
Two-Dimensional Transition Metal-Hexaaminobenzene Monolayer Single-Atom Catalyst for Electrocatalytic Carbon Dioxide Reduction
 ,
,
Abstract
1. Introduction
2. Computational Methods
3. Results and Discussion
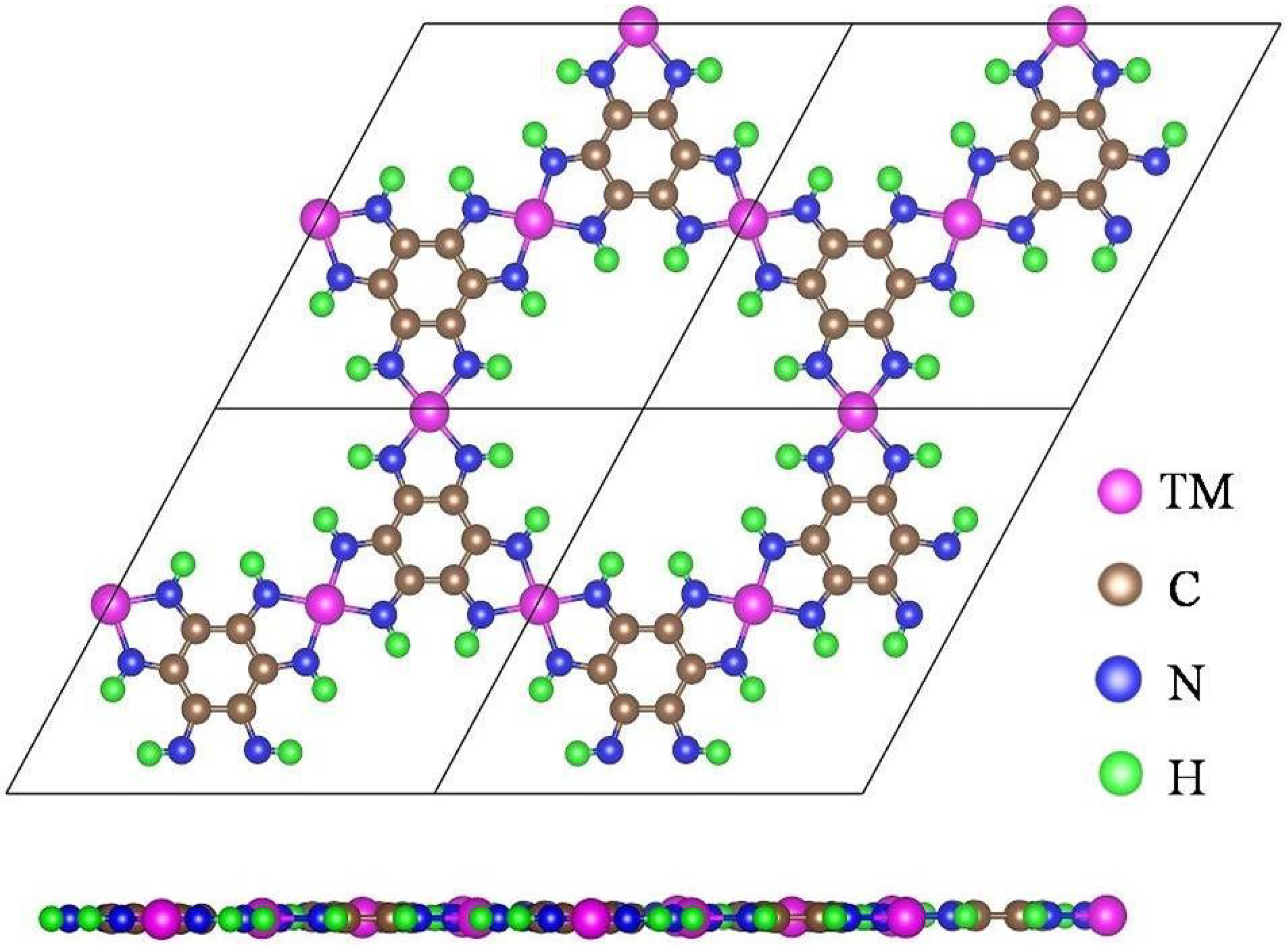
3.1. Structural Features and Properties of the TM-HAB Monolayer
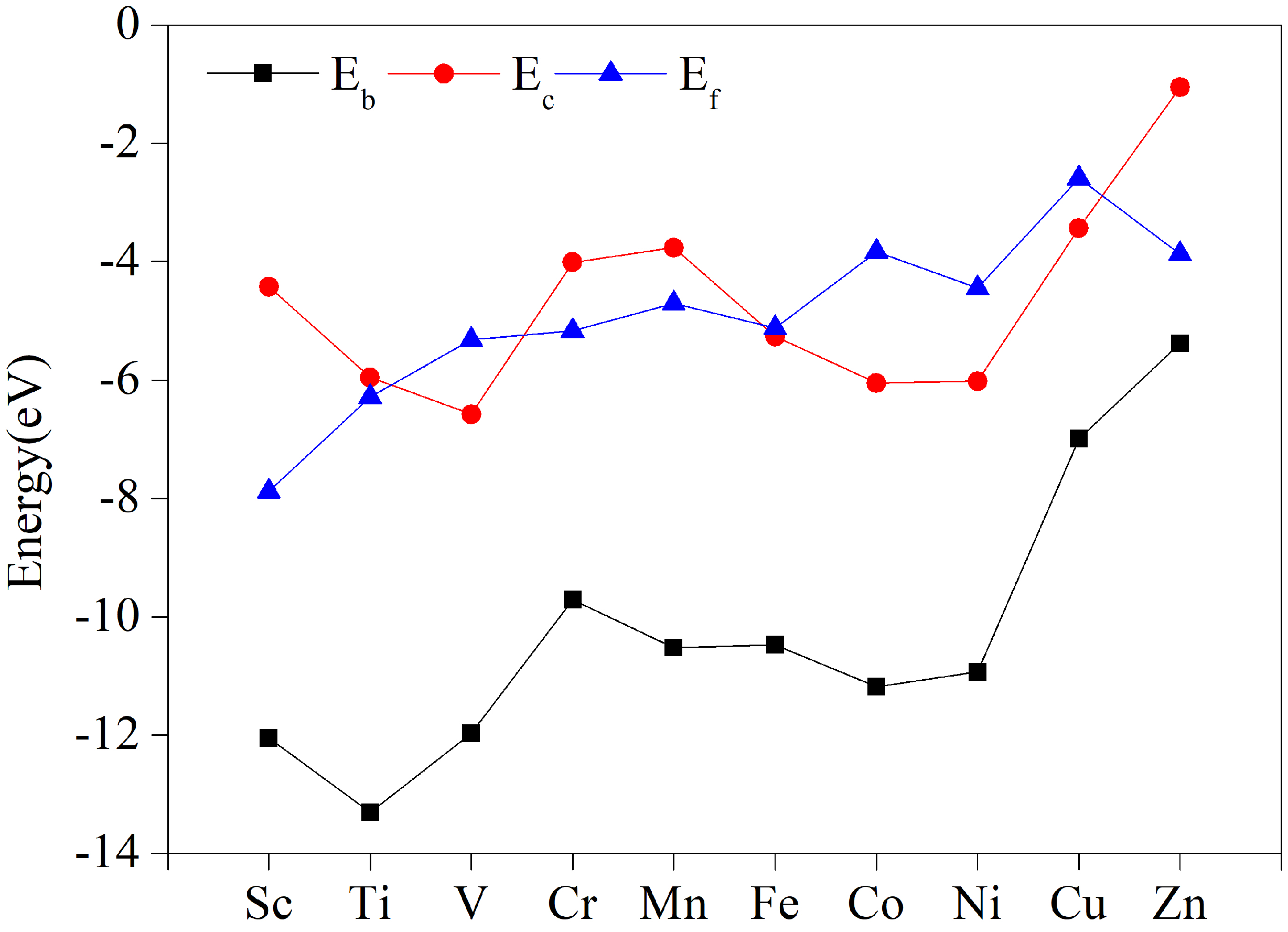
3.2. Stabilization of TM-HAB Monolayer
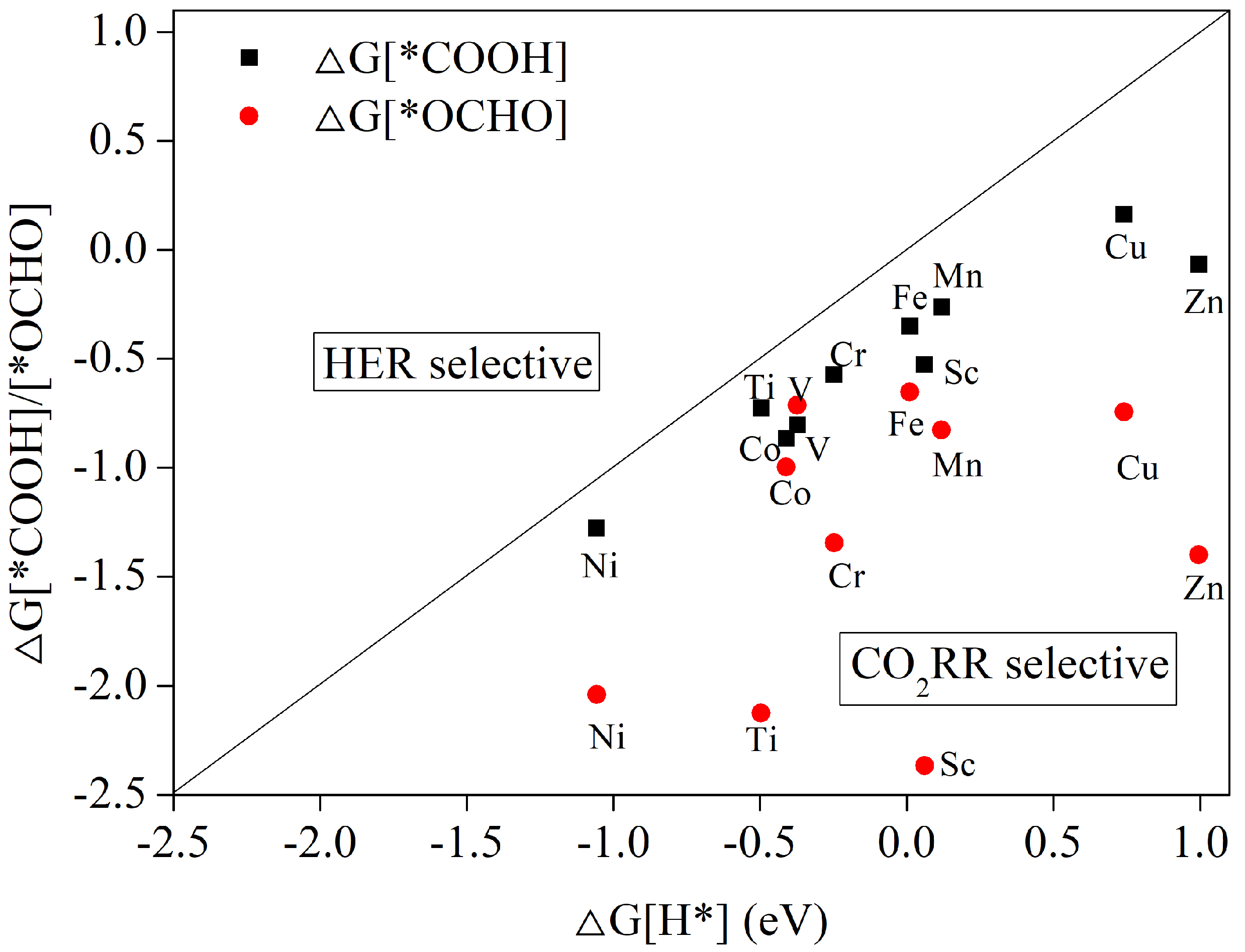
3.3. The First Hydrogenation Step: Selectivity for CO2RR vs. HER
3.4. Possible Product Pathways and Adsorption Energy
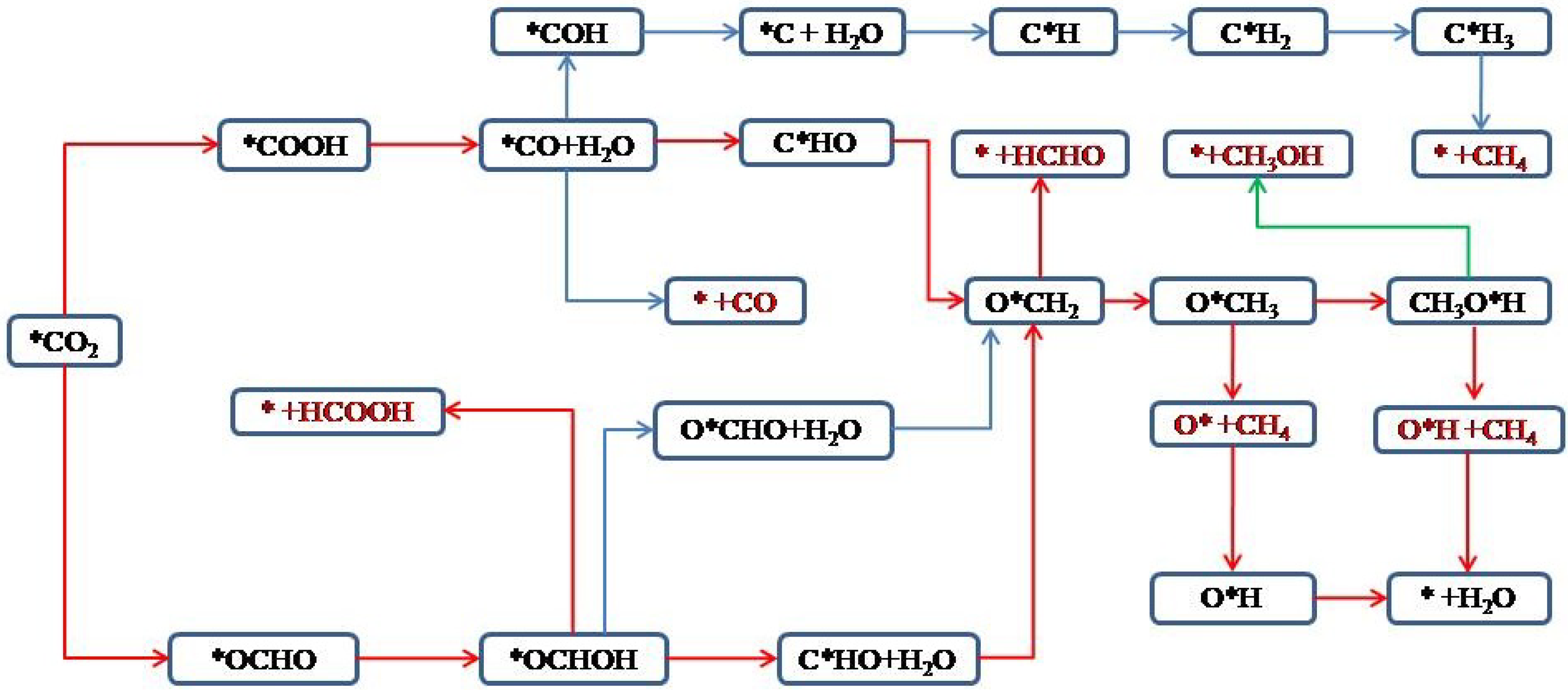
3.5. Reaction Pathways for CO2 Electrochemical Reduction
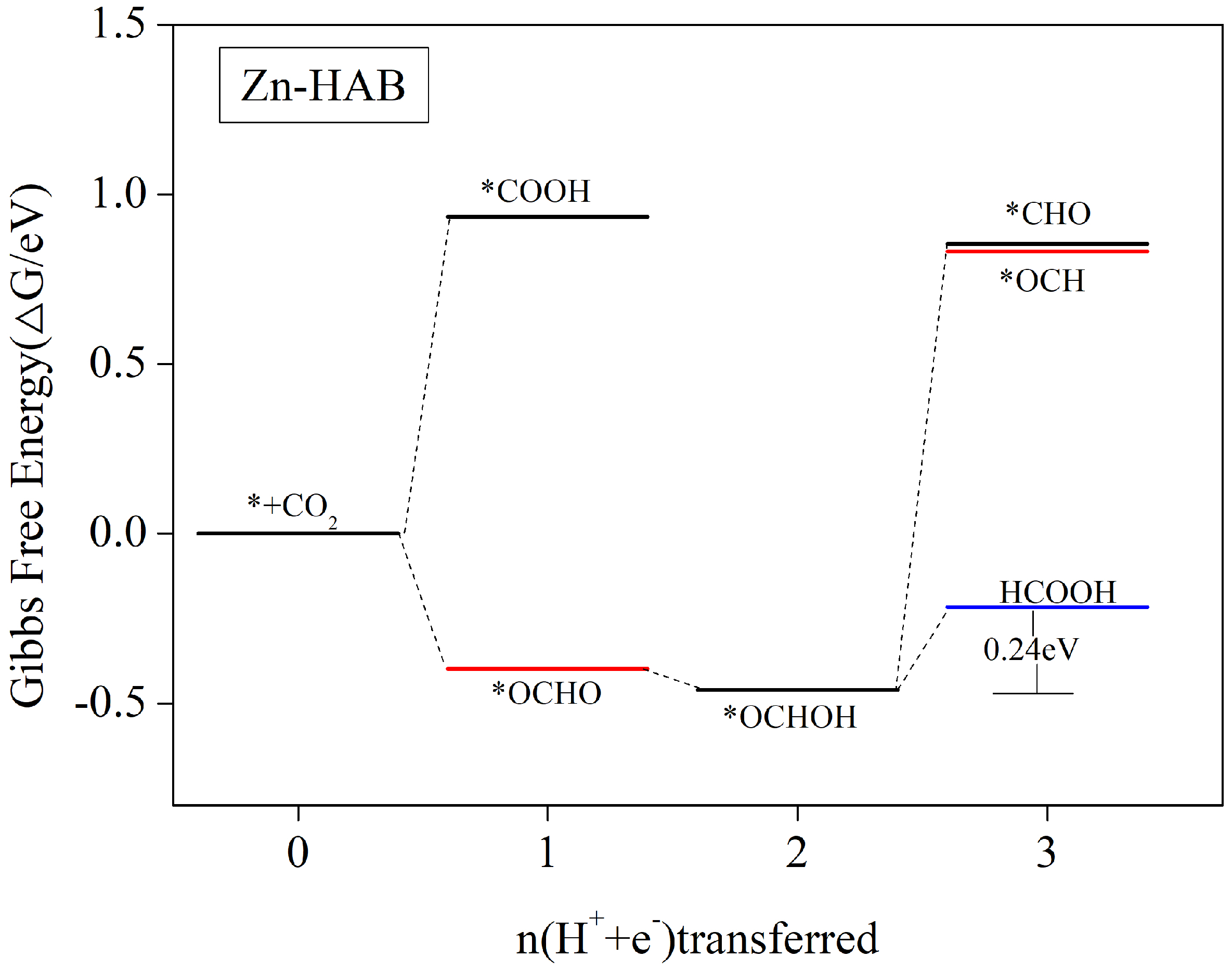
3.5.1. HCOOH as the Main Catalytic Product
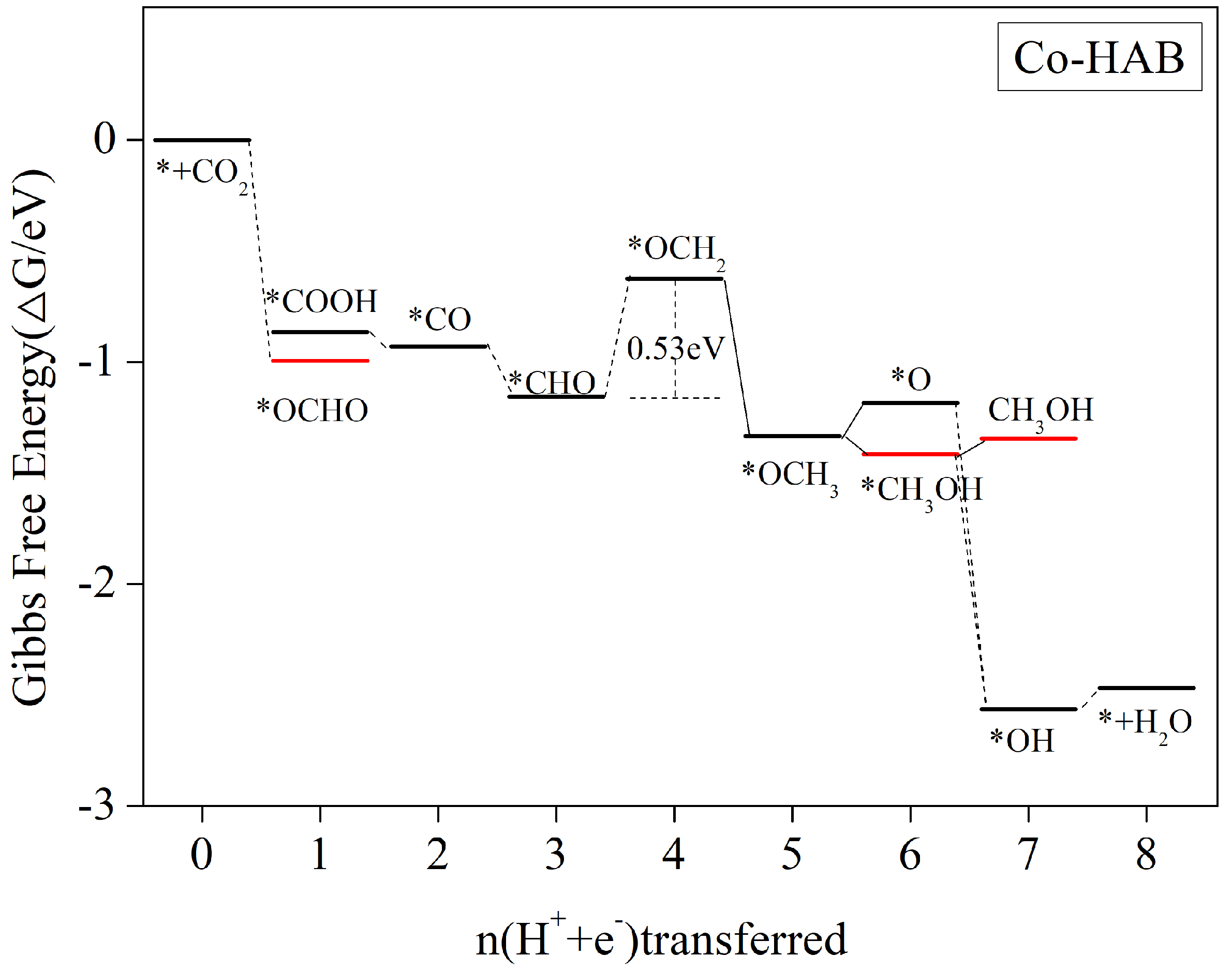
3.5.2. CH3OH and CH4 Are Produced Simultaneously as the Main Reduction Products
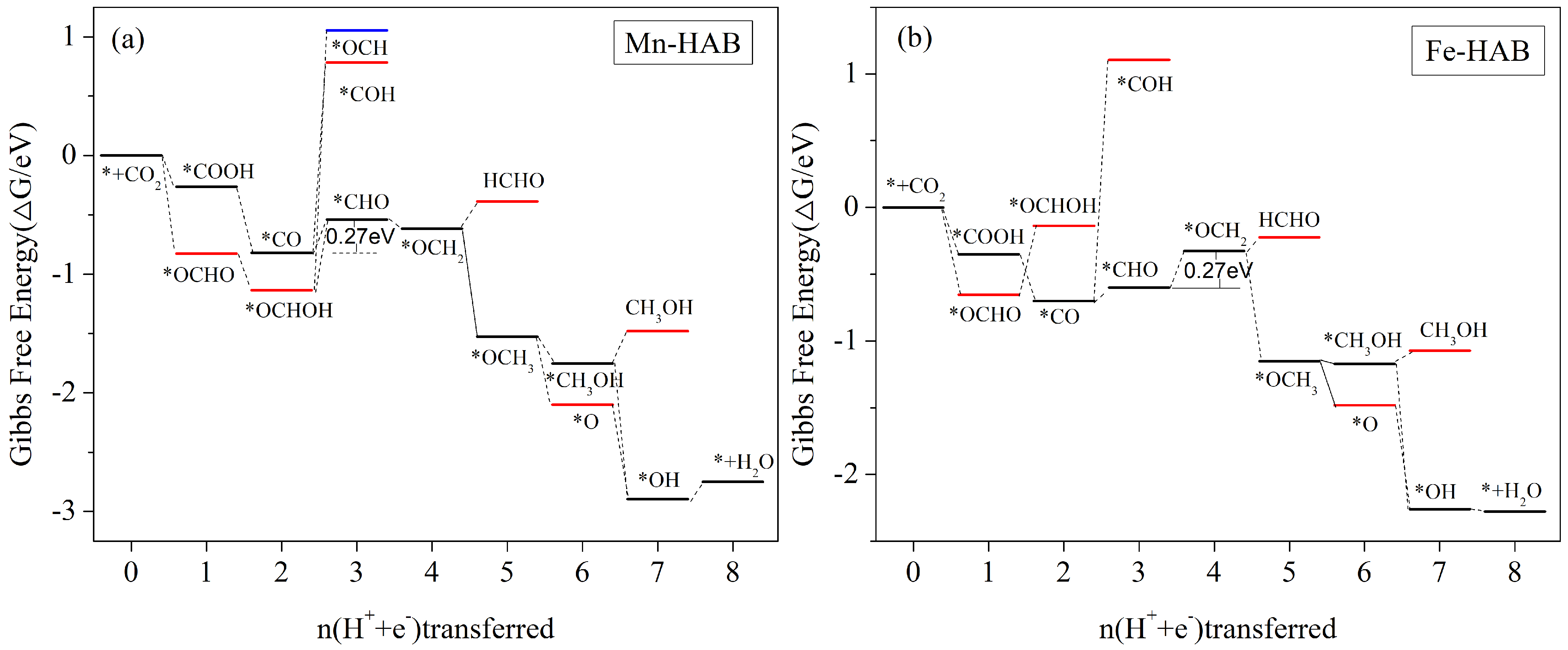
3.5.3. HCHO, CH3OH, and CH4 Are Produced Simultaneously as the Main Reduction Products
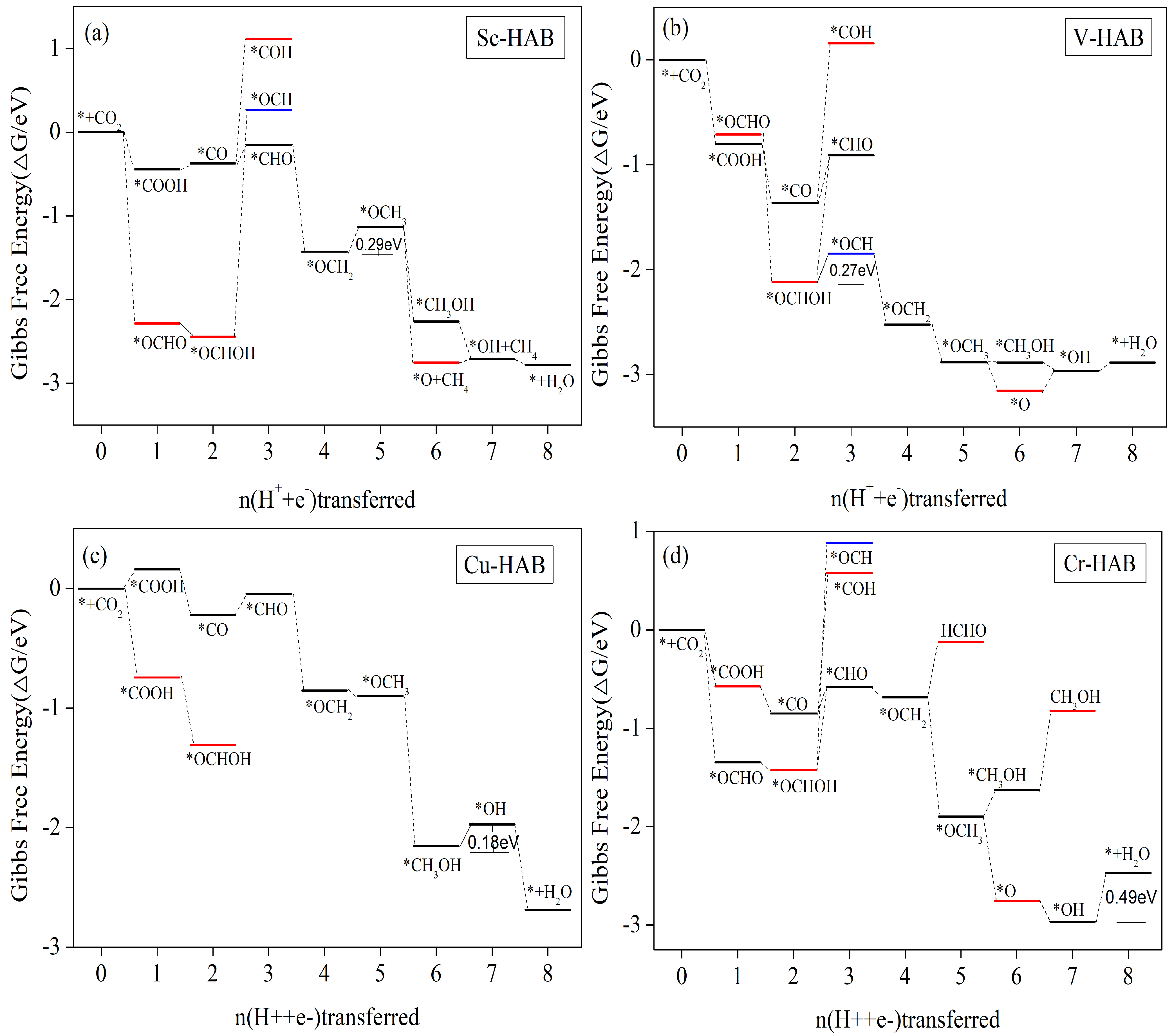
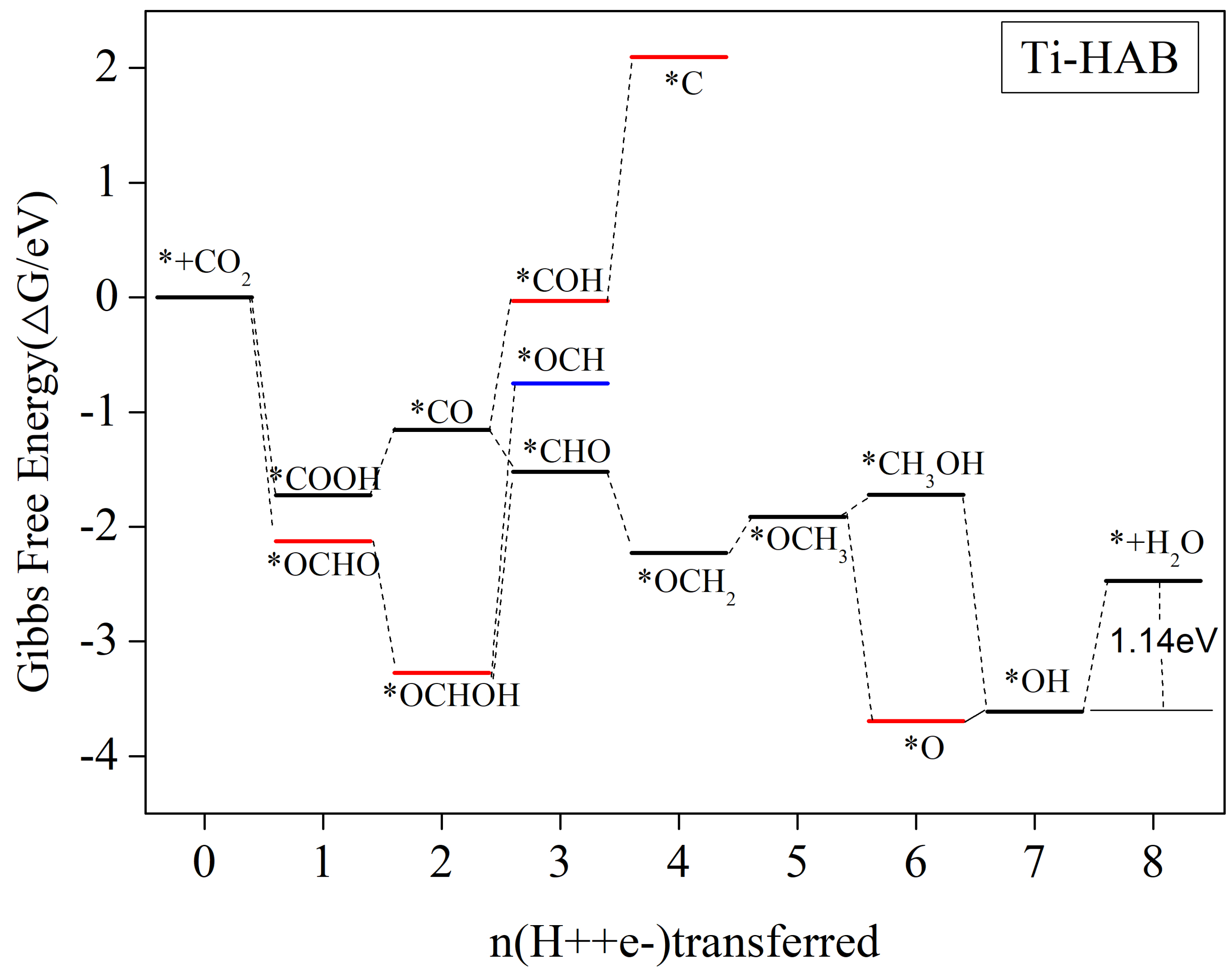
3.5.4. CH4 as the Main Catalytic Product
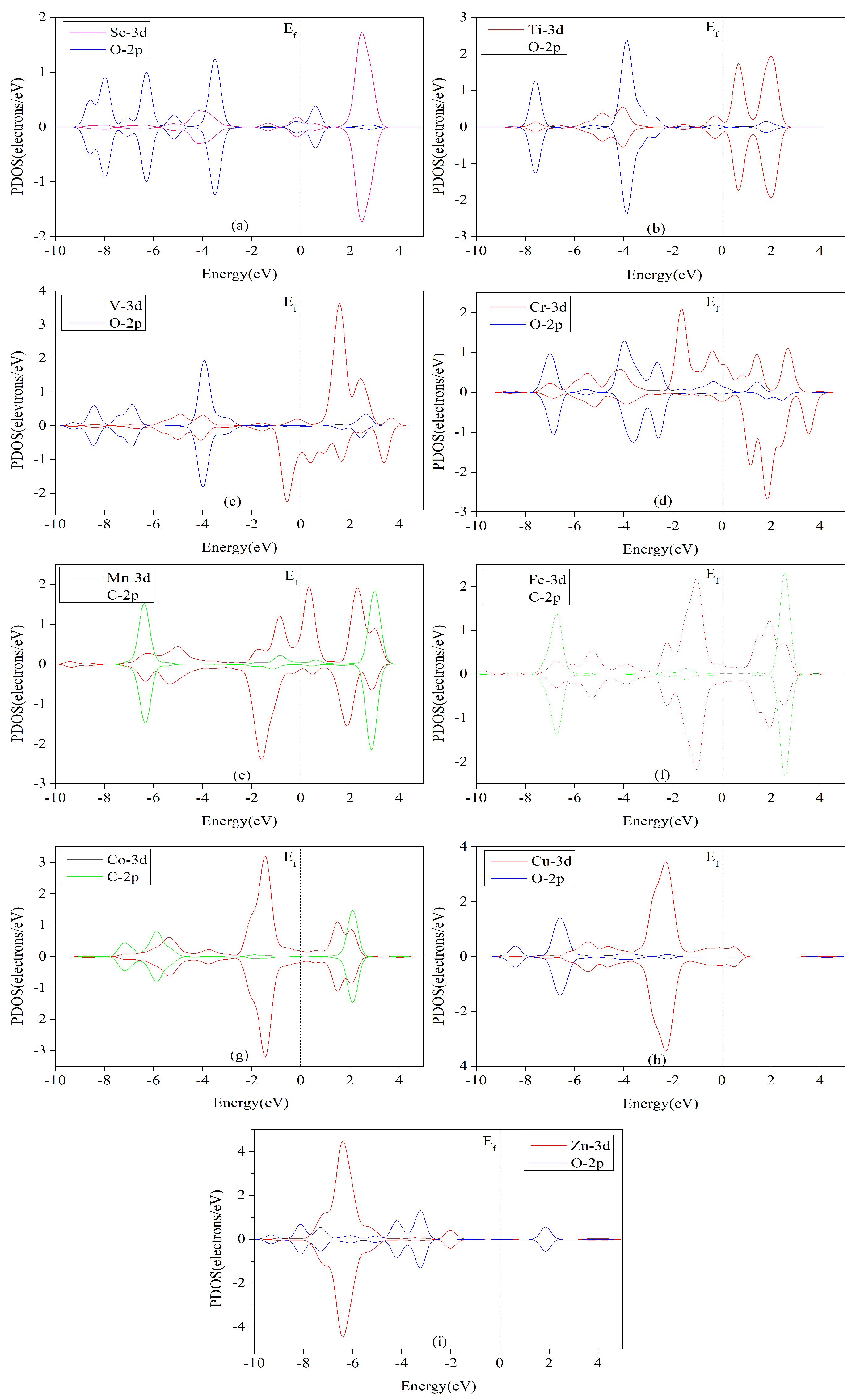
3.6. Electronic Structure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- He, M.; Sun, Y.; Han, B. Green carbon science: Scientific basis for the integration of efficient carbon resource processing, utilization and recycling. Angew. Chem. Int. Ed. 2013, 52, 9620–9633. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, D.M.; Smit, B.; Long, J.R. Carbon dioxide capture: Prospects for new materials. Angew. Chem. Int. Ed. 2010, 49, 6058–6082. [Google Scholar] [CrossRef] [PubMed]
- Pires, J.C.M.; Martins, F.G.; Alvim-Ferraz, M.C.M.; Simões, M. Recent developments on carbon capture and storage: An overview. Chem. Eng. Res. Des. 2011, 88, 1446–1460. [Google Scholar] [CrossRef]
- Whipple, D.; Kenis, P. Prospects of CO2 utilization via direct heterogeneous electrochemical reduction. J. Phys. Chem. Lett. 2010, 1, 3451–3458. [Google Scholar] [CrossRef]
- Kim, S.K.; Zhang, Y.J.; Bergstrom, H.; Michalsky, R.; Peterson, A. Understanding the Low-Overpotential Production of CH4 from CO2 on Mo2C Catalysts. ACS Catal. 2016, 6, 2003–2013. [Google Scholar] [CrossRef]
- Li, N.; Chen, X.; Ong, W.J.; MacFarlane, D.R.; Zhao, X.; Cheetham, A.K.; Sun, C. Understanding of Electrochemical Mechanisms for CO2 Capture and Conversion into Hydrocarbon Fuels in Transition-Metal Carbides (MXenes). ACS Nano 2017, 11, 10825–10833. [Google Scholar] [CrossRef]
- Liu, C.; Yang, B.; Tyo, E.; Seifert, S.; DeBartolo, J.; von Issendorff, B.; Curtiss, L.A. Carbon Dioxide Conversion to Methanol over Size-Selected Cu4 Clusters at Low Pressures. J. Am. Chem. Soc. 2015, 137, 8676–8679. [Google Scholar] [CrossRef]
- Zaman, S.F.; Pasupulety, N.; Al-Zahrani, A.A.; Daous, M.A.; Al-Shahrani, S.S.; Inokawa, H.; Driss, H. Efficient homogeneous catalysis in the reduction of CO2 to CO. J. Am. Chem. Soc. 2005, 127, 17196–17197. [Google Scholar] [CrossRef]
- Bai, S.; Jiang, J.; Zhang, Q.; Xiong, Y. Steering charge kinetics in photocatalysis: Intersection of materials syntheses, characterization techniques and theoretical simulations. Chem. Soc. Rev. 2015, 44, 2893–2939. [Google Scholar] [CrossRef]
- Costentin, C.; Robert, M.; Savéant, J.M. Catalysis of the electrochemical reduction of carbon dioxide. Chem. Soc. Rev. 2013, 42, 2423–2436. [Google Scholar] [CrossRef]
- Xu, S.W.; Lu, Y.; Li, J.; Jiang, Z.Y.; Wu, H. Efficient Conversion of CO2 to Methanol Catalyzed by Three Dehydrogenases Co-encapsulated in an Alginate–Silica (ALG–SiO2) Hybrid Gel. Ind. Eng. Chem. Res. 2006, 45, 4567–4573. [Google Scholar] [CrossRef]
- Lu, Q.; Rosen, J.; Zhou, Y.; Hutchings, G.S.; Kimmel, Y.C.; Chen, J.G.; Jiao, F. A selective and efficient electrocatalyst for carbon dioxide reduction. Nat. Commun. 2014, 5, 3242. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Li, Z.; Searles, D.J.; Chen, Y.; Lu, G.M.; Du, A. Charge-Controlled Switchable CO2 Capture on Boron Nitride Nanomaterials. J. Am. Chem. Soc. 2013, 135, 8246–8253. [Google Scholar] [CrossRef]
- Qin, G.Q.; Du, A.J.; Sun, Q. Charge- and Electric-Field-Controlled Switchable Carbon Dioxide Capture and Gas Separation on a C2N Monolayer. Energy Technol. 2018, 6, 205–212. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, M.; Li, Z.; Du, A.; Searles, D.J. Carbon Dioxide Capture and Gas Separation on B80 Fullerene. J. Phys. Chem. C 2014, 118, 2170–2177. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, M.; Li, Z.; Du, A.; Searles, D.J. A computational study of carbon dioxide adsorption on solid boron. Phys. Chem. Chem. Phys. 2014, 16, 12695–12702. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, M.; Li, Z.; Ma, Y.; Du, A. CO2 capture and gas separation on boron carbon nanotubes. Chem. Phys. Lett. 2013, 575, 59–66. [Google Scholar] [CrossRef]
- Qin, G.; Cui, Q.; Wang, W.; Li, P.; Du, A.; Sun, Q. First-Principles Study of Electrocatalytically Reversible CO2 Capture on Graphene-like C3N. ChemPhysChem 2018, 19, 2788–2795. [Google Scholar] [CrossRef]
- Peterson, A.A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Nørskov, J.K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 2010, 3, 1311–1315. [Google Scholar] [CrossRef]
- Gao, D.; Zhang, Y.; Zhou, Z.; Cai, F.; Zhao, X.; Huang, W.; Li, Y.; Zhu, J.; Liu, P.; Yang, F.; et al. Enhancing CO2 Electroreduction with the Metal–Oxide Interface. J. Am. Chem. Soc. 2017, 139, 5652–5655. [Google Scholar] [CrossRef]
- Back, S.; Lim, J.; Kim, N.Y.; Kim, Y.H.; Jung, Y. Single-atom catalysts for CO2 electroreduction with significant activity and selectivity improvements. Chem. Sci. 2017, 8, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, H.; He, J.; Liu, H.; Song, L.; Li, L.; Luo, J. Highly Active, Durable Ultrathin MoTe2 Layers for the Electroreduction of CO2 to CH4. Small 2018, 14, 1704049. [Google Scholar] [CrossRef] [PubMed]
- Li, P.Z.; Wang, X.J.; Liu, J.; Phang, H.S.; Li, Y.; Zhao, Y. Highly Effective Carbon Fixation via Catalytic Conversion of CO2 by an Acylamide-Containing Metal–Organic Framework. Chem. Mater. 2017, 29, 9256–9261. [Google Scholar] [CrossRef]
- Zhang, W.; Zheng, W. Single Atom Excels as the Smallest Functional Material. Adv. Funct. Mater. 2016, 26, 2988–2993. [Google Scholar] [CrossRef]
- Ling, C.; Li, Q.; Du, A.; Wang, J. Computation-Aided Design of Single-Atom Catalysts for One-Pot CO2 Capture, Activation, and Conversion. ACS Appl. Mater. Interfaces 2018, 10, 36866–36872. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhao, J.; Cai, Q. CO2 electroreduction performance of a single transition metal atom supported on porphyrin-like graphene: A computational study. Phys. Chem. Chem. Phys. 2017, 19, 23113–23121. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Zhang, T. Singleatom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef]
- Lin, J.; Wang, A.; Qiao, B.; Liu, X.; Yang, X.; Wang, X.; Liang, J.; Li, J.; Liu, J.; Zhang, T. Remarkable Performance of Ir1/FeOx Single-Atom Catalyst in Water Gas Shift Reaction. J. Am. Chem. Soc. 2013, 135, 15314–15317. [Google Scholar] [CrossRef]
- Yang, X.F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-Atom Catalysts: A New Frontier in Heterogeneous Catalysis. Accounts Chem. Res. 2013, 46, 1740–1748. [Google Scholar] [CrossRef]
- Liu, N.; Huang, W.; Zhang, X.; Tang, L.; Wang, L.; Wang, Y.; Wu, M. Ultrathin graphene oxide encapsulated in uniform MIL-88A(Fe) for enhanced visible light-driven photodegradation of RhB. Appl. Catal. B Environ. 2018, 221, 119–128. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Y.; Huang, W.; Yang, Y.; Wang, Y.; He, C.; Tang, L. g-C3N4/UiO-66 nanohybrids with enhanced photocatalytic activities for the oxidation of dye under visible light irradiation. Mater. Res. Bull. 2018, 99, 349–358. [Google Scholar] [CrossRef]
- Huang, W.; Liu, N.; Zhang, X.; Wu, M.; Tang, L. Metal organic framework g-C3N4/MIL-53(Fe) heterojunctions with enhanced photocatalytic activity for Cr(VI) reduction under visible light. Appl. Surf. Sci. 2017, 425, 107–116. [Google Scholar] [CrossRef]
- Li, J.R.; Kuppler, R.J.; Zhou, H.C. Selective gas adsorption and separation in metal–organic frameworks. Chem. Soc. Rev. 2009, 38, 1477–1504. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Y.; Song, L.; Chen, J.; Yang, Y.; Wang, Y. Enhanced adsorption performance of gaseous toluene on defective UiO-66 metal organic framework: Equilibrium and kinetic studies. J. Hazard. Mater. 2019, 365, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, Y.; Lv, X.; Wang, Y.; Liu, N.; Chen, D.; Cui, L. Adsorption/desorption kinetics and breakthrough of gaseous toluene for modified microporous-mesoporous UiO-66 metal organic framework. J. Hazard. Mater. 2019, 366, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lv, X.; Shi, X.; Yang, Y.; Yang, Y. Enhanced hydrophobic UiO-66 (University of Oslo 66) metal-organic framework with high capacity and selectivity for toluene capture from high humid air. J. Colloid Interface Sci. 2019, 539, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Huang, W.; Tang, M.; Yin, C.; Gao, B.; Li, Z.; Tang, L.; Lei, J.; Cui, L.; Zhang, X. In-situ fabrication of needle-shaped MIL-53(Fe) with 1T-MoS2 and study on its enhanced photocatalytic mechanism of ibuprofen. Chem. Eng. J. 2019, 359, 254–264. [Google Scholar] [CrossRef]
- Cui, Q.; Qin, G.; Wang, W.; Geethalakshmi, K.R.; Du, A.; Sun, Q. Mo-based 2D MOF as a highly efficient electrocatalyst for reduction of N2 to NH3: A density functional theory study. J. Mater. Chem. A 2019, 7, 14510–14518. [Google Scholar] [CrossRef]
- Zhang, X.; Hou, F.; Yang, Y.; Wang, Y.; Liu, N.; Chen, D.; Yang, Y. A facile synthesis for cauliflower like CeO2 catalysts from Ce-BTC precursor and their catalytic performance for CO oxidation. Appl. Surf. Sci. 2017, 423, 771–779. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, X.; Song, L.; Hou, F.; Yang, Y.; Wang, Y.; Liu, N. Enhanced catalytic performance for CO oxidation and preferential CO oxidation over CuO/CeO2 catalysts synthesized from metal organic framework: Effects of preparation methods. Int. J. Hydrog. Energy 2018, 43, 18279–18288. [Google Scholar] [CrossRef]
- Zhang, X.; Li, H.; Hou, F.; Yang, Y.; Dong, H.; Liu, N.; Wang, Y.; Cui, L. Synthesis of highly efficient Mn2O3 catalysts for CO oxidation derived from Mn-MIL-100. Appl. Surf. Sci. 2017, 411, 27–33. [Google Scholar] [CrossRef]
- Yang, Y.; Dong, H.; Wang, Y.; He, C.; Wang, Y.; Zhang, X. Synthesis of octahedral like Cu-BTC derivatives derived from MOF calcined under different atmosphere for application in CO oxidation. J. Solid State Chem. 2018, 258, 582–587. [Google Scholar] [CrossRef]
- Kattel, S.; Wang, G. Reaction Pathway for Oxygen Reduction on FeN4 Embedded Graphene. J. Phys. Chem. Lett. 2014, 5, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Li, X.F.; Li, Q.K.; Cheng, J.; Liu, L.; Yan, Q.; Wu, Y.; Zhang, X.H.; Wang, Z.Y.; Qiu, Q.; Luo, Y. Conversion of Dinitrogen to Ammonia by FeN3-Embedded Graphene. J. Am. Chem. Soc. 2016, 138, 8706–8709. [Google Scholar] [CrossRef]
- Mahmood, J.; Li, F.; Kim, C.; Choi, H.J.; Gwon, O.; Jung, S.M.; Seo, J.M.; Cho, S.J.; Ju, Y.W.; Jeong, H.Y.; et al. Fe@C2N: A highly-efficient indirect-contact oxygen reduction catalyst. Nano Energy 2018, 44, 304–310. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, Y.; Yan, L.; Zhao, J.; Su, Z. Electrochemical reduction of carbon dioxide on the two–dimensional M3(Hexaiminotriphenylene)2 sheet: A computational study. Appl. Surf. Sci. 2019, 467, 98–103. [Google Scholar] [CrossRef]
- Lahiri, N.; Lotfizadeh, N.; Tsuchikawa, R.; Deshpande, V.V.; Louie, J. Hexaaminobenzene as a building block for a Family of 2D Coordination Polymers. J. Am. Chem. Soc. 2017, 139, 19–22. [Google Scholar] [CrossRef]
- Park, J.; Chen, Z.; Flores, R.A.; Wallnerstrom, G.; Kulkarni, A.; Nørskov, J.K.; Bao, Z. Two-Dimensional Conductive Ni-HAB as a Catalyst for the Electrochemical Oxygen Reduction Reaction. ACS Appl. Mater. Interfaces 2020, 12, 39074–39081. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ernzerhof, M. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Sun, C.; Du, A.; Dou, S.; Li, Z. In-plane graphene/boron-nitride heterostructures as an efficient metal-free electrocatalyst for the oxygen reduction reaction. Nanoscale 2016, 8, 14084–14091. [Google Scholar] [CrossRef] [PubMed]
- Qin, G.; Du, A.; Sun, Q. A theoretical insight into a feasible strategy for the fabrication of borophane. Phys. Chem. Chem. Phys. 2018, 20, 16216–16221. [Google Scholar] [CrossRef] [PubMed]
- Qin, G.; Cui, Q.; Yun, B.; Sun, L.; Du, A.; Sun, Q. High capacity and reversible hydrogen storage on two dimensional C2N monolayer membrane. Int. J. Hydrogen Energy 2018, 43, 9895–9901. [Google Scholar] [CrossRef]
- Delley, B. Hardness conserving semilocal pseudopotentials. Phys. Rev. B 2002, 66, 155125. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 1993, 2, 799–805. [Google Scholar] [CrossRef]
- Back, S.; Min, S.Y.; Jung, Y. Active Sites of Au and Ag Nanoparticle Catalysts for CO2 Electroreduction to CO. Am. Chem. Soc. 2015, 5, 5089–5096. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Logadottir, A.; Nørskov, J.K. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 2005, 319, 178–184. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Liu, J.H.; Yang, L.M.; Ganz, E. Electrochemical reduction of CO2 by single atom catalyst TM–TCNQ monolayers. J. Mater. Chem. A 2019, 7, 3805–3814. [Google Scholar] [CrossRef]
- Santos-Lorenzo, J.; San José-Velado, R.; Albo, J.; Beobide, G.; Castaño, P.; Castillo, O.; Luque, A.; Pérez-Yáñez, S. A straightforward route to obtain zirconium based metal-organic gels. Microporous Mesoporous Mater. 2019, 284, 128–132. [Google Scholar] [CrossRef]
- Shi, C.; Chan, K.; Yoo, J.S.; Nørskov, J.K. Barriers of Electrochemical CO2 Reduction on Transition Metals. Org. Process. Res. Dev. 2016, 20, 1424–1430. [Google Scholar] [CrossRef]
- Li, M.; Yan, C.; Ramachandran, R.; Lan, Y.; Dai, H.; Shan, H.; Meng, X.; Cui, D.; Wang, F.; Xu, Z.X. Non-peripheral octamethyl-substituted cobalt phthalocyanine nanorods supported on N-doped reduced graphene oxide achieve efficient electrocatalytic CO2 reduction to CO. Chem. Eng. J. 2022, 430, 133050. [Google Scholar] [CrossRef]
- Merino-Garcia, I.; Albo, J.; Krzywda, P.; Mul, G.; Irabien, A. Bimetallic Cu-based hollow fibre electrodes for CO2 electroreduction. Catal. Today 2020, 346, 34–39. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TM-HAB | QTM | Spin-TM | QN/e | RTM-N/Å |
|---|---|---|---|---|
| Sc | 0.708 | 0.000 | −0.259 | 2.110 |
| Ti | 0.588 | 0.000 | −0.236 | 1.962 |
| V | 0.349 | −1.935 | −0.202 | 1.959 |
| Cr | 0.446 | 0.000 | −0.222 | 1.929 |
| Mn | 0.321 | −3.225 | −0.203 | 1.876 |
| Fe | 0.132 | 0.000 | −0.162 | 1.924 |
| Co | 0.049 | 0.000 | −0.153 | 1.837 |
| Ni | 0.052 | 0.000 | −0.147 | 1.838 |
| Cu | 0.324 | 0.000 | −0.208 | 1.945 |
| Zn | 0.394 | 0.000 | −0.220 | 2.032 |
| TM-HAB | CO | HCOOH | HCHO | CH3OH | CH4 |
|---|---|---|---|---|---|
| Sc-HAB | −1.263 | −1.432 | −0.998 | −1.361 | −0.100 |
| Ti-HAB | −2.767 | −2.013 | −2.461 | −2.334 | −0.848 |
| V-HAB | −1.910 | −0.804 | −1.296 | −1.258 | −0.125 |
| Cr-HAB | −2.209 | −1.558 | −0.161 | −0.838 | −0.109 |
| Mn-HAB | −1.617 | −0.081 | −0.073 | −0.274 | −0.243 |
| Fe-HAB | −0.165 | −0.073 | −0.070 | −0.123 | −0.071 |
| Co-HAB | −1.794 | −0.895 | −1.008 | −0.781 | −0.525 |
| Ni-HAB | −2.309 | −1.994 | −1.825 | −2.275 | −2.044 |
| Cu-HAB | −0.547 | 0.053 | 0.068 | −0.473 | −0.553 |
| Zn-HAB | −0.218 | −0.109 | −0.151 | −0.185 | −0.106 |
| TM-HAB | PDS | UL/V | Main Products and Corresponding Overpotentials (/V) |
|---|---|---|---|
| Sc-HAB | *OCH2 + H2O + H+ + e− → *OCH3 + H2O | −0.29 | CH4(0.46) |
| Ti-HAB | *OH + CH4 + H2O + H+ + e− → * + CH4 + 2H2O | −1.14 | CH4(1.31) |
| V-HAB | *OCHOH + H+ + e− → *OCH + H2O | −0.27 | CH4(0.44) |
| Cr-HAB | *OH + CH4 + H2O + H+ + e− → * + CH4 + 2H2O | −0.27 | CH4(0.66) |
| Mn-HAB | *CO + H2O + H+ + e− → *CHO + 2H2O | −0.27 | HCHO(0.2),CH3OH(0.29),CH4(0.44) |
| Fe-HAB | *CO + H2O + H+ + e− → *CHO + 2H2O | −0.27 | HCHO(0.2),CH3OH(0.29),CH4(0.44) |
| Co-HAB | *CHO + H2O + H+ + e− → *OCH2 + 2H2O | −0.53 | CH3OH(0.55),CH4(0.70) |
| Cu-HAB | *CH3OH + H2O + H+ + e− → *OH + CH4 + H2O | −0.18 | CH4(0.35) |
| Zn-HAB | *OCHOH → * + HCOOH | 0.24 | HCOOH(0.01) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, X.; Tu, Z.; Yuan, Y.; Liao, L.; Xiao, C.; Wen, Y.; Xiong, K. Two-Dimensional Transition Metal-Hexaaminobenzene Monolayer Single-Atom Catalyst for Electrocatalytic Carbon Dioxide Reduction. Nanomaterials 2022, 12, 4005. https://doi.org/10.3390/nano12224005
Zeng X, Tu Z, Yuan Y, Liao L, Xiao C, Wen Y, Xiong K. Two-Dimensional Transition Metal-Hexaaminobenzene Monolayer Single-Atom Catalyst for Electrocatalytic Carbon Dioxide Reduction. Nanomaterials. 2022; 12(22):4005. https://doi.org/10.3390/nano12224005
Chicago/Turabian StyleZeng, Xianshi, Zongxing Tu, Yanli Yuan, Luliang Liao, Chuncai Xiao, Yufeng Wen, and Kai Xiong. 2022. "Two-Dimensional Transition Metal-Hexaaminobenzene Monolayer Single-Atom Catalyst for Electrocatalytic Carbon Dioxide Reduction" Nanomaterials 12, no. 22: 4005. https://doi.org/10.3390/nano12224005
APA StyleZeng, X., Tu, Z., Yuan, Y., Liao, L., Xiao, C., Wen, Y., & Xiong, K. (2022). Two-Dimensional Transition Metal-Hexaaminobenzene Monolayer Single-Atom Catalyst for Electrocatalytic Carbon Dioxide Reduction. Nanomaterials, 12(22), 4005. https://doi.org/10.3390/nano12224005