
Pharmacokinetics of PEGylated Gold Nanoparticles: In Vitro—In Vivo Correlation

 ,
,  , ,
, ,  ,
,  ,
,  and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Gold Nanoparticles
2.2. Cell Lines
2.3. Cell Treatment
2.4. Biodistribution and Accumulation of PEG-AuNPs In Vivo
2.5. Quantification of PEG-AuNPs in the Cells, Blood, and Organs
2.6. Data Treatment and Statistics
3. Results and Discussion
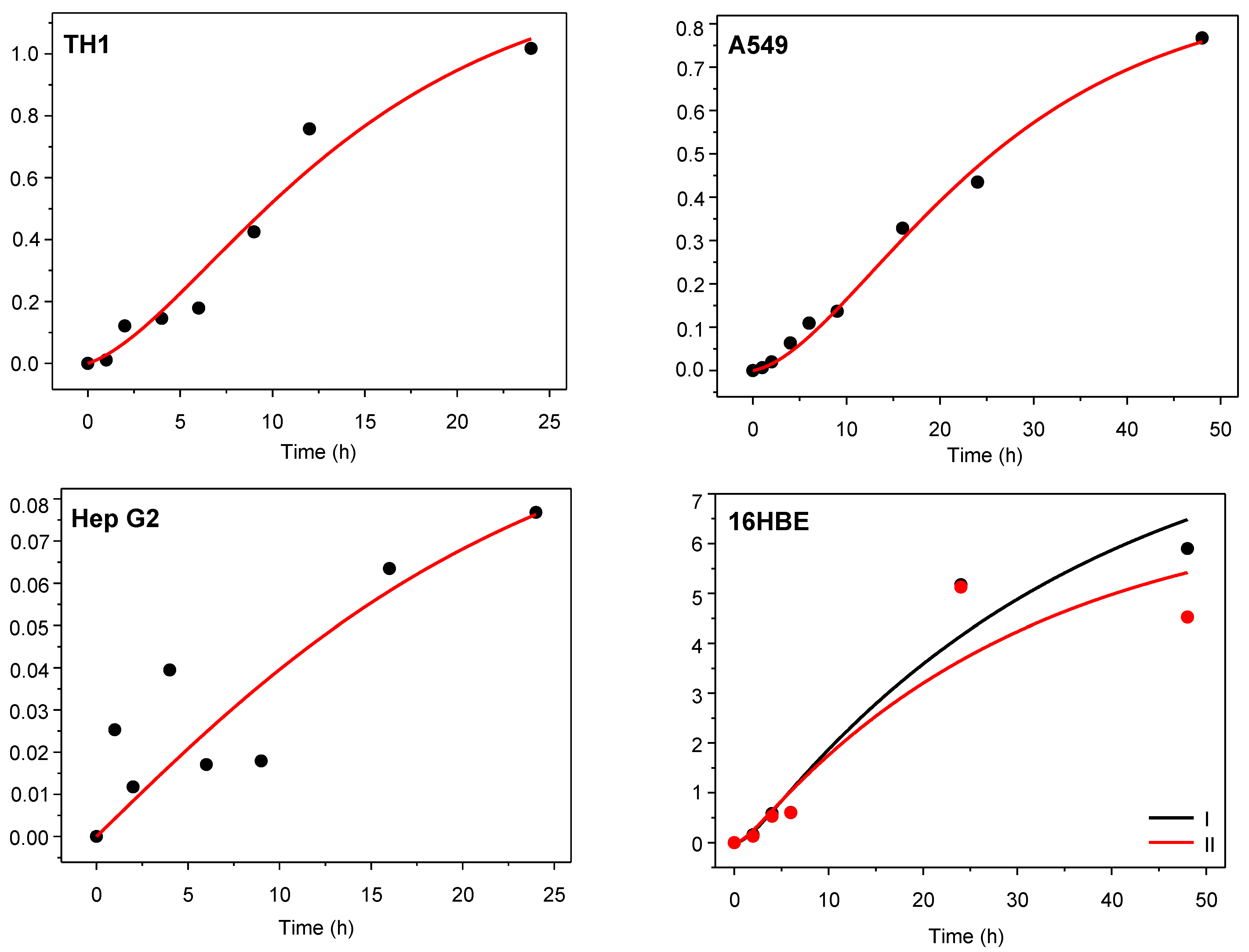
3.1. In Vitro Pharmacokinetic Study
3.2. Kinetic Analysis of Experiments with Cell Cultures
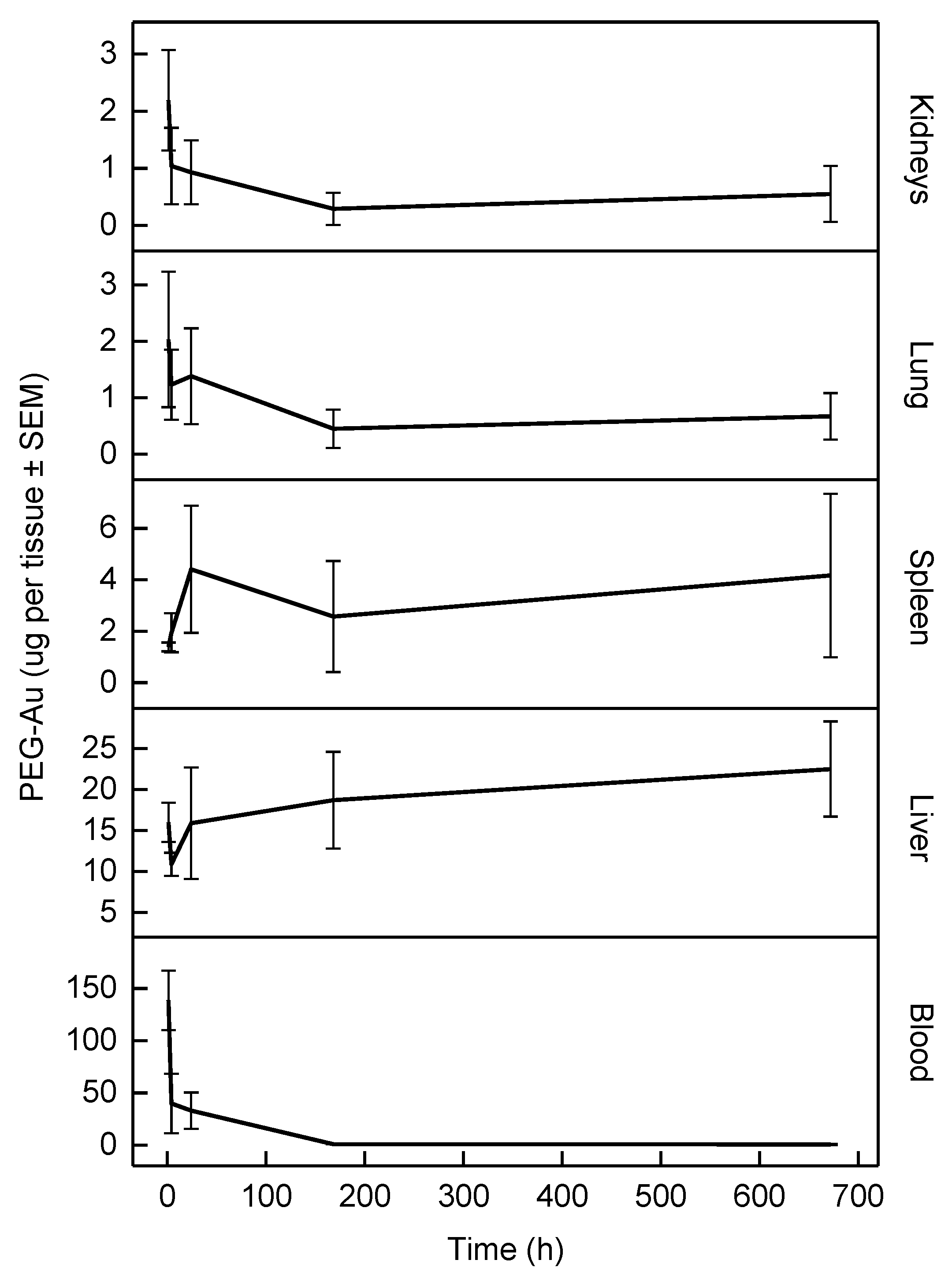
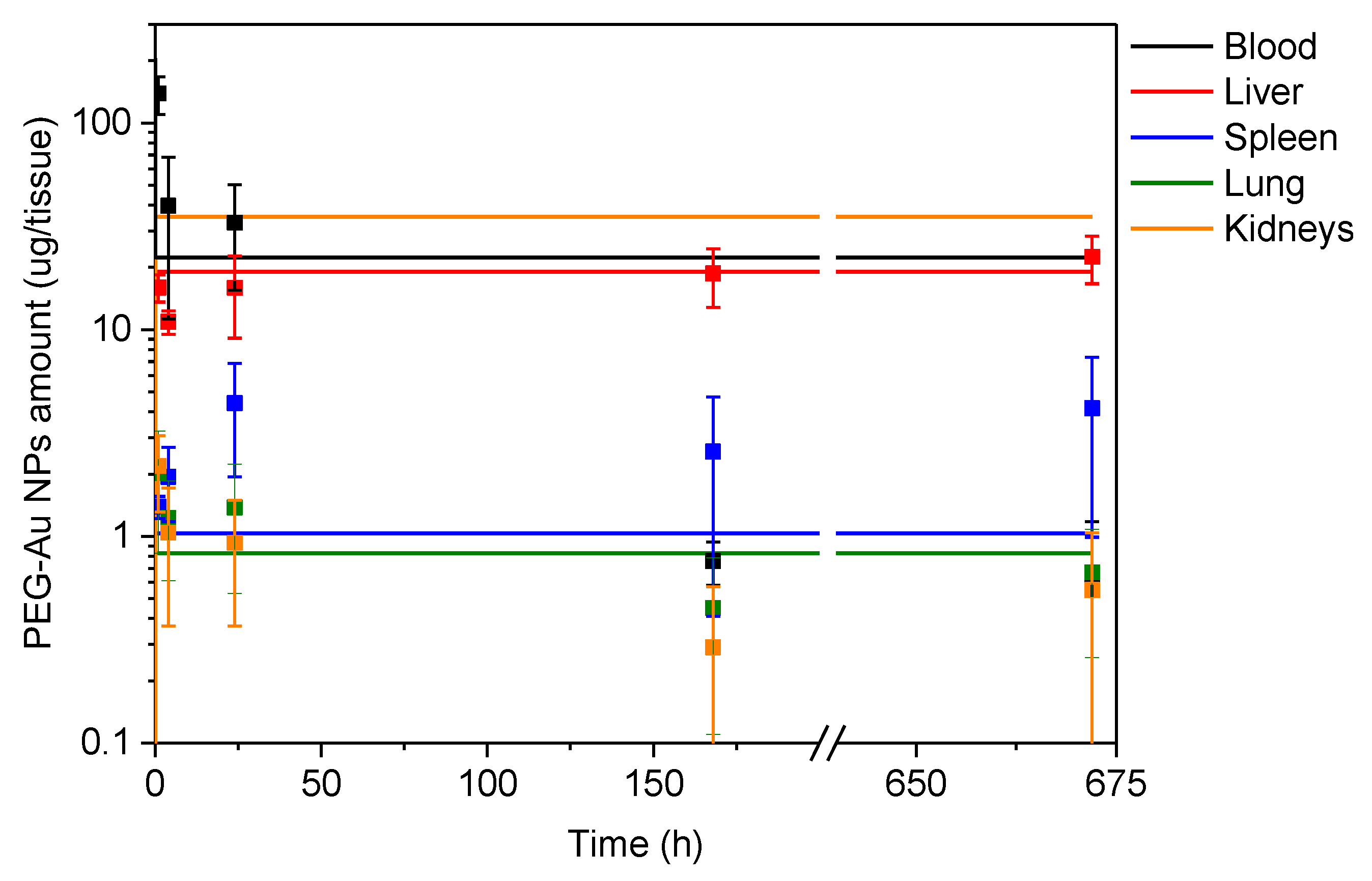
3.3. In Vivo Pharmacokinetic Study
3.4. Transfer of Parameters from In Vitro to In Vivo
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Braakhuis, H.M.; Kloet, S.K.; Kezic, S.; Kuper, F.; Park, M.V.D.Z.; Bellmann, S.; Van der Zande, M.; Le Gac, S.; Krystek, P.; Peters, R.J.B.; et al. Progress and future of in vitro models to study translocation of nanoparticles. Arch. Toxicol. 2015, 89, 1469–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riviere, J.E. Of mice, men and nanoparticle biocoronas: Are in vitro to in vivo correlations and interspecies extrapolations realistic? Nanomedicine 2013, 8, 1357–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monopoli, M.P.; Åberg, C.; Salvati, A.; Dawson, K.A. Biomolecular coronas provide the biological identity of nanosized materials. Nat. Nanotechnol. 2012, 7, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Shilo, M.; Sharon, A.; Baranes, K.; Motiei, M.; Lellouche, J.-P.M.; Popovtzer, R. The effect of nanoparticle size on the probability to cross the blood-brain barrier: An in-vitro endothelial cell model. J. Nanobiotechnol. 2015, 13, 19. [Google Scholar] [CrossRef] [Green Version]
- Hajal, C.; Campisi, M.; Mattu, C.; Chiono, V.; Kamm, R.D. In vitro models of molecular and nano-particle transport across the blood-brain barrier. Biomicrofluidics 2018, 12, 042213. [Google Scholar] [CrossRef]
- Dohle, E.; Singh, S.; Nishiguchi, A.; Fischer, T.; Wessling, M.; Möller, M.; Sader, R.; Kasper, J.; Ghanaati, S.; Kirkpatrick, C.J. Human Co- and Triple-Culture Model of the Alveolar-Capillary Barrier on a Basement Membrane Mimic. Tissue Eng. Part C Methods 2018, 24, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Dekali, S.; Gamez, C.; Kortulewski, T.; Blazy, K.; Rat, P.; Lacroix, G. Assessment of an in vitro model of pulmonary barrier to study the translocation of nanoparticles. Toxicol. Rep. 2014, 1, 157–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasper, J.Y.; Hermanns, M.I.; Kraegeloh, A.; Roth, W.; Kirkpatrick, C.J.; Unger, R.E. In Vitro Entero-Capillary Barrier Exhibits Altered Inflammatory and Exosomal Communication Pattern after Exposure to Silica Nanoparticles. Int. J. Mol. Sci. 2019, 20, 3301. [Google Scholar] [CrossRef] [Green Version]
- Mieremet, A.; Rietveld, M.; Absalah, S.; Van Smeden, J.; Bouwstra, J.A.; El Ghalbzouri, A. Improved epidermal barrier formation in human skin models by chitosan modulated dermal matrices. PLoS ONE 2017, 12, e0174478. [Google Scholar] [CrossRef] [Green Version]
- Sanches, P.L.; Geaquinto, L.R.D.O.; Cruz, R.; Schuck, D.C.; Lorencini, M.; Granjeiro, J.M.; Ribeiro, A.R.L. Toxicity Evaluation of TiO2 Nanoparticles on the 3D Skin Model: A Systematic Review. Front. Bioeng. Biotechnol. 2020, 8, 575. [Google Scholar] [CrossRef] [PubMed]
- Correia Carreira, S.; Walker, L.; Paul, K.; Saunders, M. The toxicity, transport and uptake of nanoparticles in the in vitro BeWo b30 placental cell barrier model used within NanoTEST. Nanotoxicology 2015, 9, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Blundell, C.; Tess, E.R.; Schanzer, A.S.R.; Coutifaris, C.; Su, E.J.; Parry, S.; Huh, D. A microphysiological model of the human placental barrier. Lab Chip 2016, 16, 3065–3073. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-H.; Riviere, J.E.; Monteiro-Riviere, N.A.; Lin, Z. Probabilistic risk assessment of gold nanoparticles after intravenous administration by integrating in vitro and in vivo toxicity with physiologically based pharmacokinetic modeling. Nanotoxicology 2018, 12, 453–469. [Google Scholar] [CrossRef] [PubMed]
- Teeguarden, J.G.; Hinderliter, P.M.; Orr, G.; Thrall, B.D.; Pounds, J. Particokinetics In Vitro: Dosimetry Considerations for In Vitro Nanoparticle Toxicity Assessments. Toxicol. Sci. 2007, 95, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Hobson, J.J.; Al-Khouja, A.; Curley, P.; Meyers, D.; Flexner, C.; Siccardi, M.; Owen, A.; Meyers, C.F.; Rannard, S.P. Semi-solid prodrug nanoparticles for long-acting delivery of water-soluble antiretroviral drugs within combination HIV therapies. Nat. Commun. 2019, 10, 1413. [Google Scholar] [CrossRef]
- Shelley, M.L.; Wagner, A.J.; Hussain, S.M.; Bleckmann, C. Modeling the In Vivo Case with In Vitro Nanotoxicity Data. Int. J. Toxicol. 2008, 27, 359–367. [Google Scholar] [CrossRef]
- Cheng, W.; Ng, C.A. A Permeability-Limited Physiologically Based Pharmacokinetic (PBPK) Model for Perfluorooctanoic acid (PFOA) in Male Rats. Environ. Sci. Technol. 2017, 51, 9930–9939. [Google Scholar] [CrossRef]
- Sharma, R.P.; Kumar, V.; Schuhmacher, M.; Kolodkin, A.; Westerhoff, H.V. Development and evaluation of a harmonized whole body physiologically based pharmacokinetic (PBPK) model for flutamide in rats and its extrapolation to humans. Environ. Res. 2020, 182, 108948. [Google Scholar] [CrossRef]
- Khlebtsov, N.; Dykman, L. Biodistribution and toxicity of engineered gold nanoparticles: A review of in vitro and in vivo studies. Chem. Soc. Rev. 2011, 40, 1647–1671. [Google Scholar] [CrossRef]
- De Jong, W.H.; Hagens, W.I.; Krystek, P.; Burger, M.C.; Sips, A.J.; Geertsma, R. Particle size-dependent organ distribution of gold nanoparticles after intravenous administration. Biomaterials 2008, 29, 1912–1919. [Google Scholar] [CrossRef]
- Semmler-Behnke, M.; Kreyling, W.G.; Lipka, J.; Fertsch, S.; Wenk, A.; Takenaka, S.; Schmid, G.; Brandau, W. Biodistribution of 1.4- and 18-nm Gold Particles in Rats. Small 2008, 4, 2108–2111. [Google Scholar] [CrossRef] [PubMed]
- Alric, C.; Miladi, I.; Kryza, D.; Taleb, J.; Lux, F.; Bazzi, R.; Billotey, C.; Janier, M.; Perriat, P.; Roux, S.; et al. The biodistribution of gold nanoparticles designed for renal clearance. Nanoscale 2013, 5, 5930–5939. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Monteiro-Riviere, N.A.; Kannan, R.; Riviere, J.E. A computational framework for interspecies pharmacokinetics, exposure and toxicity assessment of gold nanoparticles. Nanomedicine 2016, 11, 107–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozics, K.; Sramkova, M.; Kopecka, K.; Begerova, P.; Manova, A.; Krivosikova, Z.; Sevcikova, Z.; Liskova, A.; Rollerova, E.; Dubaj, T.; et al. Pharmacokinetics, Biodistribution, and Biosafety of PEGylated Gold Nanoparticles In Vivo. Nanomaterials 2021, 11, 1702. [Google Scholar] [CrossRef]
- Sramkova, M.; Kozics, K.; Masanova, V.; Uhnakova, I.; Razga, F.; Nemethova, V.; Mazancova, P.; Kapka-Skrzypczak, L.; Kruszewski, M.; Novotova, M.; et al. Kidney nanotoxicity studied in human renal proximal tubule epithelial cell line TH1. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2019, 845, 403017. [Google Scholar] [CrossRef]
- Kohl, Y.; Biehl, M.; Spring, S.; Hesler, M.; Ogourtsov, V.; Todorovic, M.; Owen, J.; Elje, E.; Kopecka, K.; Moriones, O.H.; et al. Microfluidic In Vitro Platform for (Nano)Safety and (Nano)Drug Efficiency Screening. Small 2021, 17, 2006012. [Google Scholar] [CrossRef]
- Hirn, S.; Semmler-Behnke, M.; Schleh, C.; Wenk, A.; Lipka, J.; Schäffler, M.; Takenaka, S.; Möller, W.; Schmid, G.; Simon, U.; et al. Particle size-dependent and surface charge-dependent biodistribution of gold nanoparticles after intravenous administration. Eur. J. Pharm. Biopharm. 2011, 77, 407–416. [Google Scholar] [CrossRef] [Green Version]
- Bailly, A.-L.; Correard, F.; Popov, A.; Tselikov, G.; Chaspoul, F.; Appay, R.; Al-Kattan, A.; Kabashin, A.V.; Braguer, D.; Esteve, M.-A. In vivo evaluation of safety, biodistribution and pharmacokinetics of laser-synthesized gold nanoparticles. Sci. Rep. 2019, 9, 12890. [Google Scholar] [CrossRef] [Green Version]
- Gabelova, A.; Kozics, K.; Kapka-Skrzypczak, L.; Kruszewski, M.; Sramkova, M. Nephrotoxicity: Topical issue. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2019, 845, 402988. [Google Scholar] [CrossRef]
- Du, B.; Yu, M.; Zheng, J. Transport and interactions of nanoparticles in the kidneys. Nat. Rev. Mater. 2018, 3, 358–374. [Google Scholar] [CrossRef]
- Nair, A.V.; Keliher, E.J.; Core, A.B.; Brown, D.; Weissleder, R. Characterizing the Interactions of Organic Nanoparticles with Renal Epithelial Cells in Vivo. ACS Nano 2015, 9, 3641–3653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, I.; Vranic, S.; Boland, S.; Courtois, A.; Baeza-Squiban, A. Development of an in vitro model of human bronchial epithelial barrier to study nanoparticle translocation. Toxicol. Vitr. 2015, 29, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, P.J.; Ferrick, B.; Rybakovsky, E.; Thomas, S.; Mullin, J.M. Epithelial barrier function properties of the 16HBE14o- human bronchial epithelial cell culture model. Biosci. Rep. 2020, 40, 20201532. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.P.; Delp, M.; Lindstedt, S.L.; Rhomberg, L.R.; Beliles, R.P. Physiological Parameter Values for Physiologically Based Pharmacokinetic Models. Toxicol. Ind. Health 1997, 13, 407–484. [Google Scholar] [CrossRef]
- Dogra, P.; Butner, J.D.; Chuang, Y.-L.; Caserta, S.; Goel, S.; Brinker, C.J.; Cristini, V.; Wang, Z. Mathematical modeling in cancer nanomedicine: A review. Biomed. Microdevices 2019, 21, 40. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, H.H.; Holt-Casper, D.; Grainger, D.W.; Ghandehari, H. Nanoparticle uptake: The phagocyte problem. Nano Today 2015, 10, 487–510. [Google Scholar] [CrossRef] [Green Version]
- Bachler, G.; Von Goetz, N.; Hungerbühler, K. A physiologically based pharmacokinetic model for ionic silver and silver nanoparticles. Int. J. Nanomed. 2013, 8, 3365–3382. [Google Scholar] [CrossRef] [Green Version]
- Bachler, G.; Von Goetz, N.; Hungerbühler, K. Using physiologically based pharmacokinetic (PBPK) modeling for dietary risk assessment of titanium dioxide (TiO2) nanoparticles. Nanotoxicology 2015, 9, 373–380. [Google Scholar] [CrossRef]
- Carlander, U.; Li, D.; Jolliet, O.; Emond, C.; Johanson, G. Toward a general physiologically-based pharmacokinetic model for intravenously injected nanoparticles. Int. J. Nanomed. 2016, 11, 625–640. [Google Scholar] [CrossRef] [Green Version]
- Tyner, K.; Bancos, S.; Stevens, D. Effect of silica and gold nanoparticles on macrophage proliferation, activation markers, cytokine production, and phagocytosis in vitro. Int. J. Nanomed. 2014, 10, 183–206. [Google Scholar] [CrossRef] [Green Version]
- Meindl, C.; Öhlinger, K.; Ober, J.; Roblegg, E.; Fröhlich, E. Comparison of fluorescence-based methods to determine nanoparticle uptake by phagocytes and non-phagocytic cells in vitro. Toxicology 2017, 378, 25–36. [Google Scholar] [CrossRef]
- Nicolete, R.; Dos Santos, D.F.; Faccioli, L.H. The uptake of PLGA micro or nanoparticles by macrophages provokes distinct in vitro inflammatory response. Int. Immunopharmacol. 2011, 11, 1557–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Sampling Time (h) | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 4 | 6 | 9 | 16 | 24 | 48 | |
| TH1 | 4.87 | 7.49 | 11.75 | 13.71 | 19.79 | 26.35 | 32.51 | 160.96 1 |
| A549 | 1.2 | 4.07 | 5.81 | 9.95 | 10.74 | 14.18 | 16.68 | 30.94 |
| Hep G2 | 1.48 | 0.56 | 2.06 | 0.8 | 0.85 | 2.14 | 3.2 | – |
| 16HBE I 2 | – | 1.07 | 4.02 | 3.80 | – | – | 3.07 | 2.83 |
| 16HBE II 2 | – | 0.79 | 3.50 | 3.77 | – | – | 3.04 | 2.33 |
| Cell Line | R2 | kin (h−1) | kout (h−1) | Rc:m = kin/kout |
|---|---|---|---|---|
| TH1 | 0.96 | 1.28 ± 0.30 | 0.171 ± 0.056 | 7.48 |
| A549 | 0.99 | 0.400 ± 0.033 | 0.0598 ± 0.0078 | 6.69 |
| Hep G2 | 0.67 | 0.051 ± 0.024 | 0.063 ± 0.060 | 0.810 |
| 16HBE I | 0.95 | 0.40 ± 0.60 | 0.66 ± 1.03 | 0.606 |
| 16HBE II | 0.86 | 0.50 ± 1.45 | 0.94 ± 2.79 | 0.532 |
| Organ/Tissue | Mass 1 | Blood Flow 2 |
|---|---|---|
| Blood | 6.40 | – |
| Liver | 3.66 | 18.3 |
| Spleen | 0.20 | 0.85 |
| Lung 3 | 0.50 | 2.10 |
| Kidneys | 0.73 | 14.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubaj, T.; Kozics, K.; Sramkova, M.; Manova, A.; Bastús, N.G.; Moriones, O.H.; Kohl, Y.; Dusinska, M.; Runden-Pran, E.; Puntes, V.; et al. Pharmacokinetics of PEGylated Gold Nanoparticles: In Vitro—In Vivo Correlation. Nanomaterials 2022, 12, 511. https://doi.org/10.3390/nano12030511
Dubaj T, Kozics K, Sramkova M, Manova A, Bastús NG, Moriones OH, Kohl Y, Dusinska M, Runden-Pran E, Puntes V, et al. Pharmacokinetics of PEGylated Gold Nanoparticles: In Vitro—In Vivo Correlation. Nanomaterials. 2022; 12(3):511. https://doi.org/10.3390/nano12030511
Chicago/Turabian StyleDubaj, Tibor, Katarina Kozics, Monika Sramkova, Alena Manova, Neus G. Bastús, Oscar H. Moriones, Yvonne Kohl, Maria Dusinska, Elise Runden-Pran, Victor Puntes, and et al. 2022. "Pharmacokinetics of PEGylated Gold Nanoparticles: In Vitro—In Vivo Correlation" Nanomaterials 12, no. 3: 511. https://doi.org/10.3390/nano12030511
APA StyleDubaj, T., Kozics, K., Sramkova, M., Manova, A., Bastús, N. G., Moriones, O. H., Kohl, Y., Dusinska, M., Runden-Pran, E., Puntes, V., Nelson, A., Gabelova, A., & Simon, P. (2022). Pharmacokinetics of PEGylated Gold Nanoparticles: In Vitro—In Vivo Correlation. Nanomaterials, 12(3), 511. https://doi.org/10.3390/nano12030511