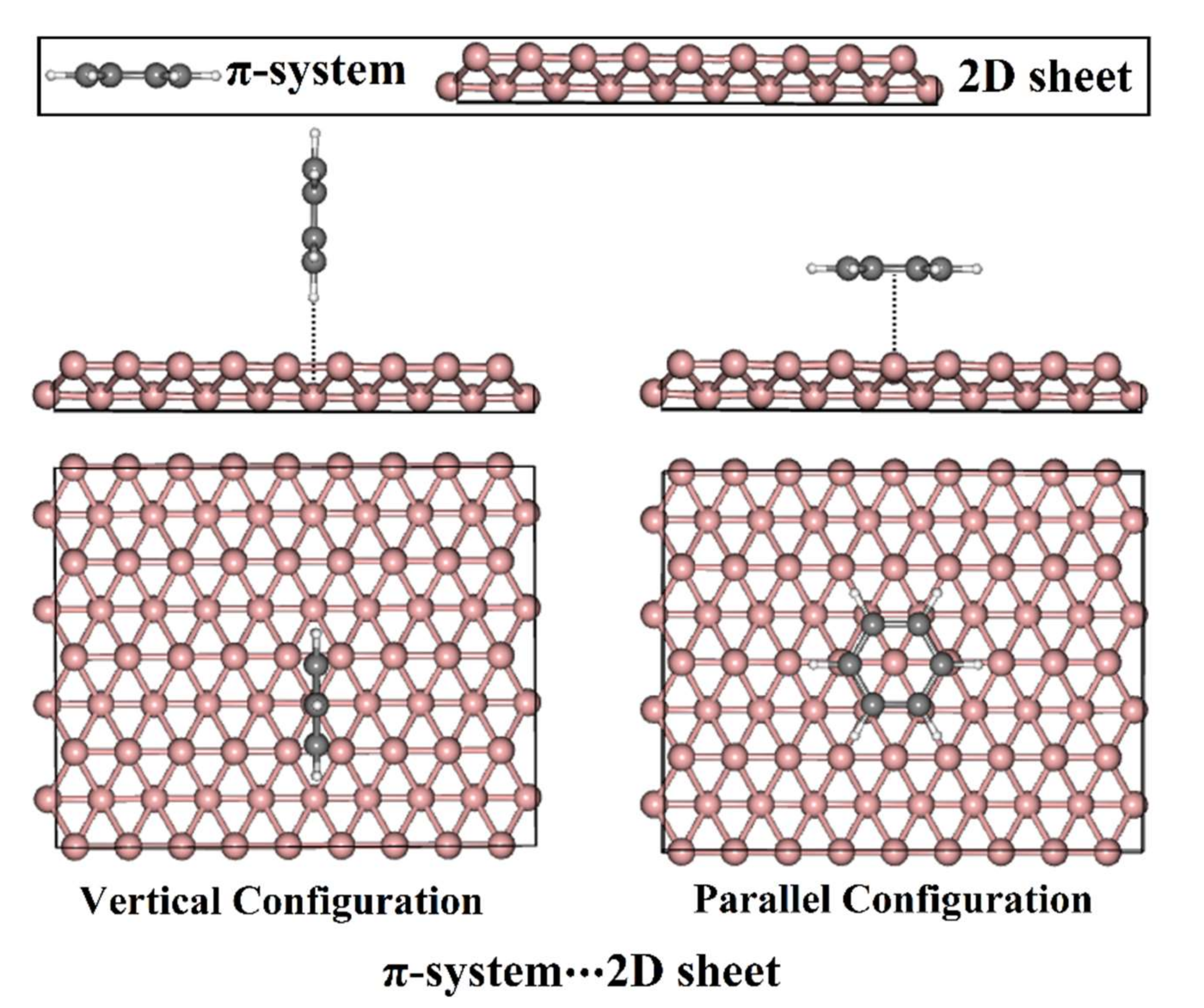
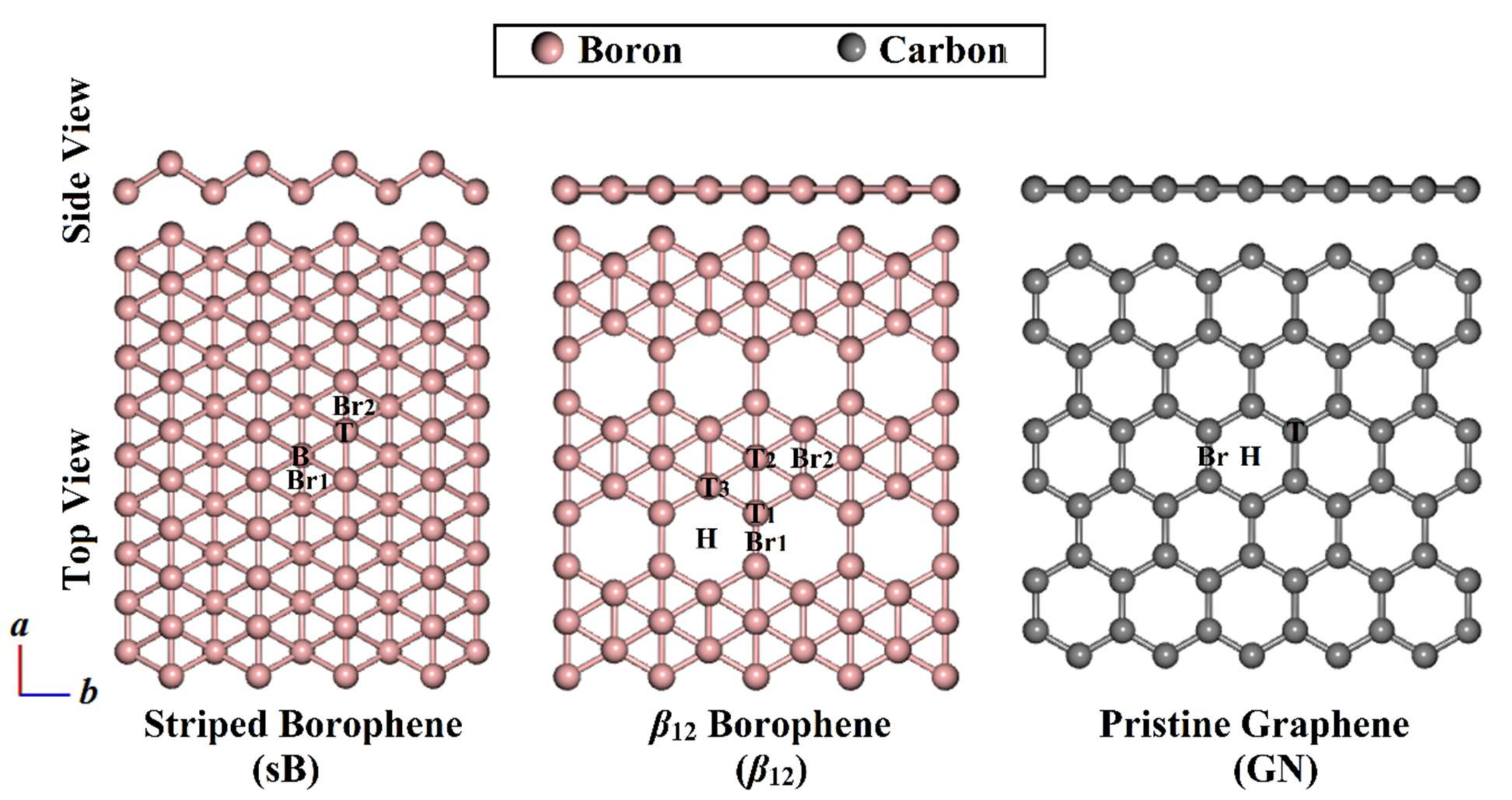
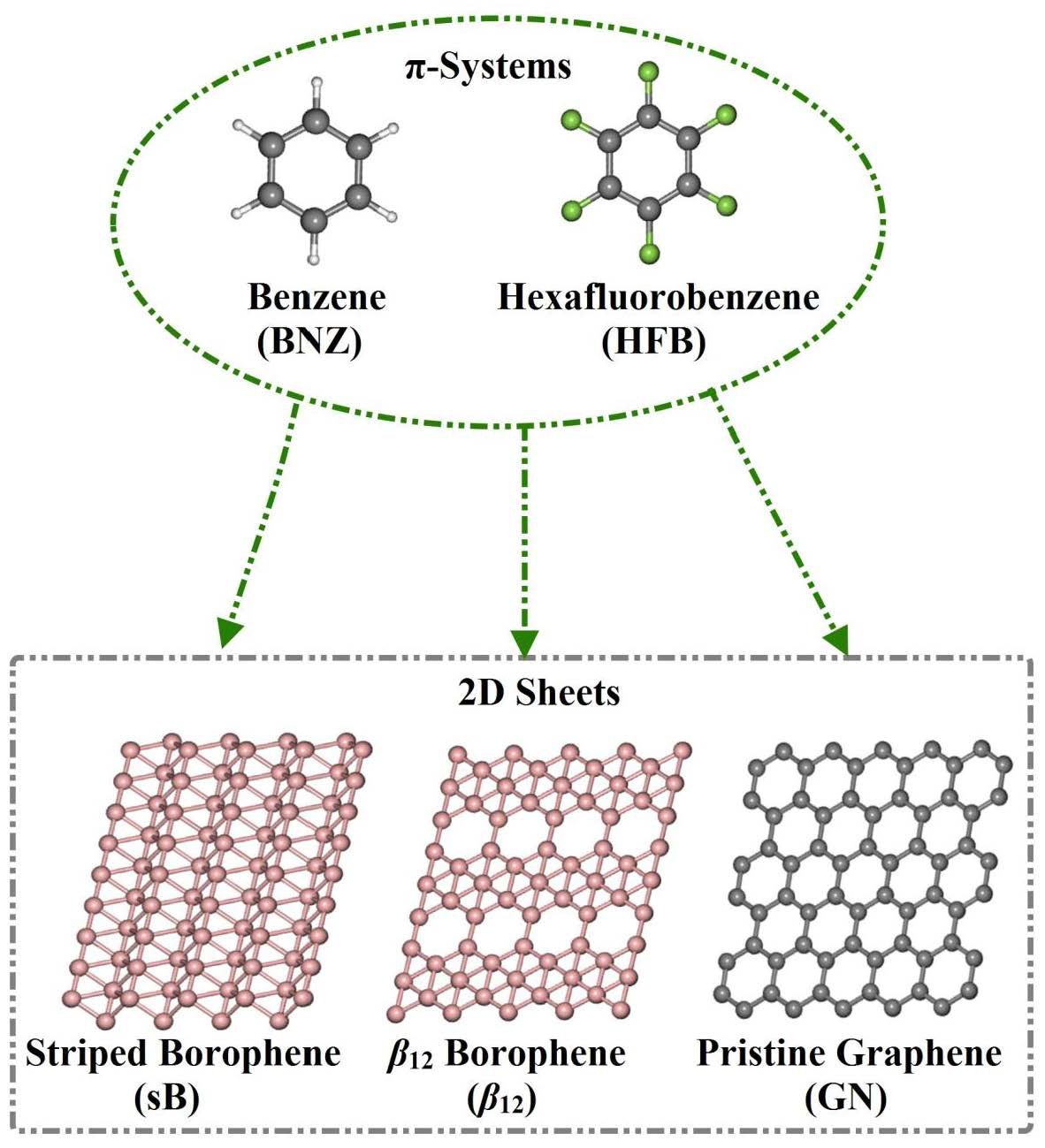
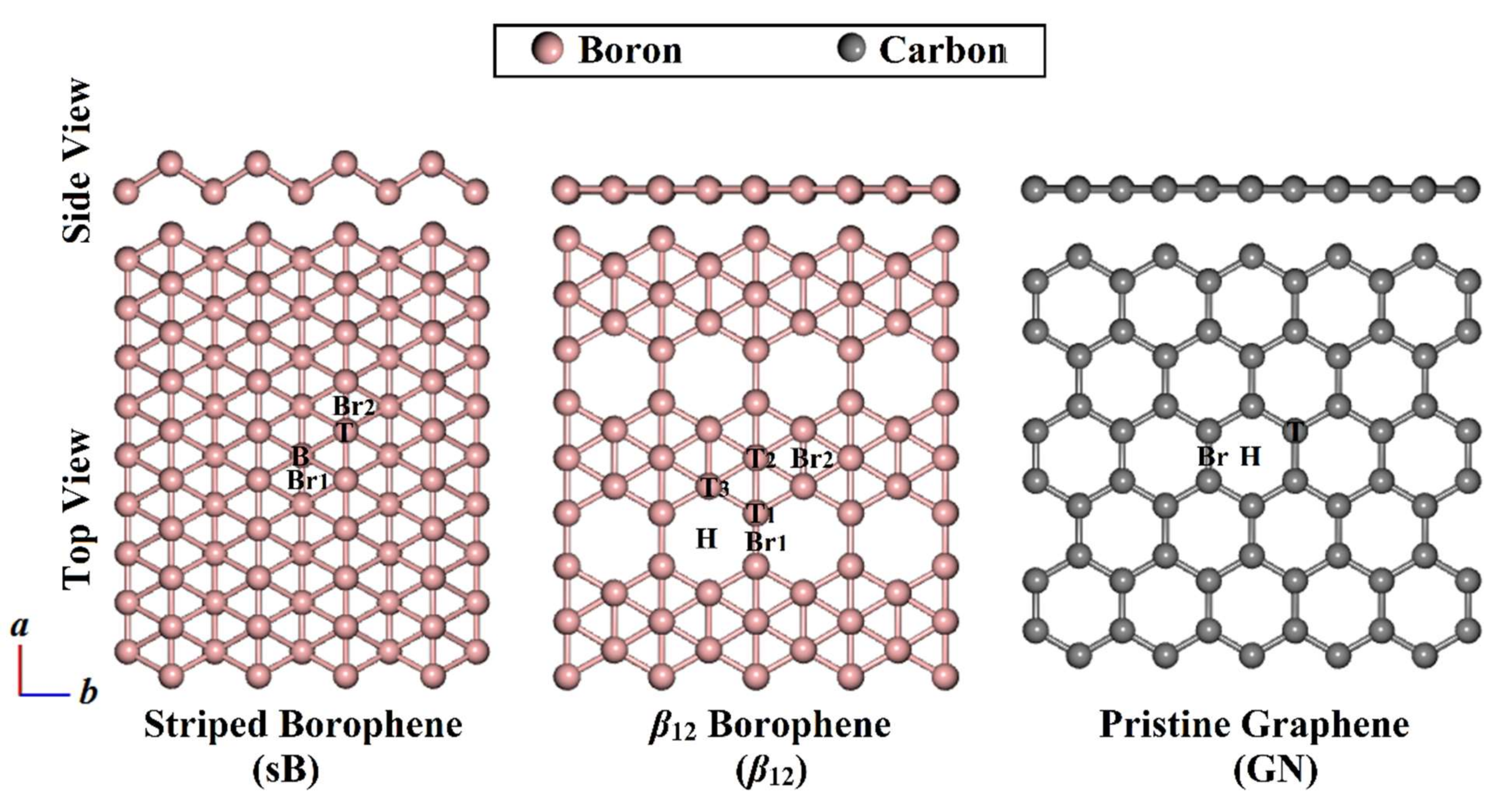
3.1. Geometric Structures
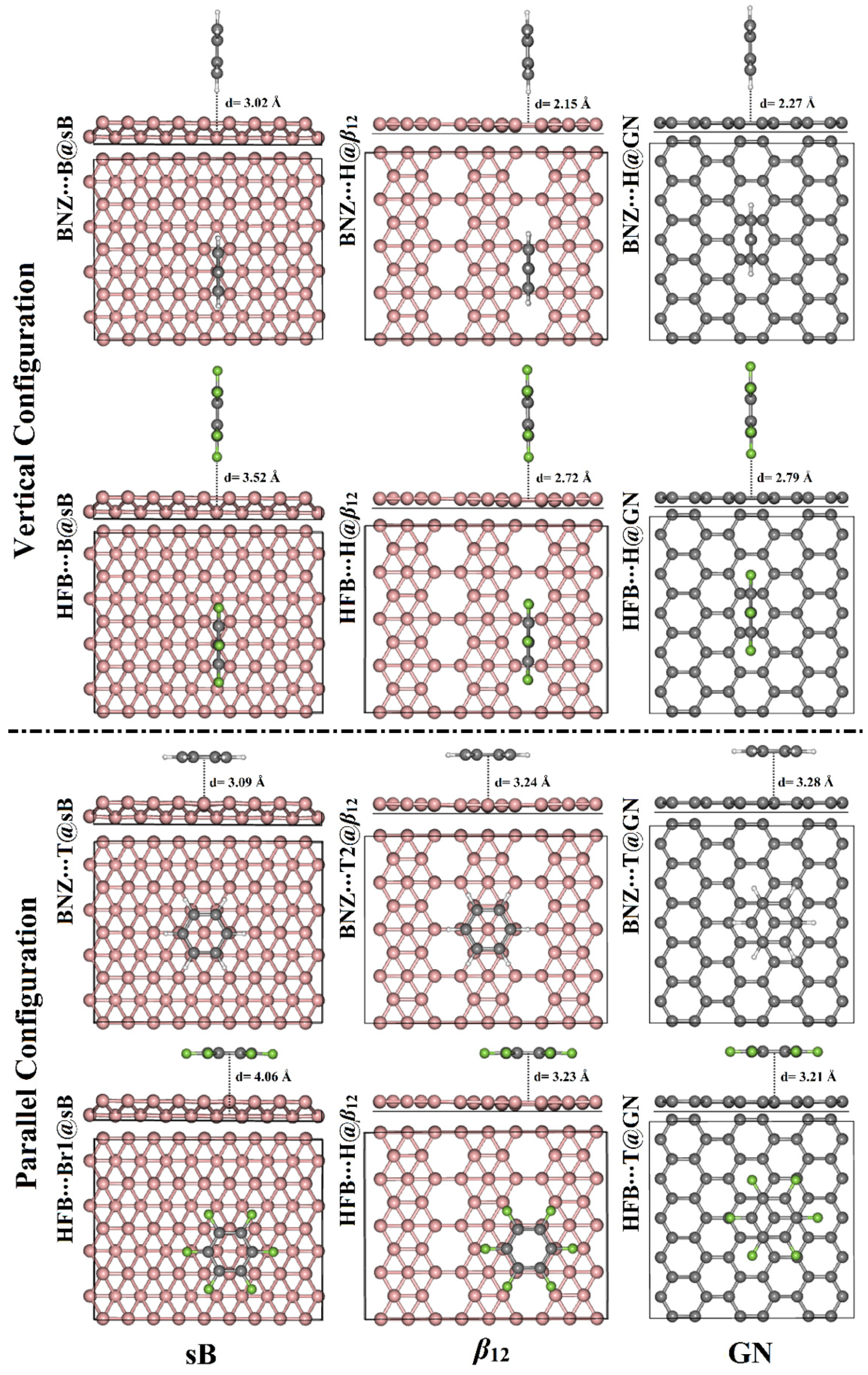
3.2. Adsorption Energy Calculations
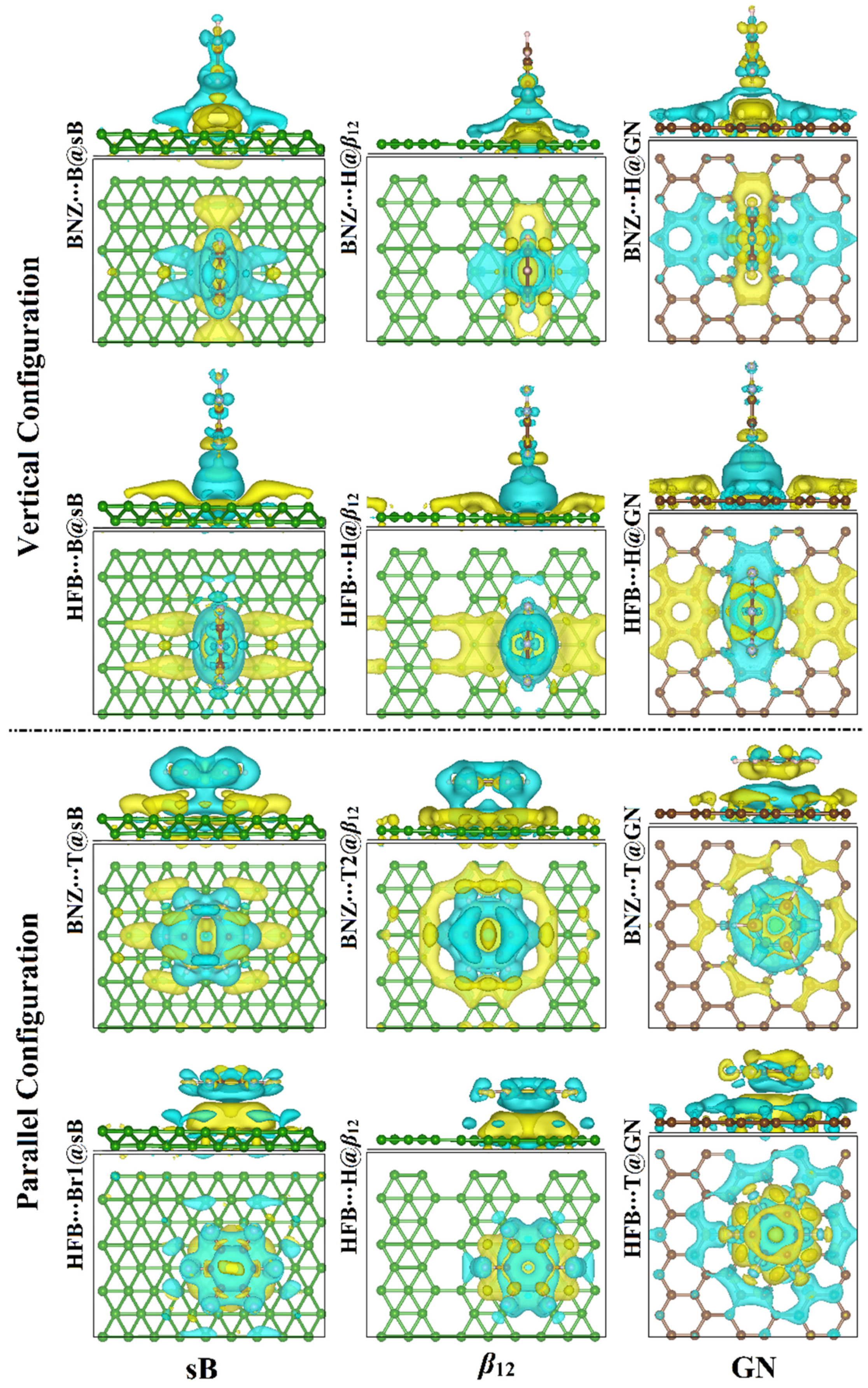
3.3. Charge Transfer Calculations
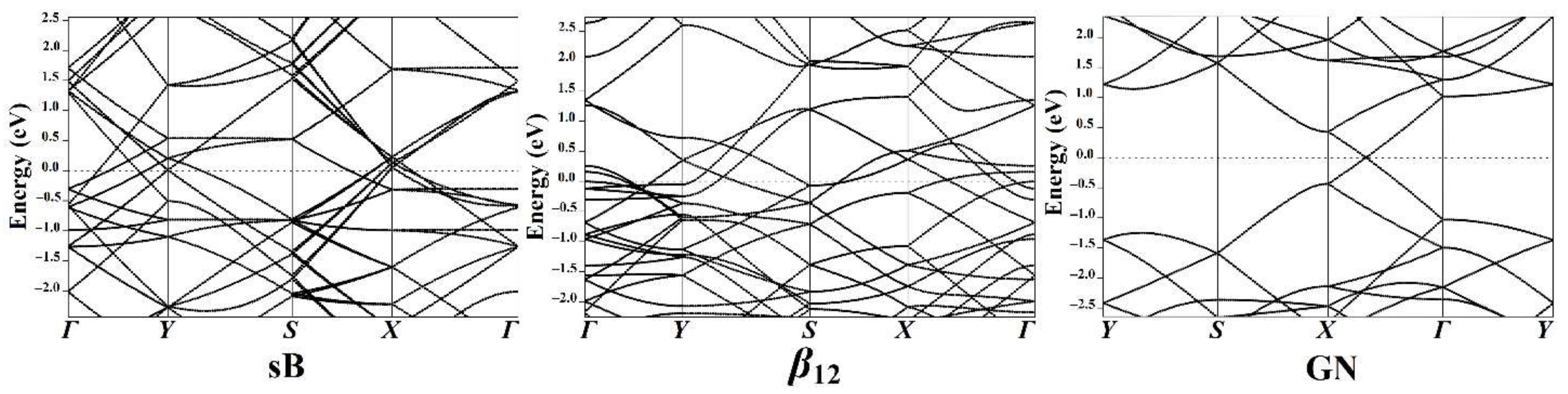
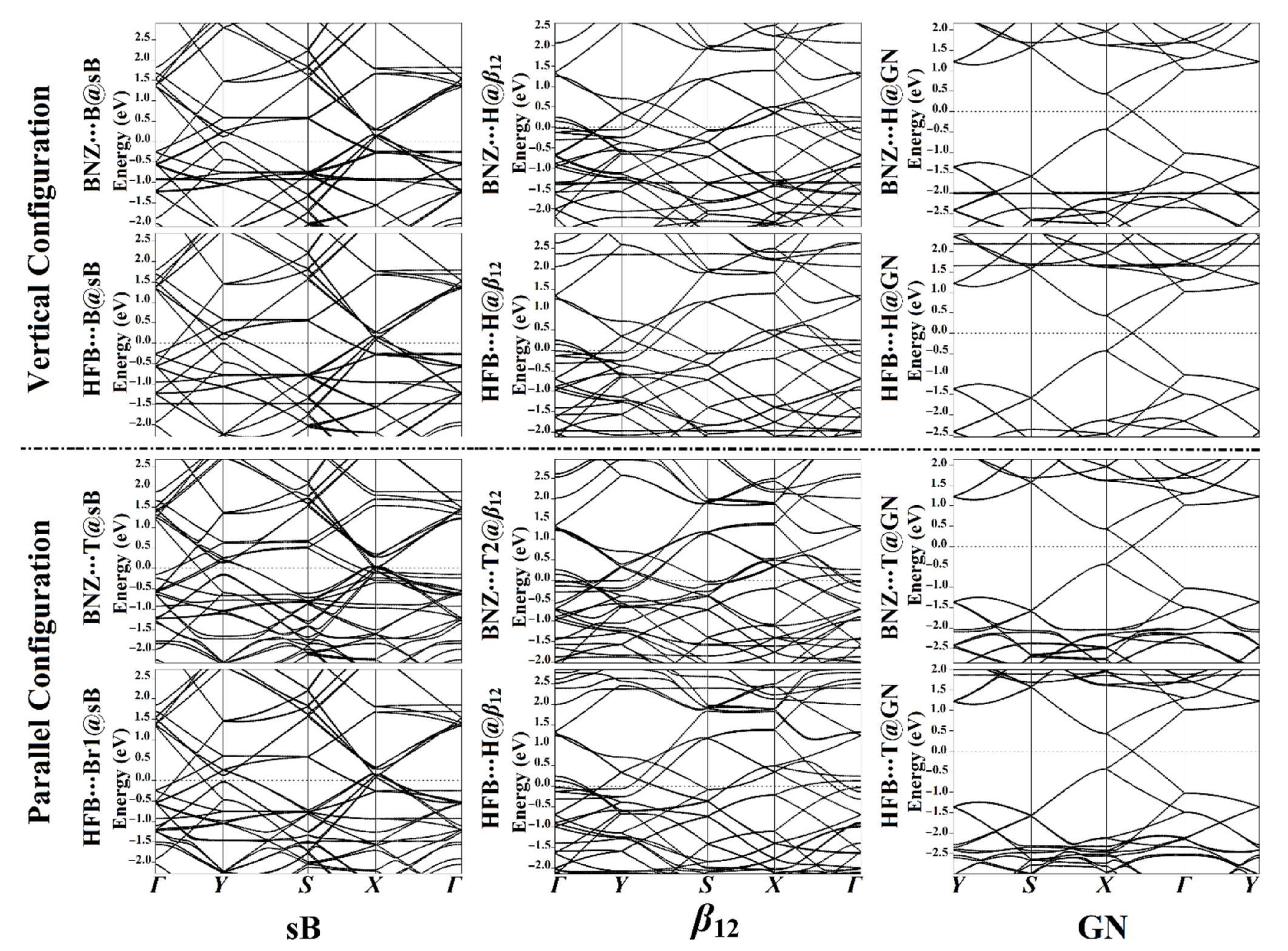
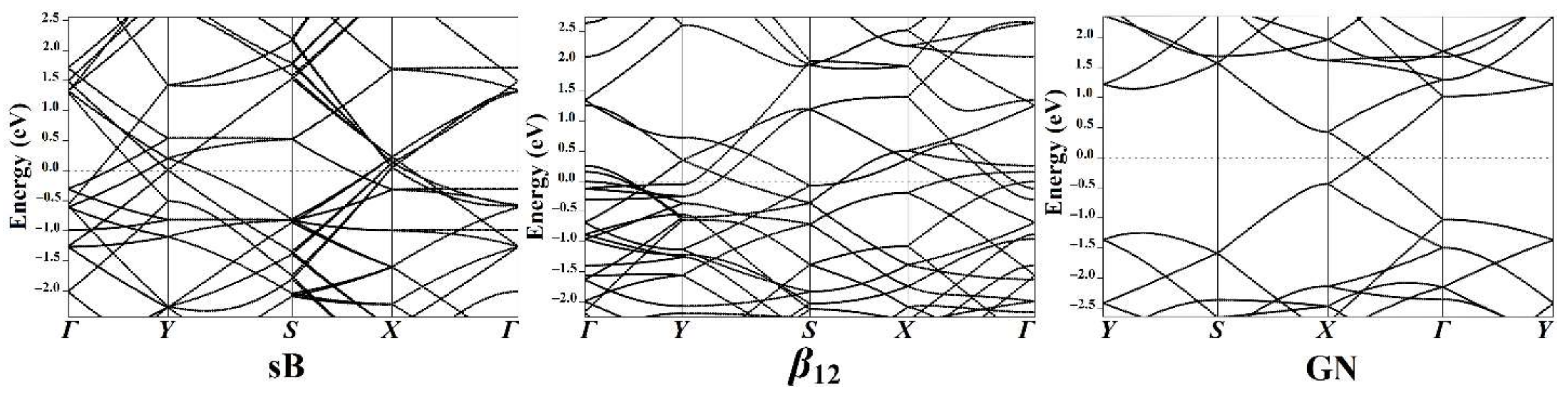
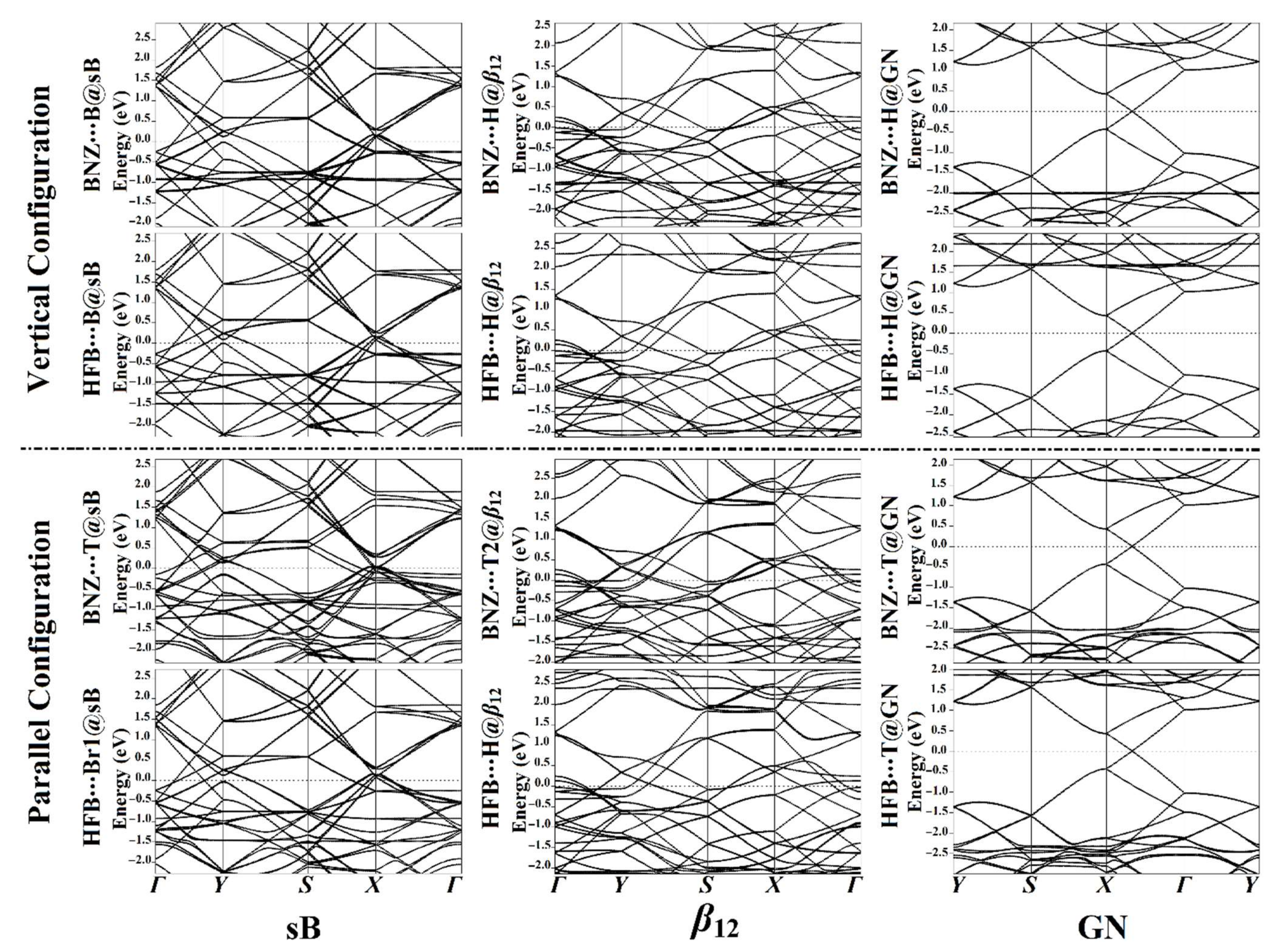
3.4. Band Structure Calculations
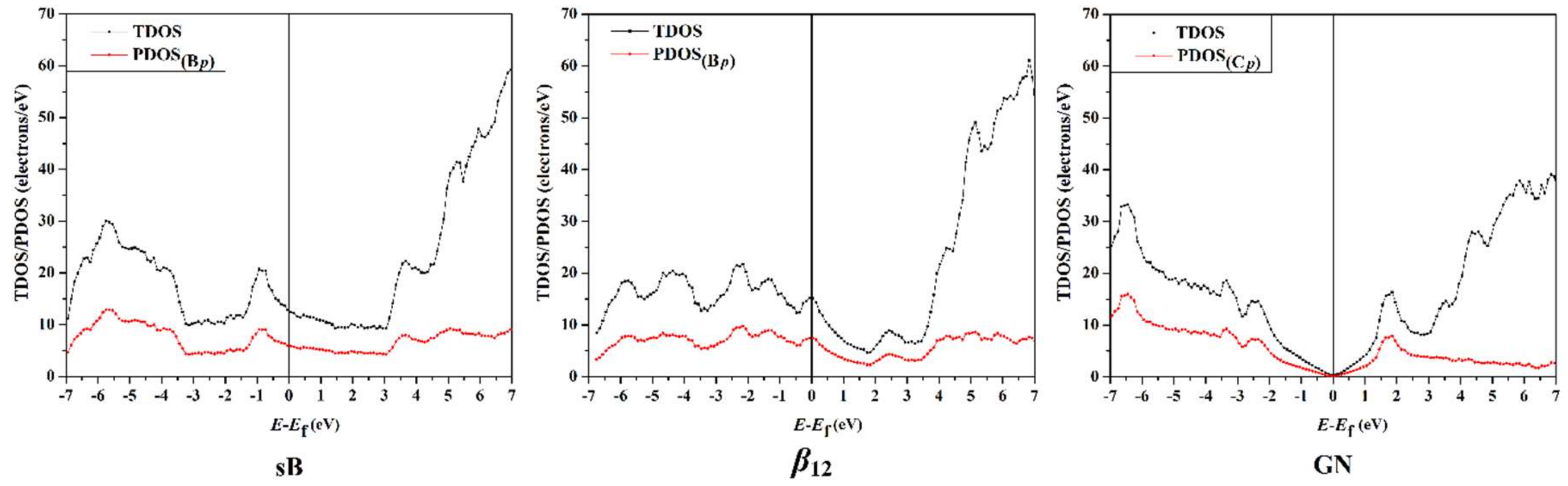
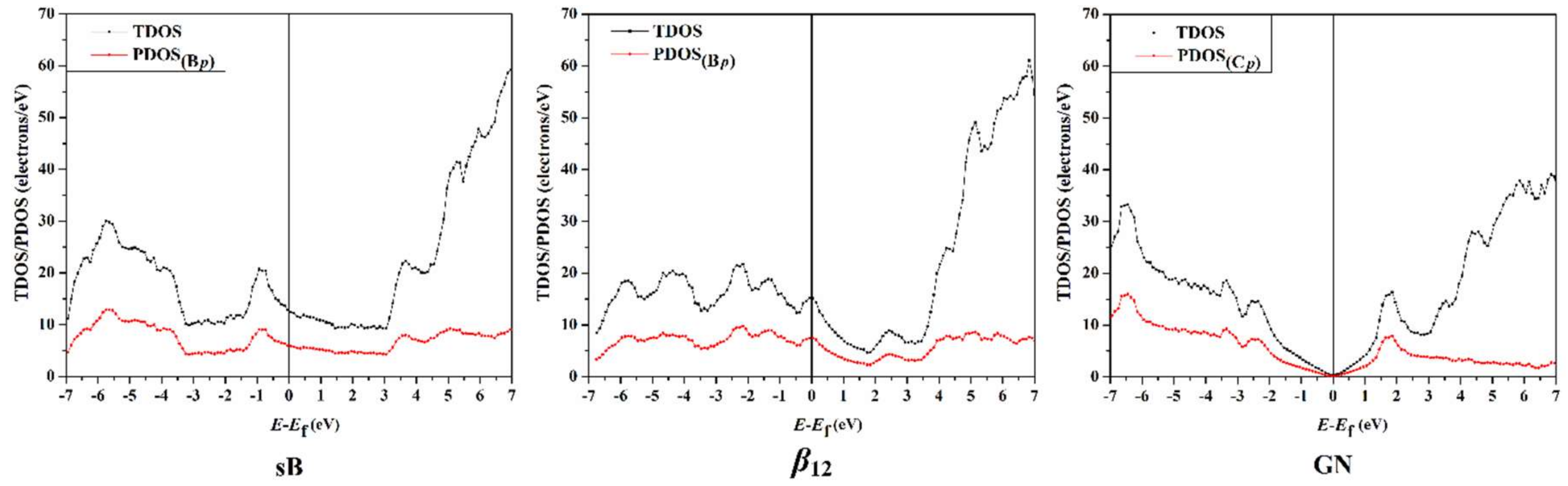
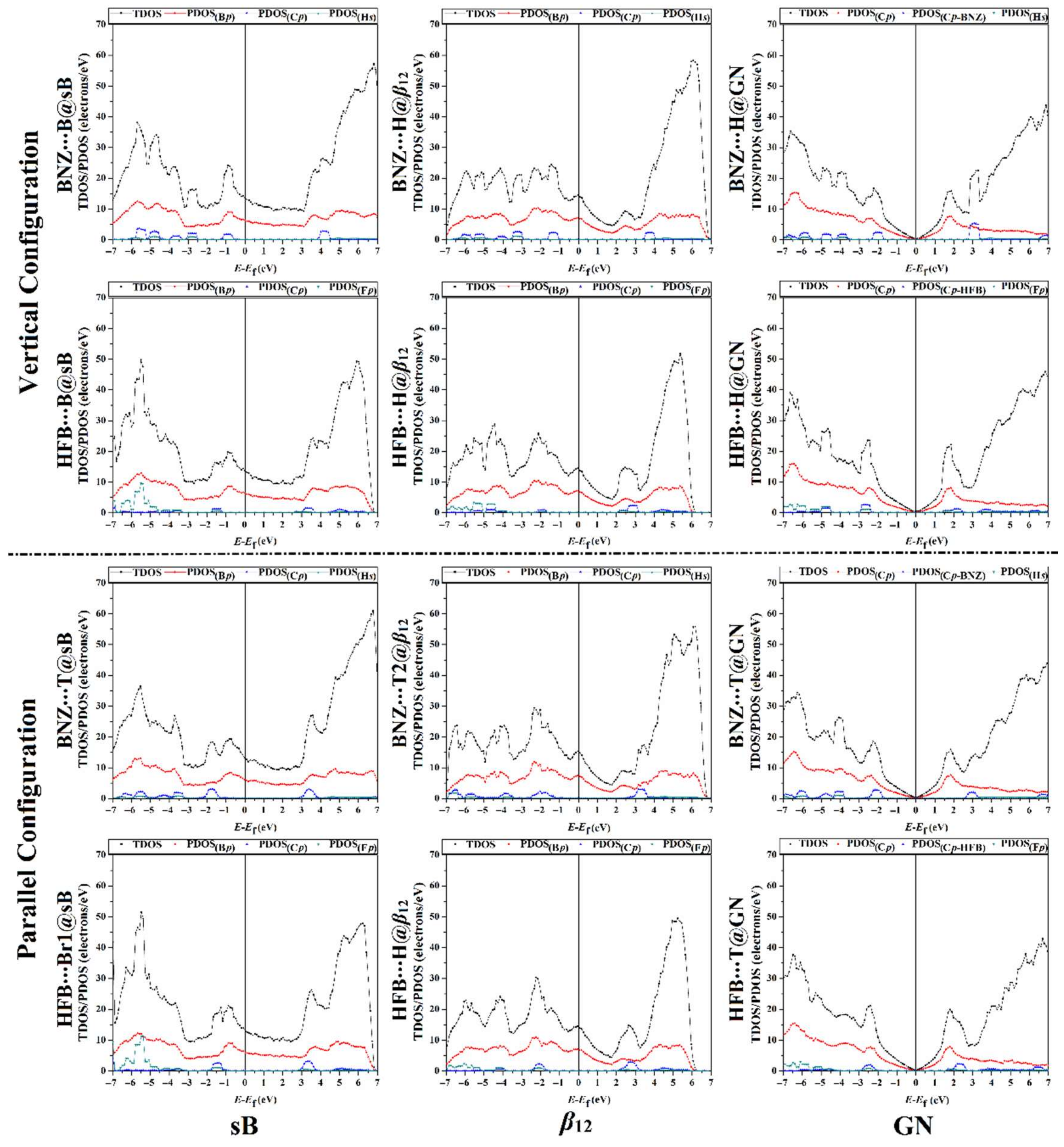
3.5. Density of State Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jariwala, D.; Sangwan, V.K.; Lauhon, L.J.; Marks, T.J.; Hersam, M.C. Carbon nanomaterials for electronics, optoelectronics, photovoltaics, and sensing. Chem. Soc. Rev. 2013, 42, 2824–2860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yung, K.C.; Wu, W.M.; Pierpoint, M.P.; Kusmartsev, F.V. Introduction to graphene electronics—A new era of digital transistors and devices. Contemp. Phys. 2013, 54, 233–251. [Google Scholar] [CrossRef] [Green Version]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Li, Y.; Tan, X.; Peng, R.; Liu, Z. Behavior and toxicity of graphene and its functionalized derivatives in biological systems. Small 2013, 9, 1492–1503. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Wu, L.; Qu, X. New horizons for diagnostics and therapeutic applications of graphene and graphene oxide. Adv. Mater. 2013, 25, 168–186. [Google Scholar] [CrossRef]
- Liu, Y.; Dong, X.; Chen, P. Biological and chemical sensors based on graphene materials. Chem. Soc. Rev. 2012, 41, 2283–2307. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, F.; Zhang, Z.; Chen, N.; Qu, L. Dimension-tailored functional graphene structures for energy conversion and storage. Nanoscale 2013, 5, 3112–3126. [Google Scholar] [CrossRef]
- Sun, Y.Q.; Wu, Q.O.; Shi, G.Q. Graphene based new energy materials. Energy Environ. Sci. 2011, 4, 1113–1132. [Google Scholar] [CrossRef]
- Mahmood, N.; Zhang, C.Z.; Yin, H.; Hou, Y.L. Graphene-based nanocomposites for energy storage and conversion in lithium batteries, supercapacitors and fuel cells. J. Mater. Chem. A 2014, 2, 15–32. [Google Scholar] [CrossRef] [Green Version]
- Novoselov, K.S.; Fal’ko, V.I.; Colombo, L.; Gellert, P.R.; Schwab, M.G.; Kim, K. A roadmap for graphene. Nature 2012, 490, 192–200. [Google Scholar] [CrossRef]
- Leenaerts, O.; Partoens, B.; Peeters, F.M. Adsorption of H2O, NH3, CO, NO2, and NO on graphene: A first-principles study. Phys. Rev. B Condens. Matter 2008, 77, 125416. [Google Scholar] [CrossRef] [Green Version]
- Elgengehi, S.M.; El-Taher, S.; Ibrahim, M.A.A.; Desmarais, J.K.; El-Kelany, K.E. Graphene and graphene oxide as adsorbents for cadmium and lead heavy metals: A theoretical investigation. Appl. Surf. Sci. 2020, 507, 145038. [Google Scholar] [CrossRef]
- Varghese, S.S.; Lonkar, S.; Singh, K.K.; Swaminathan, S.; Abdala, A. Recent advances in graphene based gas sensors. Sens. Actuators B-Chem. 2015, 218, 160–183. [Google Scholar] [CrossRef]
- Raccichini, R.; Varzi, A.; Wei, D.; Passerini, S. Critical insight into the relentless progression toward graphene and graphene-containing materials for lithium-ion battery anodes. Adv. Mater. 2017, 29, 1603421. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Wen, Z.H.; Cui, S.M.; Ci, S.Q.; Mao, S.; Chen, J.H. An advanced nitrogen-doped graphene/cobalt-embedded porous carbon polyhedron hybrid for efficient catalysis of oxygen reduction and water splitting. Adv. Funct. Mater. 2015, 25, 872–882. [Google Scholar] [CrossRef]
- Ayatollahi, A.; Roknabadi, M.R.; Behdani, M.; Shahtahmassebi, N.; Sanyal, B. Adsorption characteristics of amino acids on graphene and germanene using dispersion-corrected density functional theory. Phys. E Low-Dimens. Syst. Nanostruct. 2021, 127, 114498. [Google Scholar] [CrossRef]
- Li, K.; Li, N.; Yan, N.N.; Wang, T.Y.; Zhang, Y.T.; Song, Q.; Li, H.J. Adsorption of small hydrocarbons on pristine, N-doped and vacancy graphene by DFT study. Appl. Surf. Sci. 2020, 515, 146028. [Google Scholar] [CrossRef]
- Kulish, V.V. Surface reactivity and vacancy defects in single-layer borophene polymorphs. Phys. Chem. Chem. Phys. 2017, 19, 11273–11281. [Google Scholar] [CrossRef]
- Cahangirov, S.; Topsakal, M.; Akturk, E.; Sahin, H.; Ciraci, S. Two- and one-dimensional honeycomb structures of silicon and germanium. Phys. Rev. Lett. 2009, 102, 236804. [Google Scholar] [CrossRef] [Green Version]
- Derivaz, M.; Dentel, D.; Stephan, R.; Hanf, M.C.; Mehdaoui, A.; Sonnet, P.; Pirri, C. Continuous germanene layer on Al(111). Nano Lett. 2015, 15, 2510–2516. [Google Scholar] [CrossRef]
- Popov, I.; Seifert, G.; Tomanek, D. Designing electrical contacts to MoS2 monolayers: A computational study. Phys. Rev. Lett. 2012, 108, 156802. [Google Scholar] [CrossRef] [Green Version]
- Qiao, J.; Kong, X.; Hu, Z.X.; Yang, F.; Ji, W. High-mobility transport anisotropy and linear dichroism in few-layer black phosphorus. Nat. Commun. 2014, 5, 4475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.H.; Kalantar-Zadeh, K.; Kis, A.; Coleman, J.N.; Strano, M.S. Electronics and optoelectronics of two-dimensional transition metal dichalcogenides. Nat. Nanotechnol. 2012, 7, 699–712. [Google Scholar] [CrossRef]
- Radisavljevic, B.; Radenovic, A.; Brivio, J.; Giacometti, V.; Kis, A. Single-layer MoS2 transistors. Nat. Nanotechnol. 2011, 6, 147–150. [Google Scholar] [CrossRef]
- Aufray, B.; Kara, A.; Vizzini, S.; Oughaddou, H.; Leandri, C.; Ealet, B.; Le Lay, G. Graphene-like silicon nanoribbons on Ag(110): A possible formation of silicene. Appl. Phys. Lett. 2010, 96, 183102. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Zhuang, J.; Liu, H.; Xu, X.; Eilers, S.; Wu, K.; Cheng, P.; Zhao, J.; Pi, X.; See, K.W.; et al. Tuning the band gap in silicene by oxidation. ACS Nano 2014, 8, 10019–10025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannix, A.J.; Zhou, X.F.; Kiraly, B.; Wood, J.D.; Alducin, D.; Myers, B.D.; Liu, X.; Fisher, B.L.; Santiago, U.; Guest, J.R.; et al. Synthesis of borophenes: Anisotropic, two-dimensional boron polymorphs. Science 2015, 350, 1513–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, B.; Zhang, H.; Shao, H.Z.; Xu, Y.F.; Zhang, R.J.; Zhua, H.Y. The electronic, optical, and thermodynamic properties of borophene from first-principles calculations. J. Mater. Chem. C 2016, 4, 3592–3598. [Google Scholar] [CrossRef] [Green Version]
- Feng, B.; Zhang, J.; Zhong, Q.; Li, W.; Li, S.; Li, H.; Cheng, P.; Meng, S.; Chen, L.; Wu, K. Experimental realization of two-dimensional boron sheets. Nat. Chem. 2016, 8, 563–568. [Google Scholar] [CrossRef] [Green Version]
- Ranjan, P.; Lee, J.M.; Kumar, P.; Vinu, A. Borophene: New sensation in flatland. Adv. Mater. 2020, 32, e2000531. [Google Scholar] [CrossRef]
- Zhou, X.F.; Dong, X.; Oganov, A.R.; Zhu, Q.; Tian, Y.J.; Wang, H.T. Semimetallic two-dimensional boron allotrope with massless dirac fermions. Phys. Rev. Lett. 2014, 112, 085502. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.R.; Lu, Z.H.; Wu, M.C.; Ciucci, F.; Zhao, T.S. Borophene: A promising anode material offering high specific capacity and high rate capability for lithium-ion batteries. Nano Energy 2016, 23, 97–104. [Google Scholar] [CrossRef]
- Novotny, M.; Dominguez-Gutierrez, F.J.; Krstic, P. A computational study of hydrogen detection by borophene. J. Mater. Chem. C 2017, 5, 5426–5433. [Google Scholar] [CrossRef]
- Chen, X.F.; Wang, L.F.Z.; Zhang, W.T.; Zhang, J.L.; Yuan, Y.Q. Ca-decorated borophene as potential candidates for hydrogen storage: A first-principle study. Int. J. Hydrog. Energy 2017, 42, 20036–20045. [Google Scholar] [CrossRef]
- Xu, S.G.; Zhao, Y.J.; Liao, J.H.; Yang, X.B.; Xu, H. The nucleation and growth of borophene on the Ag (111) surface. Nano Res. 2016, 9, 2616–2622. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.B.; Ding, Y.; Ni, J. Ab initio prediction of stable boron sheets and boron nanotubes: Structure, stability, and electronic properties. Phys. Rev. B Condens. Matter 2008, 77, 041402. [Google Scholar] [CrossRef]
- Li, X.B.; Xie, S.Y.; Zheng, H.; Tian, W.Q.; Sun, H.B. Boron based two-dimensional crystals: Theoretical design, realization proposal and applications. Nanoscale 2015, 7, 18863–18871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Yang, Y.; Gao, G.; Yakobson, B.I. Two-dimensional boron monolayers mediated by metal substrates. Angew. Chem. Int. Ed. Engl. 2015, 54, 13022–13026. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Ismail-Beigi, S. Novel precursors for boron nanotubes: The competition of two-center and three-center bonding in boron sheets. Phys. Rev. Lett. 2007, 99, 115501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Gao, J.; Zhao, J. From boron cluster to two-dimensional boron sheet on Cu(111) surface: Growth mechanism and hole formation. Sci. Rep. 2013, 3, 3238. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Mu, Y.; Bai, H.; Chen, Q.; Li, S.D. Binary nature of monolayer boron sheets from ab initio global searches. J. Chem. Phys. 2013, 138, 024701. [Google Scholar] [CrossRef] [Green Version]
- Peng, B.; Zhang, H.; Shao, H.Z.; Ning, Z.Y.; Xu, Y.F.; Ni, G.; Lu, H.L.; Zhang, D.W.; Zhu, H.Y. Stability and strength of atomically thin borophene from first principles calculations. Mater. Res. Lett. 2017, 5, 399–407. [Google Scholar] [CrossRef]
- Wang, H.; Li, Q.; Gao, Y.; Miao, F.; Zhou, X.-F.; Wan, X.G. Strain effects on borophene: Ideal strength, negative Possion’s ratio and phonon instability. New J. Phys. 2016, 18, 073016. [Google Scholar] [CrossRef]
- He, C.; Cheng, J.; Zhang, X.; Douthwaite, M.; Pattisson, S.; Hao, Z. Recent advances in the catalytic oxidation of volatile organic compounds: A review based on pollutant sorts and sources. Chem. Rev. 2019, 119, 4471–4568. [Google Scholar] [CrossRef]
- Scire, S.; Liotta, L.F. Supported gold catalysts for the total oxidation of volatile organic compounds. Appl. Catal. B-Environ. 2012, 125, 222–246. [Google Scholar] [CrossRef]
- Woodruff, T.J.; Axelrad, D.A.; Caldwell, J.; Morello-Frosch, R.; Rosenbaum, A. Public health implications of 1990 air toxics concentrations across the United States. Environ. Health Perspect. 1998, 106, 245–251. [Google Scholar] [CrossRef]
- Cohen, A.J.; Pope, C.A. 3rd. Lung cancer and air pollution. Environ. Health Perspect. 1995, 103 (Suppl. 8), 219–224. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Yao, Q.S.; Li, Q.Y.; Xiao, C.Y.; Huang, C.X.; Kan, E.J. Tuning electronic and magnetic properties of two-dimensional ferromagnetic semiconductor CrI3 through adsorption of benzene. J. Phys. Chem. C 2020, 124, 22143–22149. [Google Scholar] [CrossRef]
- AlZahrani, A.Z. First-principles study on the structural and electronic properties of graphene upon benzene and naphthalene adsorption. Appl. Surf. Sci. 2010, 257, 807–810. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Zhou, K.G.; Xie, K.F.; Zeng, J.; Zhang, H.L.; Peng, Y. Tuning the electronic structure and transport properties of graphene by noncovalent functionalization: Effects of organic donor, acceptor and metal atoms. Nanotechnology 2010, 21, 065201. [Google Scholar] [CrossRef]
- Chakarova-Kack, S.D.; Schroder, E.; Lundqvist, B.I.; Langreth, D.C. Application of van der Waals density functional to an extended system: Adsorption of benzene and naphthalene on graphite. Phys. Rev. Lett. 2006, 96, 146107. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.S.; Yu, C.M. DFT study of the adsorption of C6H6 and C6H5OH molecules on stanene nanosheets: Applications to sensor devices. Phys. E Low-Dimens. Syst. Nanostruct. 2021, 127, 114533. [Google Scholar] [CrossRef]
- Sabokdast, S.; Horri, A.; Azar, Y.T.; Momeni, M.; Tavakoli, M.b. Adsorption of adenine molecule on χ3 borophene nanosheets: A density functional theory study. Phys. E Low-Dimens. Syst. Nanostruct. 2020, 119, 114026. [Google Scholar] [CrossRef]
- Li, Q.X.; Liu, H.Y.; Tian, Y.G.; Guo, J.H.; Chen, G.; Lee, J.Y. Methylation detection and DNA sequencing based on adsorption of nucleobases on silicene nanoribbon. J. Phys. Chem. C 2020, 124, 10823–10831. [Google Scholar] [CrossRef]
- Wang, Y.P.; Ji, W.X.; Zhang, C.W.; Li, S.S.; Li, F.; Li, P.; Ren, M.J.; Chen, X.L.; Yuan, M.; Wang, P.J. Enhanced band gap opening in germanene by organic molecule adsorption. Mater. Chem. Phys. 2016, 173, 379–384. [Google Scholar] [CrossRef]
- Li, W.; Jiang, Q.; Li, D.; Ao, Z.; An, T. Density functional theory investigation on selective adsorption of VOCs on borophene. Chin. Chem. Lett. 2021, 32, 2803–2806. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Buongiorno Nardelli, M.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B Condens. Matter 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Marzari, N.; Vanderbilt, D.; De Vita, A.; Payne, M.C. Thermal contraction and disordering of the Al(110) surface. Phys. Rev. Lett. 1999, 82, 3296–3299. [Google Scholar] [CrossRef] [Green Version]
- Henkelman, G.; Arnaldsson, A.; Jonsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comp. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory, 1st ed.; Clarendon Press: Oxford, UK, 1990; Volume XVIII, p. 438. [Google Scholar]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2D Sheet | Adsorption Site a | π-System | |||||
|---|---|---|---|---|---|---|---|
| Benzene (BNZ) | Hexafluorobenzene (HFB) | ||||||
| Ead (kcal/mol) | d (Å) | Qtb (e) | Ead (kcal/mol) | d (Å) | Qtb (e) | ||
| Vertical Configuration c | |||||||
| sB | T | −4.35 | 2.57 | −0.0154 | −3.99 | 2.95 | −0.0275 |
| B | −5.66 | 3.02 | −0.0136 | −4.83 | 3.52 | −0.0318 | |
| Br1 | −5.64 | 3.02 | −0.0132 | −4.79 | 3.53 | −0.0318 | |
| Br2 | −4.40 | 2.54 | −0.0169 | −4.00 | 2.93 | −0.0274 | |
| β12 | T1 | −5.07 | 2.54 | −0.0176 | −3.94 | 3.02 | −0.0219 |
| T2 | −4.35 | 2.62 | −0.0157 | −4.39 | 2.94 | −0.0219 | |
| T3 | −4.84 | 2.53 | −0.0165 | −4.22 | 2.96 | −0.0220 | |
| H | −6.40 | 2.15 | −0.0199 | −4.54 | 2.72 | −0.0274 | |
| Br1 | −5.65 | 2.38 | −0.0177 | −4.20 | 2.90 | −0.0261 | |
| Br2 | −4.95 | 2.47 | −0.0157 | −4.50 | 2.91 | −0.0215 | |
| GN | T | −4.30 | 2.52 | −0.0067 | −3.74 | 2.89 | −0.0124 |
| H | −5.04 | 2.27 | −0.0058 | −3.97 | 2.79 | −0.0117 | |
| Br | −4.47 | 2.45 | −0.0074 | −3.76 | 2.88 | −0.0121 | |
| Parallel Configuration c | |||||||
| sB | T | −14.93 | 3.09 | 0.0735 | −14.66 | 3.23 | −0.0733 |
| B | −11.46 | 4.00 | 0.0469 | −15.74 | 4.04 | −0.0812 | |
| Br1 | −11.43 | 4.03 | 0.0454 | −15.76 | 4.06 | −0.0813 | |
| Br2 | −14.50 | 3.01 | 0.0572 | −14.75 | 3.18 | −0.0730 | |
| β12 | T1 | −11.45 | 3.26 | 0.0184 | −16.18 | 3.27 | −0.0672 |
| T2 | −12.16 | 3.24 | 0.0436 | −16.05 | 3.27 | −0.0520 | |
| T3 | −10.50 | 3.30 | 0.0066 | −15.80 | 3.27 | −0.0652 | |
| H | −11.73 | 3.23 | 0.0385 | −16.55 | 3.23 | −0.0534 | |
| Br1 | −11.55 | 3.25 | 0.0184 | −16.19 | 3.22 | −0.0608 | |
| Br2 | −10.77 | 3.30 | 0.0099 | −15.66 | 3.27 | −0.0645 | |
| GN | T | −9.97 | 3.28 | 0.0065 | −14.34 | 3.21 | −0.0378 |
| H | −8.78 | 3.40 | 0.0080 | −12.51 | 3.33 | −0.0325 | |
| Br | −9.89 | 3.28 | 0.0050 | −14.08 | 3.22 | −0.0357 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, M.A.A.; Mahmoud, A.H.M.; Soliman, K.A.; Mekhemer, G.A.H.; Ahmed, M.N.; Shawky, A.M.; Abourehab, M.A.S.; Elkaeed, E.B.; Soliman, M.E.S.; Moussa, N.A.M. Borophene and Pristine Graphene 2D Sheets as Potential Surfaces for the Adsorption of Electron-Rich and Electron-Deficient π-Systems: A Comparative DFT Study. Nanomaterials 2022, 12, 1028. https://doi.org/10.3390/nano12061028
Ibrahim MAA, Mahmoud AHM, Soliman KA, Mekhemer GAH, Ahmed MN, Shawky AM, Abourehab MAS, Elkaeed EB, Soliman MES, Moussa NAM. Borophene and Pristine Graphene 2D Sheets as Potential Surfaces for the Adsorption of Electron-Rich and Electron-Deficient π-Systems: A Comparative DFT Study. Nanomaterials. 2022; 12(6):1028. https://doi.org/10.3390/nano12061028
Chicago/Turabian StyleIbrahim, Mahmoud A. A., Amna H. M. Mahmoud, Kamal A. Soliman, Gamal A. H. Mekhemer, Muhammad Naeem Ahmed, Ahmed M. Shawky, Mohammed A. S. Abourehab, Eslam B. Elkaeed, Mahmoud E. S. Soliman, and Nayra A. M. Moussa. 2022. "Borophene and Pristine Graphene 2D Sheets as Potential Surfaces for the Adsorption of Electron-Rich and Electron-Deficient π-Systems: A Comparative DFT Study" Nanomaterials 12, no. 6: 1028. https://doi.org/10.3390/nano12061028
APA StyleIbrahim, M. A. A., Mahmoud, A. H. M., Soliman, K. A., Mekhemer, G. A. H., Ahmed, M. N., Shawky, A. M., Abourehab, M. A. S., Elkaeed, E. B., Soliman, M. E. S., & Moussa, N. A. M. (2022). Borophene and Pristine Graphene 2D Sheets as Potential Surfaces for the Adsorption of Electron-Rich and Electron-Deficient π-Systems: A Comparative DFT Study. Nanomaterials, 12(6), 1028. https://doi.org/10.3390/nano12061028