

Stable Configurations of DOXH Interacting with Graphene: Heuristic Algorithm Approach Using NSGA-II and U-NSGA-III


Abstract
1. Introduction
2. Methodology
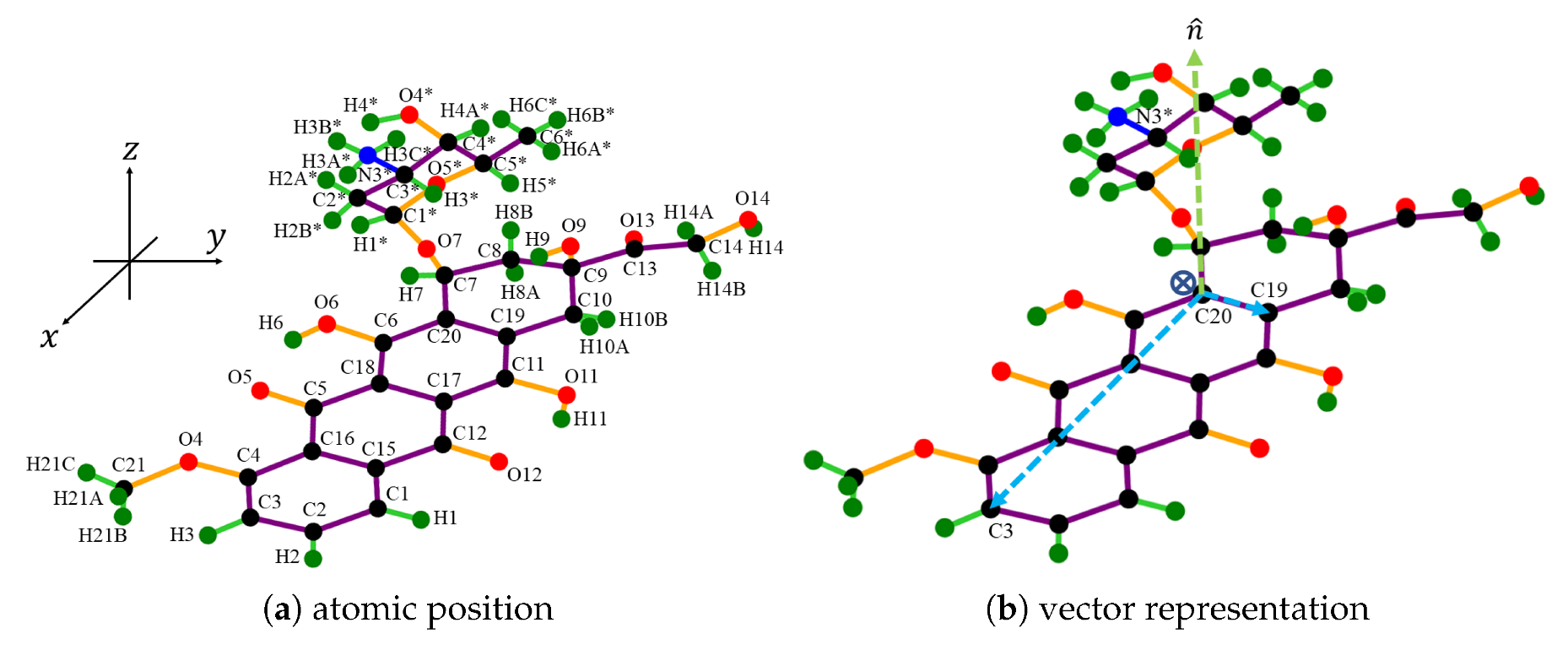
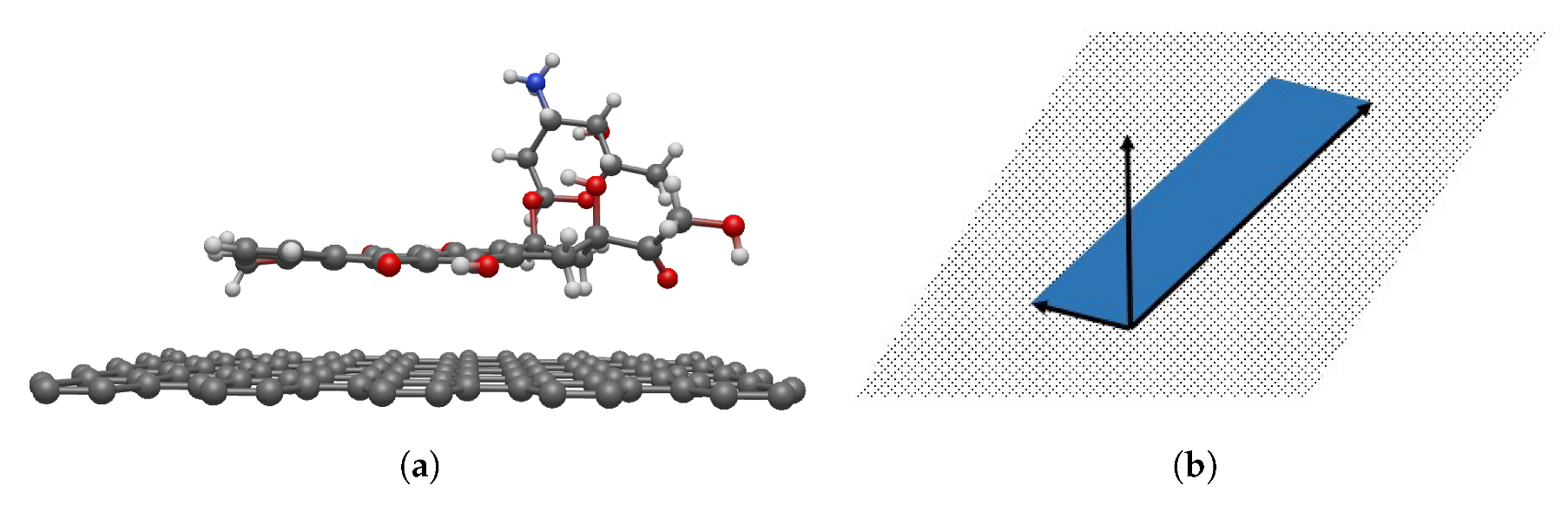
2.1. Molecular Description
2.2. Interaction Energy and Parameter Values
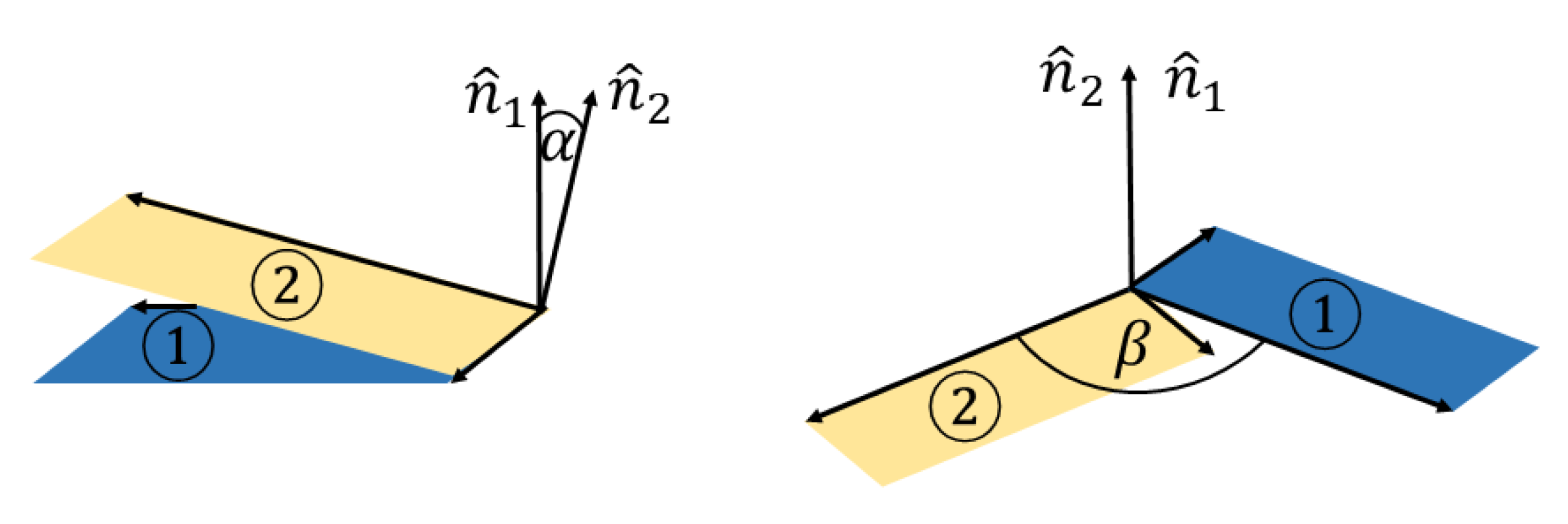
2.3. Optimization Setting
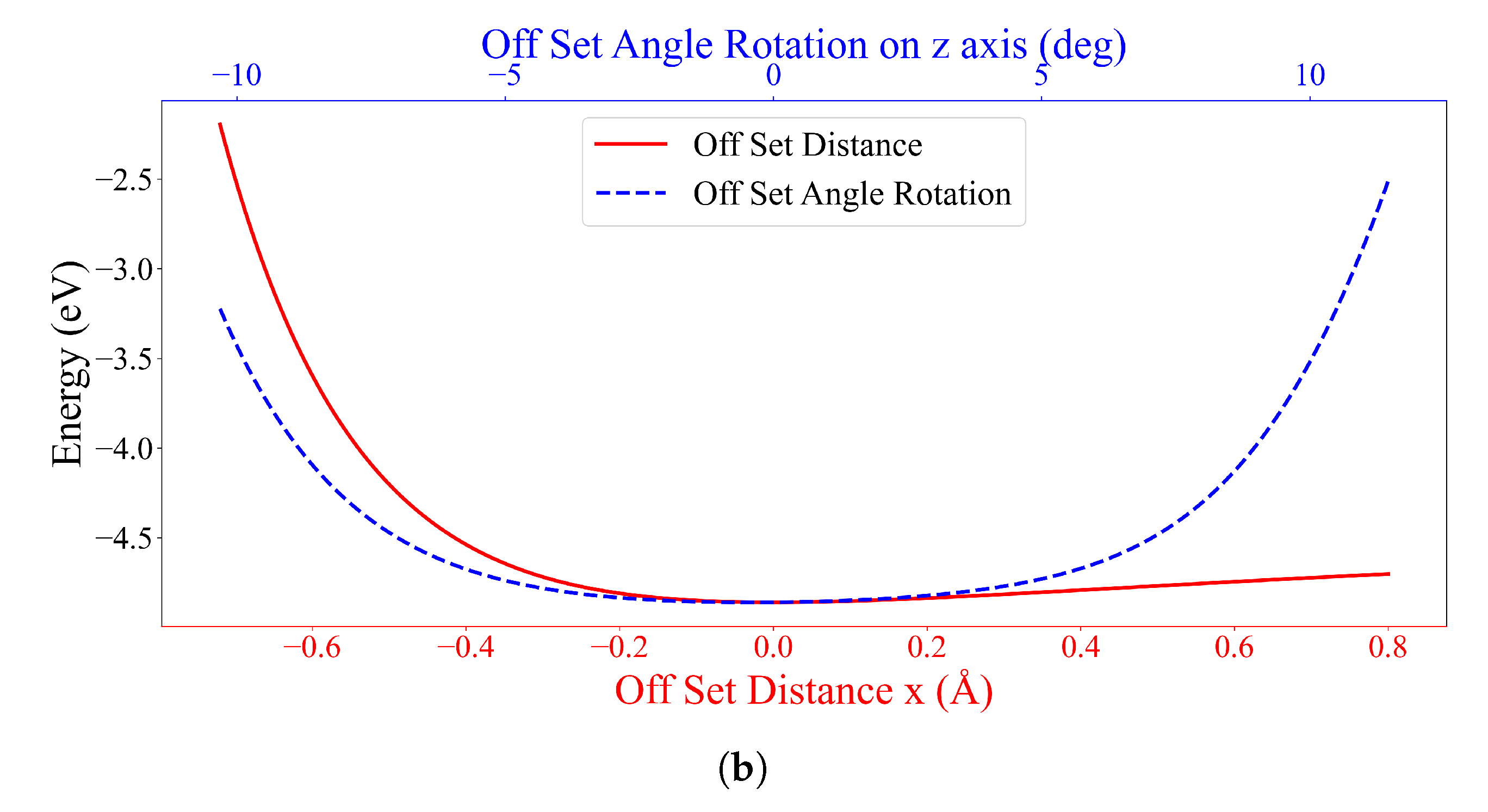
3. Numerical Results
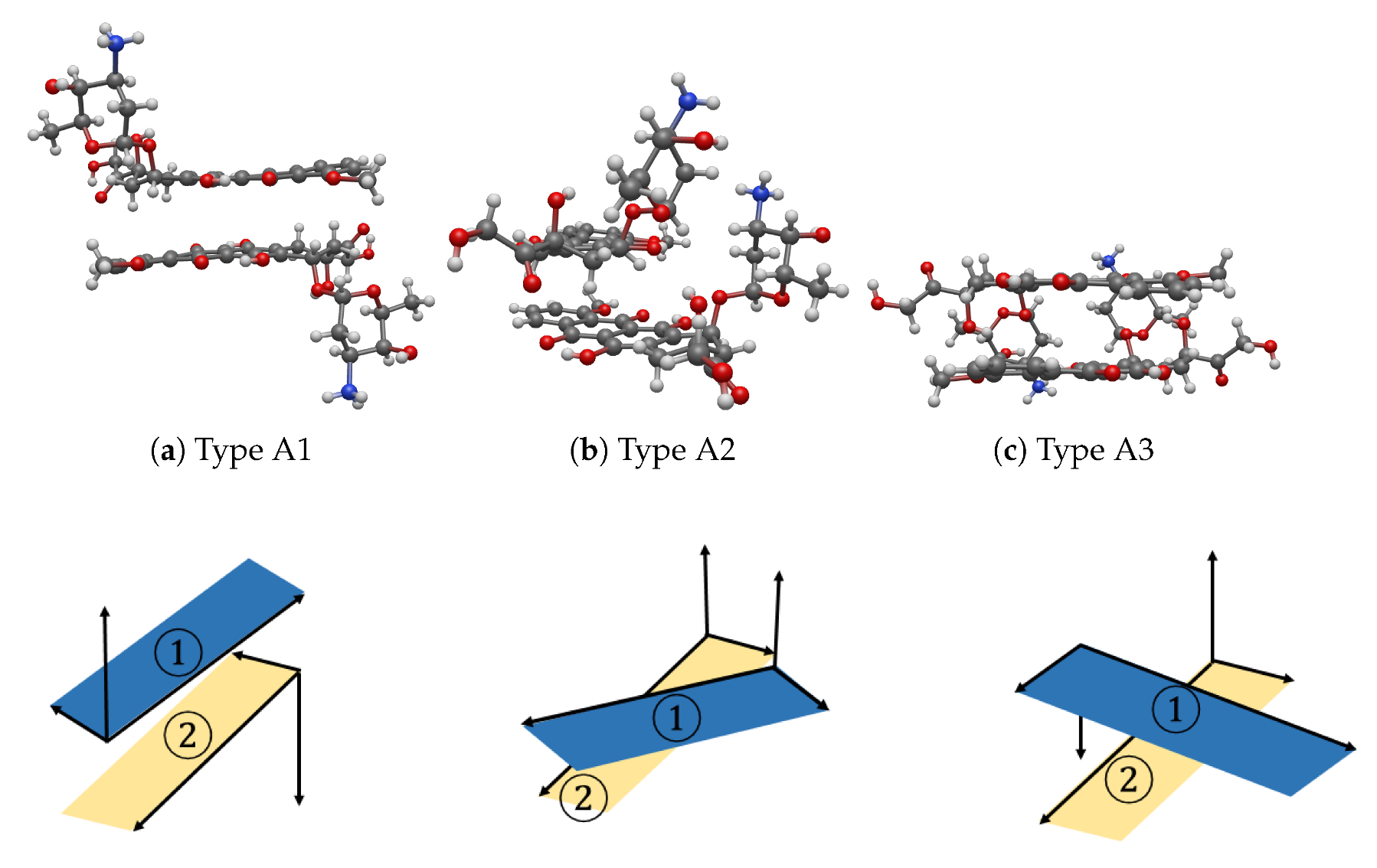
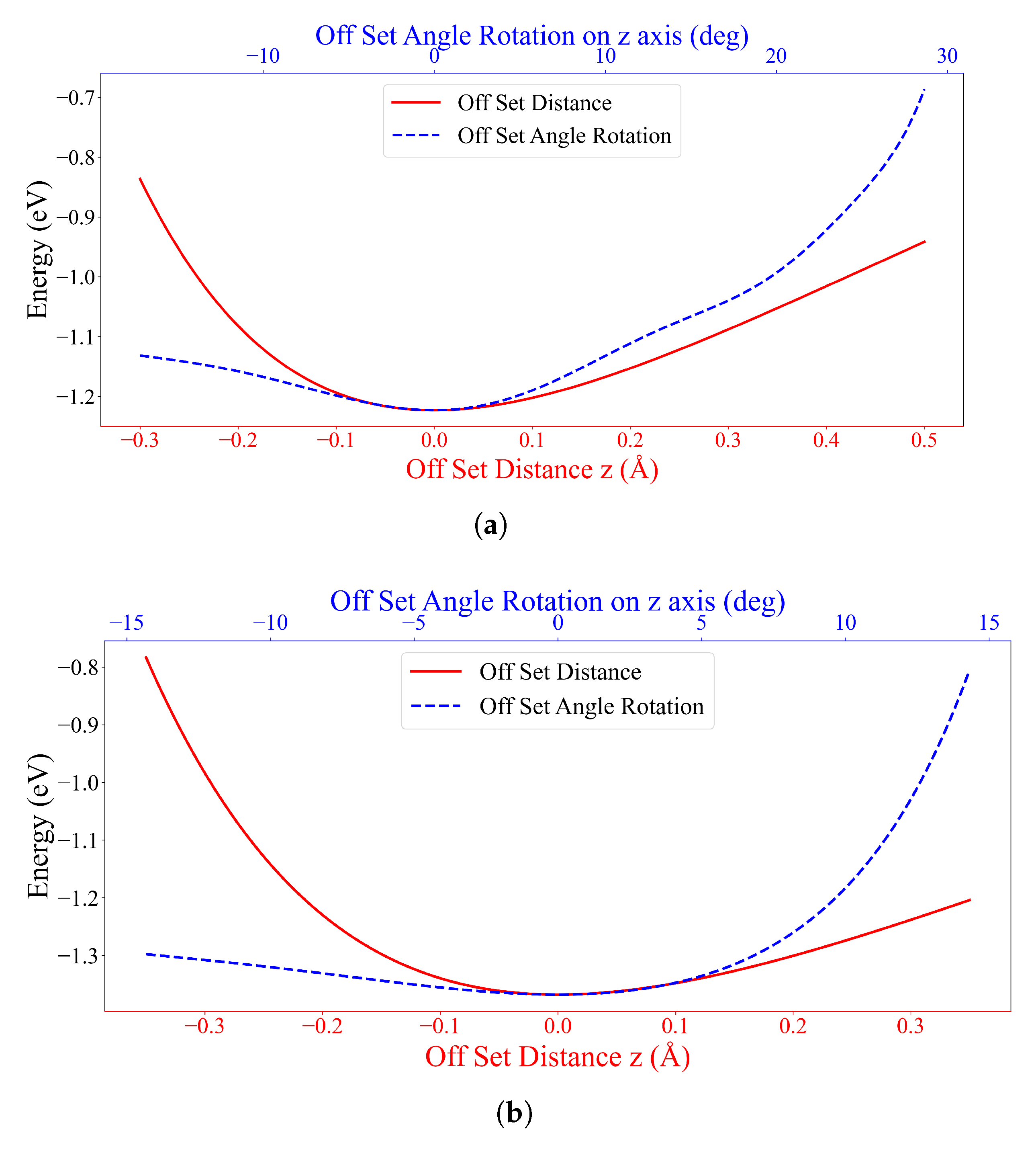
3.1. Interaction between Two DOXHs
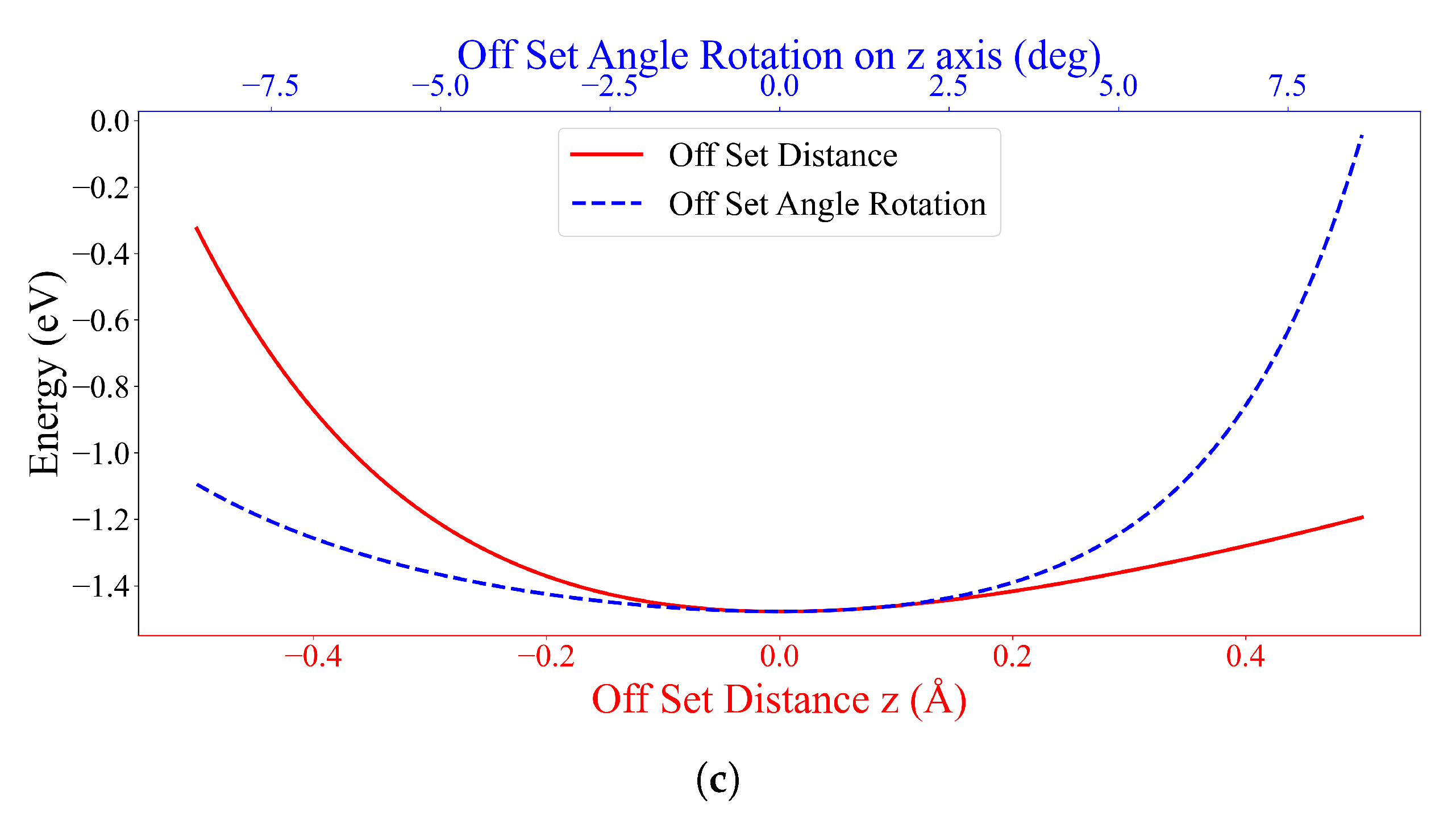
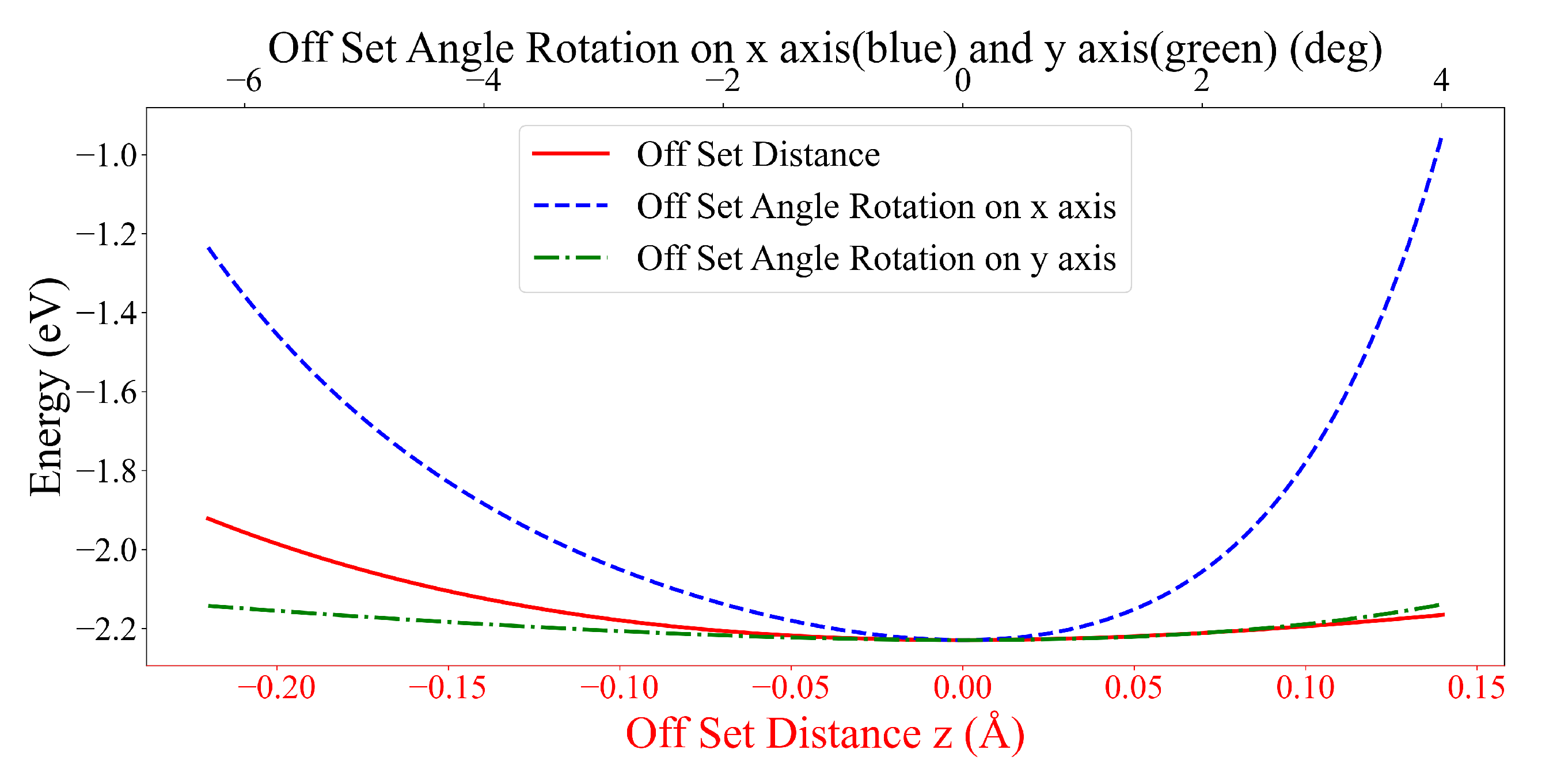
3.2. Interaction between DOXH and Graphene
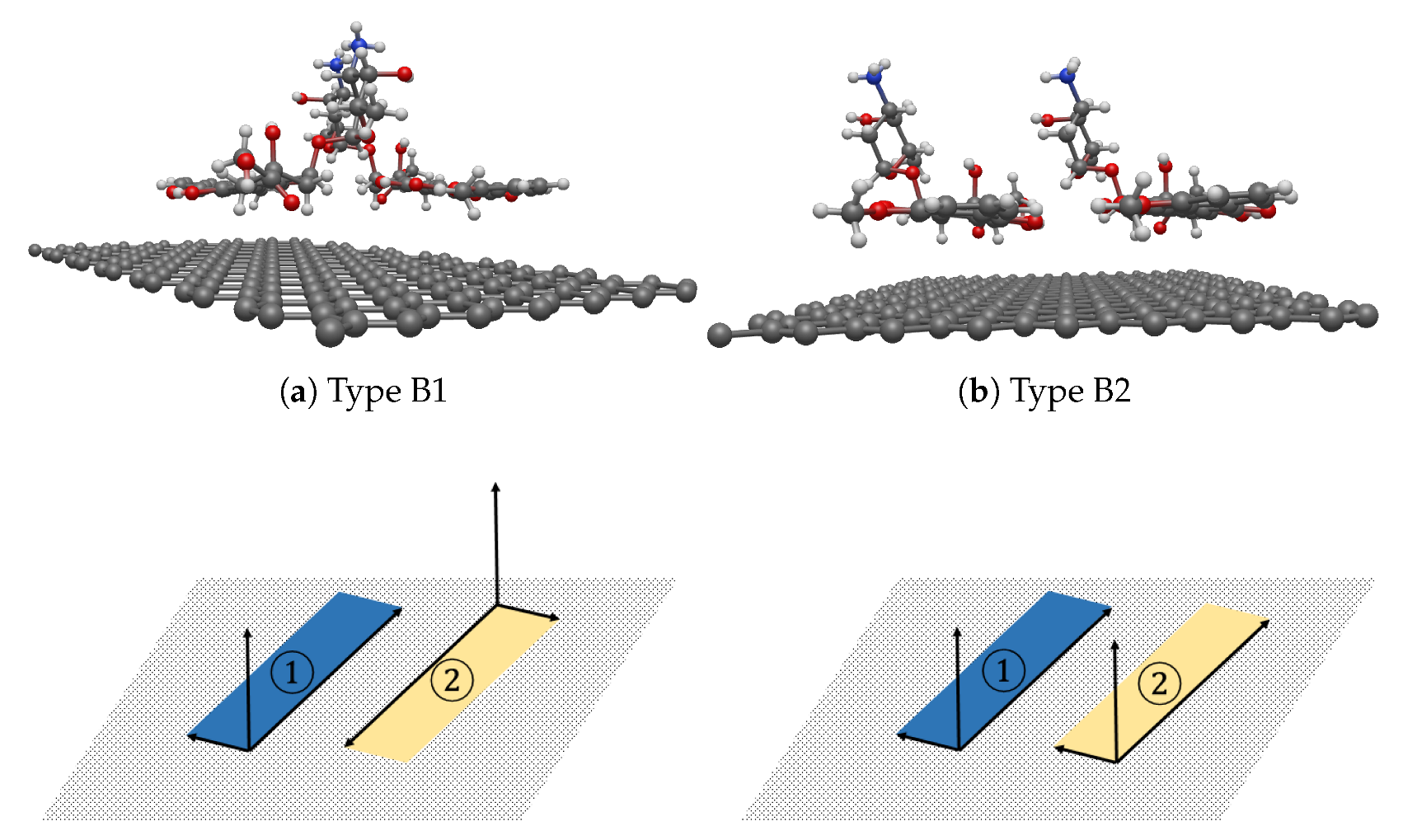
3.3. Interaction between two DOXHs and Graphene
4. Discussion
5. Summary
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bianco, A.; Kostarelos, K.; Prato, M. Applications of carbon nanotubes in drug delivery. Curr. Opin. Chem. Biol. 2005, 9, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, K.; Davis, C.; Sherlock, S.; Cao, Q.; Chen, X.; Dai, H. Drug Delivery with Carbon Nanotubes for In vivo Cancer Treatment. Cancer Res. 2008, 68, 6652–6660. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, D.; Shanmugam, R.; Pitchiah, S.; Murugan, P.; Chinnathambi, A.; Alharbi, S.A.; Durairaj, K.; Sundramoorthy, A.K. Potential Applications of Halloysite Nanotubes as Drug Carriers: A Review. J. Nanomater. 2022, 2022, 1068536. [Google Scholar] [CrossRef]
- Patil, Y.B.; Toti, U.S.; Khdair, A.; Ma, L.; Panyam, J. Single-step surface functionalization of polymeric nanoparticles for targeted drug delivery. Biomaterials 2009, 30, 859–866. [Google Scholar] [CrossRef]
- Tian, T.; Zhang, H.X.; He, C.P.; Fan, S.; Zhu, Y.L.; Qi, C.; Huang, N.P.; Xiao, Z.D.; Lu, Z.H.; Tannous, B.A.; et al. Surface functionalized exosomes as targeted drug delivery vehicles for cerebral ischemia therapy. Biomaterials 2018, 150, 137–149. [Google Scholar] [CrossRef]
- Liu, J.; Cui, L.; Losic, D. Graphene and graphene oxide as new nanocarriers for drug delivery applications. Acta Biomater. 2013, 9, 9243–9257. [Google Scholar] [CrossRef]
- Sun, X.; Liu, Z.; Welsher, K.; Robinson, J.T.; Goodwin, A.; Zaric, S.; Dai, H. Nano-graphene oxide for cellular imaging and drug delivery. Nano Res. 2008, 1, 203–212. [Google Scholar] [CrossRef]
- Goenka, S.; Sant, V.; Sant, S. Graphene-based nanomaterials for drug delivery and tissue engineering. J. Control. Release 2014, 173, 75–88. [Google Scholar] [CrossRef]
- Jampilek, J.; Kralova, K. Advances in Drug Delivery Nanosystems Using Graphene-Based Materials and Carbon Nanotubes. Materials 2021, 14, 1059. [Google Scholar] [CrossRef]
- Kik, K.; Wasowsk-Lukawska, M.; Oszczapowicz, I.; Szmigiero, L. Cytotoxicity and Cellular Uptake of Doxorubicin and its Formamidine Derivatives in HL60 Sensitive and HL60/MX2 Resistant Cells. Anticancer. Res. 2009, 29, 1429–1433. [Google Scholar]
- Weinberg, B.D.; Ai, H.; Blanco, E.; Anderson, J.M.; Gao, J. Antitumor efficacy and local distribution of doxorubicin via intratumoral delivery from polymer millirods. J. Biomed. Mater. Res. Part A 2007, 81, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Huang, R.; Li, J.; Liu, S.; Huang, S.; Jiang, C. Plasmid pORF-hTRAIL and doxorubicin co-delivery targeting to tumor using peptide-conjugated polyamidoamine dendrimer. Biomaterials 2011, 32, 1242–1252. [Google Scholar] [CrossRef] [PubMed]
- McNeeley, K.M.; Karathanasis, E.; Annapragada, A.V.; Bellamkonda, R.V. Masking and triggered unmasking of targeting ligands on nanocarriers to improve drug delivery to brain tumors. Biomaterials 2009, 30, 3986–3995. [Google Scholar] [CrossRef] [PubMed]
- Gillies, E.R.; Fréchet, J.M. pH-responsive copolymer assemblies for controlled release of doxorubicin. Bioconjug. Chem. 2005, 16, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Mhawi, A.A.; Fernandes, A.B.; Ottensmeyer, F.P. Low-energy-loss electron microscopy of doxorubicin in human breast cancer MCF-7 cells: Localization by color. J. Struct. Biol. 2007, 158, 80–92. [Google Scholar] [CrossRef]
- Sanchez, V.C.; Jachak, A.; Hurt, R.H.; Kane, A.B. Biological Interactions of Graphene-Family Nanomaterials: An Interdisciplinary Review. Chem. Res. Toxicol. 2012, 25, 15–34. [Google Scholar] [CrossRef]
- Vovusha, H.; Banerjee, D.; Yadav, M.K.; Perrozzi, F.; Ottaviano, L.; Sanyal, S.; Sanyal, B. Binding Characteristics of Anticancer Drug Doxorubicin with Two-Dimensional Graphene and Graphene Oxide: Insights from Density Functional Theory Calculations and Fluorescence Spectroscopy. J. Phys. Chem. C 2018, 122, 21031–21038. [Google Scholar] [CrossRef]
- Tonel, M.Z.; Martins, M.O.; Zanella, I.; Pontes, R.B.; Fagan, S.B. A first-principles study of the interaction of doxorubicin with graphene. Comput. Theor. Chem. 2017, 1115, 270–275. [Google Scholar] [CrossRef]
- Shen, J.W.; Li, J.; Dai, J.; Zhou, M.; Ren, H.; Zhang, L.; Hu, Q.; Kong, Z.; Liang, L. Molecular dynamics study on the adsorption and release of doxorubicin by chitosan-decorated graphene. Carbohydr. Polym. 2020, 248, 116809. [Google Scholar] [CrossRef]
- Song, J.; Cui, N.; Sun, S.; Lu, X.; Wang, Y.; Shi, H.; Lee, E.S.; Jiang, H.B. Controllability of Graphene Oxide Doxorubicin Loading Capacity Based on Density Functional Theory. Nanomaterials 2022, 12, 479. [Google Scholar] [CrossRef]
- Chen, J.X.; Chen, Y.G.; Kapral, R. Chemically Propelled Motors Navigate Chemical Patterns. Adv. Sci. 2018, 5, 1800028. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cao, Z.; Farimani, A.B. Efficient water desalination with graphene nanopores obtained using artificial intelligence. npj 2D Mater. Appl. 2021, 5, 66. [Google Scholar] [CrossRef]
- Villaseñor-Cavazos, F.J.; Torres-Valladares, D.; Lozano, O. Modeling and optimization of nanovector drug delivery systems: Exploring the most efficient algorithms. J. Nanoparticle Res. 2022, 24, 119. [Google Scholar] [CrossRef]
- Deb, K.; Pratap, A.; Agarwal, S.; Meyarivan, T. A Fast and Elitist Multiobjective Genetic Algorithm: NSGA-II. IEEE Trans. Evol. Comput. 2002, 6, 182–197. [Google Scholar] [CrossRef]
- Murugan, P.; Kannan, S.; Baskar, S. Application of NSGA-II Algorithm to Single-Objective Transmission Constrained Generation Expansion Planning. IEEE Trans. Power Syst. 2009, 24, 1790–1797. [Google Scholar] [CrossRef]
- Seada, H.; Deb, K. A Unified Evolutionary Optimization Procedure for Single, Multiple, and Many Objectives. IEEE Trans. Evol. Comput. 2016, 20, 358–369. [Google Scholar] [CrossRef]
- Wei, H.; Li, S.; Quan, H.; Liu, D.; Rao, S.; Li, C.; Hu, J. Unified Multi-Objective Genetic Algorithm for Energy Efficient Job Shop Scheduling. IEEE Access 2021, 9, 54542–54557. [Google Scholar] [CrossRef]
- Sumetpipat, K.; Baowan, D.; Hill, J.M. Modelling Packing Arrangements of Doxorubicin in Liposomal Molecules. J. Comput. Theor. Nanosci. 2016, 13, 8241–8248. [Google Scholar] [CrossRef]
- Putthikorn, S.; Ruengrot, P.; Baowan, D. Energy behaviour of Doxorubicin interacting with peptide nanotubes. J. Math. Chem. 2020, 58, 382–392. [Google Scholar] [CrossRef]
- Blank, J.; Deb, K. pymoo: Multi-Objective Optimization in Python. IEEE Access 2020, 8, 89497–89509. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Mathivathanan, L.; Yang, G.; Leng, F.; Raptis, R.G. Crystal structure and conformational analysis of doxorubicin nitrate. Acta Crystallogr. Sect. Crystallogr. Commun. 2018, E74, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Rappé, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A., III; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Baowan, D.; Cox, B.J.; Hilder, T.A.; Hill, J.M.; Thamwattana, N. Modelling and Mechanics of Carbon-Based Nanostructured Materials; Elsevier: Oxford, UK, 2017. [Google Scholar]
- Mirhosseini, M.M.; Rahmati, M.; Zargarian, S.S.; Khordad, R. Molecular dynamics simulation of functionalized graphene surface for high efficient loading of doxorubicin. J. Mol. Struct. 2017, 1141, 441–450. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interaction | (Å) | ( eV) | A (eV/Å) | B ( eV/Å) |
|---|---|---|---|---|
| Carbon–Carbon | 3.8510 | 4.5150 | 29.4530 | 4.8033 |
| Carbon–Oxygen | 3.6755 | 3.4130 | 16.8293 | 2.0746 |
| Carbon–Hydrogen | 3.3685 | 2.9227 | 8.5396 | 0.6237 |
| Carbon–Nitrogen | 3.7555 | 3.6601 | 20.5364 | 2.8807 |
| Algorithm | NSGA-II | U-NSGA-III | |||
|---|---|---|---|---|---|
| System | pop_size | n_offsprings | n_gen | pop_size | n_gen |
| i | 3300 | 2500 | 400 | 4400 | 400 |
| ii | 2700 | 2000 | 200 | 3600 | 200 |
| iii | 3500 | 2600 | 400 | 4000 | 400 |
| Algorithm | NSGA-II | ||||
|---|---|---|---|---|---|
| Seed | Type | ||||
| 1 | A2 | ||||
| 2 | - | ||||
| 3 | A2 | ||||
| 4 | A1 | ||||
| 5 | A1 | ||||
| 6 | A2 | ||||
| 7 | A3 | ||||
| 8 | A2 | ||||
| 9 | A2 | ||||
| 10 | A1 | ||||
| 11 | A2 | ||||
| 12 | A2 | ||||
| 13 | A2 | ||||
| 14 | - | ||||
| 15 | A2 | ||||
| Algorithm | U-NSGA-III | ||||
| Seed | Type | ||||
| 1 | A2 | ||||
| 2 | A2 | ||||
| 3 | A3 | ||||
| 4 | A2 | ||||
| 5 | A2 | ||||
| 6 | A2 | ||||
| 7 | A2 | ||||
| 8 | A3 | ||||
| 9 | A2 | ||||
| 10 | A2 | ||||
| 11 | A2 | ||||
| 12 | A2 | ||||
| 13 | A2 | ||||
| 14 | A2 | ||||
| 15 | - | ||||
| Algorithm | NSGA-II | U-NSGA-III | ||||||
|---|---|---|---|---|---|---|---|---|
| Seed | ||||||||
| 1 | ||||||||
| 2 | ||||||||
| 3 | ||||||||
| 4 | ||||||||
| 5 | ||||||||
| 6 | ||||||||
| 7 | ||||||||
| 8 | ||||||||
| 9 | ||||||||
| 10 | ||||||||
| 11 | ||||||||
| 12 | ||||||||
| 13 | ||||||||
| 14 | ||||||||
| 15 | ||||||||
| Algo. | NSGA-II | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Seed | Type | ||||||||
| 1 | B1 | ||||||||
| 2 | B2 | ||||||||
| 3 | B2 | ||||||||
| 4 | B2 | ||||||||
| 5 | B2 | ||||||||
| 6 | B2 | ||||||||
| 7 | B2 | ||||||||
| 8 | B2 | ||||||||
| 9 | B2 | ||||||||
| 10 | B1 | ||||||||
| 11 | B1 | ||||||||
| 12 | B1 | ||||||||
| 13 | B1 | ||||||||
| 14 | B2 | ||||||||
| 15 | B2 | ||||||||
| Seed | Type | ||||||||
| 1 | B2 | ||||||||
| 2 | B1 | ||||||||
| 3 | B2 | ||||||||
| 4 | B2 | ||||||||
| 5 | B1 | ||||||||
| 6 | B2 | ||||||||
| 7 | B1 | ||||||||
| 8 | B2 | ||||||||
| 9 | B1 | ||||||||
| 10 | B2 | ||||||||
| 11 | B1 | ||||||||
| 12 | B2 | ||||||||
| 13 | B2 | ||||||||
| 14 | B1 | ||||||||
| 15 | B1 | ||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sumetpipat, K.; Baowan, D. Stable Configurations of DOXH Interacting with Graphene: Heuristic Algorithm Approach Using NSGA-II and U-NSGA-III. Nanomaterials 2022, 12, 4097. https://doi.org/10.3390/nano12224097
Sumetpipat K, Baowan D. Stable Configurations of DOXH Interacting with Graphene: Heuristic Algorithm Approach Using NSGA-II and U-NSGA-III. Nanomaterials. 2022; 12(22):4097. https://doi.org/10.3390/nano12224097
Chicago/Turabian StyleSumetpipat, Kanes, and Duangkamon Baowan. 2022. "Stable Configurations of DOXH Interacting with Graphene: Heuristic Algorithm Approach Using NSGA-II and U-NSGA-III" Nanomaterials 12, no. 22: 4097. https://doi.org/10.3390/nano12224097
APA StyleSumetpipat, K., & Baowan, D. (2022). Stable Configurations of DOXH Interacting with Graphene: Heuristic Algorithm Approach Using NSGA-II and U-NSGA-III. Nanomaterials, 12(22), 4097. https://doi.org/10.3390/nano12224097