
Tensile and Viscoelastic Behavior in Nacre-Inspired Nanocomposites: A Coarse-Grained Molecular Dynamics Study


Abstract
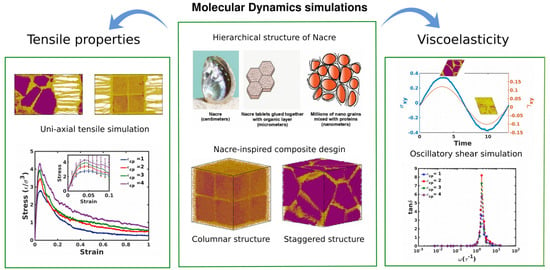
1. Introduction
2. Computational and Analysis Methods
2.1. Model Systems
2.1.1. Polymer Domain
2.1.2. Crystalline Domain
2.1.3. Creation of Composites
2.2. Equilibration Protocol
2.3. Non-Equilibrium Simulations
2.3.1. Uniaxial Tension
2.3.2. Oscillatory Shear Simulation
2.4. Analysis
2.4.1. Radius of Gyration
2.4.2. Bond Orientation Parameter
2.4.3. Polymer-Crystal Interfacial Coordination Number
2.4.4. Mean Square Displacement (MSD)
2.4.5. Polymer Chain Relaxation and Vibrational Density of States (VDOS)
3. Results and Discussion
3.1. Glass-Transition Temperature and Polymer Chain Statistics
3.2. Tensile Properties of Composites
Mechanisms Responsible for Tensile Behavior
3.3. Viscoelastic Behavior
3.3.1. Effect of Strain Amplitude
3.3.2. Effect of Deformation Frequency
3.3.3. Domain-Dependent Stress Response
3.3.4. Mechanisms Responsible for Viscoelasticity
- (a)
- Polymer chain dynamics
- (b)
- Rouse mode analysis
- (c)
- Vibrational Density of States
4. Conclusions
- The mechanical properties of biomimetic, nacre-like composites can be highly tunable for certain morphologies such as the staggered composite, especially as a function of the polymer-crystal interaction strength . The Young’s modulus, and the tensile strength increased with higher .
- Conformational analysis of polymer chains during inelastic deformation exhibited the significant role played by on the deformation behavior of composites. The weakly adsorbed polymers on the crystallites exhibited notable deformation as verified by and parameters during tensile deformation.
- The rough crystallite surface in the staggered composite dramatically arrests and delays the dynamics of polymer chains in the vicinity of the crystallites, exhibiting a notable increment in the glass-transition temperature. This, in turn, affects mechanical behavior.
- A detailed study of viscoelastic properties of the composites indicated a increment in the LVER for the staggered composite in comparison to the columnar structure. Additionally, it was observed that plays a significant role in the stiffness and dissipative characteristics of the staggered composite.
- The dissipative behavior of the nacre-like composites is very sensitive to the deformation frequency and can be tuned by tuning the microstructure. The maximum loss tangent for the staggered composite was found to be 8, which was 40 times higher than that for the columnar structure, which in turn is 34 times lower with respect to the homogeneous polymer.
- At frequencies corresponding to maximum damping, about 15–30% of the overall stress was supported by the polymer domain.
- The dynamics of polymer chains were substantially restricted in the cage-like confined regions. The mean square displacement and Rouse mode analysis of polymeric chains essentially showed a solid-like behavior, corroborating its highly confined nature. Finally, the large damping effects, especially at high deformation frequencies in nacre-like composites, are a direct consequence of the vibrational properties of the constituent atoms.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Currey, J.D. Mechanical properties of mother of pearl in tension. Proc. R. Soc. Lond. Ser. B. Biol. Sci. 1977, 196, 443–463. [Google Scholar]
- Song, F.; Soh, A.; Bai, Y. Structural and mechanical properties of the organic matrix layers of nacre. Biomaterials 2003, 24, 3623–3631. [Google Scholar] [CrossRef]
- Sun, J.; Bhushan, B. Hierarchical structure and mechanical properties of nacre: A review. Rsc Adv. 2012, 2, 7617–7632. [Google Scholar] [CrossRef]
- Checa, A.G.; Cartwright, J.H.; Willinger, M.G. Mineral bridges in nacre. J. Struct. Biol. 2011, 176, 330–339. [Google Scholar] [CrossRef]
- Barthelat, F. Architectured materials in engineering and biology: Fabrication, structure, mechanics and performance. Int. Mater. Rev. 2015, 60, 413–430. [Google Scholar] [CrossRef]
- Porter, M.M.; McKittrick, J. It’s tough to be strong: Advances. Am. Ceram. Soc. Bull. 2014, 93, 18–24. [Google Scholar]
- Zhao, H.; Yang, Z.; Guo, L. Nacre-inspired composites with different macroscopic dimensions: Strategies for improved mechanical performance and applications. NPG Asia Mater. 2018, 10, 1–22. [Google Scholar] [CrossRef]
- Xu, S.; Li, X.; Roseman, R.; Stock, A.M. Crystal Structure of the Chitin Oligasaccharide Binding Protein. Available online: https://www.rcsb.org/structure/4gf8 (accessed on 25 July 2022).
- Jackson, A.; Vincent, J.F.; Turner, R. The mechanical design of nacre. Proc. R. Soc. Lond. Ser. B. Biol. Sci. 1988, 234, 415–440. [Google Scholar]
- Katti, D.R.; Katti, K.S. Modeling microarchitecture and mechanical behavior of nacre using 3D finite element techniques Part I Elastic properties. J. Mater. Sci. 2001, 36, 1411–1417. [Google Scholar] [CrossRef]
- Katti, D.R.; Katti, K.S.; Tang, J.; Sopp, J.M.; Sarikaya, M. Effects of nanostructure on bulk mechanical properties of nacre-3D finite element modeling of the segmented/layered biocomposite. MRS Online Proc. Libr. 2001, 677, 781–786. [Google Scholar] [CrossRef]
- Katti, K.S.; Katti, D.R.; Pradhan, S.M.; Bhosle, A. Platelet interlocks are the key to toughness and strength in nacre. J. Mater. Res. 2005, 20, 1097–1100. [Google Scholar] [CrossRef]
- Katti, K.S.; Katti, D.R. Why is nacre so tough and strong? Mater. Sci. Eng. C 2006, 26, 1317–1324. [Google Scholar] [CrossRef]
- Katti, D.R.; Pradhan, S.M.; Katti, K.S. Modeling the organic-inorganic interfacial nanoasperities in a model bio-nanocomposite, nacre. Rev. Adv. Mater. Sci. 2004, 6, 162–168. [Google Scholar]
- Askarinejad, S.; Rahbar, N. Toughening mechanisms in bioinspired multilayered materials. J. R. Soc. Interface 2015, 12, 20140855. [Google Scholar] [CrossRef] [PubMed]
- Maghsoudi-Ganjeh, M.; Lin, L.; Wang, X.; Wang, X.; Zeng, X. Computational Modeling of the Mechanical Behavior of 3D Hybrid Organic–Inorganic Nanocomposites. JOM 2019, 71, 3951–3961. [Google Scholar] [CrossRef]
- Kotha, S.; Kotha, S.; Guzelsu, N. A shear-lag model to account for interaction effects between inclusions in composites reinforced with rectangular platelets. Compos. Sci. Technol. 2000, 60, 2147–2158. [Google Scholar] [CrossRef]
- Askarinejad, S.; Rahbar, N. Mechanics of bioinspired lamellar structured ceramic/polymer composites: Experiments and models. Int. J. Plast. 2018, 107, 122–149. [Google Scholar] [CrossRef]
- Nairn, J.A.; Mendels, D.A. On the use of planar shear-lag methods for stress-transfer analysis of multilayered composites. Mech. Mater. 2001, 33, 335–362. [Google Scholar] [CrossRef]
- Gao, H.; Ji, B.; Jäger, I.L.; Arzt, E.; Fratzl, P. Materials become insensitive to flaws at nanoscale: Lessons from nature. Proc. Natl. Acad. Sci. USA 2003, 100, 5597–5600. [Google Scholar] [CrossRef]
- Jackson, A.; Vincent, J.; Turner, R. A physical model of nacre. Compos. Sci. Technol. 1989, 36, 255–266. [Google Scholar] [CrossRef]
- Espinosa, H.D.; Rim, J.E.; Barthelat, F.; Buehler, M.J. Merger of structure and material in nacre and bone–Perspectives on de novo biomimetic materials. Prog. Mater. Sci. 2009, 54, 1059–1100. [Google Scholar] [CrossRef]
- Meyers, M.A.; Lin, A.Y.M.; Chen, P.Y.; Muyco, J. Mechanical strength of abalone nacre: Role of the soft organic layer. J. Mech. Behav. Biomed. Mater. 2008, 1, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Suo, Z.; Evans, A.; Yao, N.; Aksay, I.A. Deformation mechanisms in nacre. J. Mater. Res. 2001, 16, 2485–2493. [Google Scholar] [CrossRef]
- Barthelat, F.; Espinosa, H. An experimental investigation of deformation and fracture of nacre–mother of pearl. Exp. Mech. 2007, 47, 311–324. [Google Scholar] [CrossRef]
- Schäffer, T.E.; Ionescu-Zanetti, C.; Proksch, R.; Fritz, M.; Walters, D.A.; Almqvist, N.; Zaremba, C.M.; Belcher, A.M.; Smith, B.L.; Stucky, G.D.; et al. Does abalone nacre form by heteroepitaxial nucleation or by growth through mineral bridges? Chem. Mater. 1997, 9, 1731–1740. [Google Scholar] [CrossRef]
- Evans, A.; Suo, Z.; Wang, R.; Aksay, I.A.; He, M.; Hutchinson, J. Model for the robust mechanical behavior of nacre. J. Mater. Res. 2001, 16, 2475–2484. [Google Scholar] [CrossRef]
- Zuo, S.; Wei, Y. Microstructure observation and mechanical behavior modeling for limnetic nacre. Acta Mech. Sin. 2008, 24, 83–89. [Google Scholar] [CrossRef]
- Li, X.; Chang, W.C.; Chao, Y.J.; Wang, R.; Chang, M. Nanoscale structural and mechanical characterization of a natural nanocomposite material: The shell of red abalone. Nano Lett. 2004, 4, 613–617. [Google Scholar] [CrossRef]
- Bruet, B.; Qi, H.; Boyce, M.; Panas, R.; Tai, K.; Frick, L.; Ortiz, C. Nanoscale morphology and indentation of individual nacre tablets from the gastropod mollusc Trochus niloticus. J. Mater. Res. 2005, 20, 2400–2419. [Google Scholar] [CrossRef]
- Li, X.; Xu, Z.H.; Wang, R. In situ observation of nanograin rotation and deformation in nacre. Nano Lett. 2006, 6, 2301–2304. [Google Scholar] [CrossRef]
- Sumitomo, T.; Kakisawa, H.; Owaki, Y.; Kagawa, Y. Transmission electron microscopy observation of nanoscale deformation structures in nacre. J. Mater. Res. 2008, 23, 3213–3221. [Google Scholar] [CrossRef]
- Zhang, N.; Yang, S.; Xiong, L.; Hong, Y.; Chen, Y. Nanoscale toughening mechanism of nacre tablet. J. Mech. Behav. Biomed. Mater. 2016, 53, 200–209. [Google Scholar] [CrossRef]
- Smith, B.L.; Schäffer, T.E.; Viani, M.; Thompson, J.B.; Frederick, N.A.; Kindt, J.; Belcher, A.; Stucky, G.D.; Morse, D.E.; Hansma, P.K. Molecular mechanistic origin of the toughness of natural adhesives, fibres and composites. Nature 1999, 399, 761–763. [Google Scholar] [CrossRef]
- Ghosh, P.; Katti, D.R.; Katti, K.S. Impact of β-sheet conformations on the mechanical response of protein in biocomposites. Mater. Manuf. Process. 2006, 21, 676–682. [Google Scholar] [CrossRef]
- Zhang, N.; Chen, Y. Molecular origin of the sawtooth behavior and the toughness of nacre. Mater. Sci. Eng. C 2012, 32, 1542–1547. [Google Scholar] [CrossRef]
- Mohanty, B.; Katti, K.S.; Katti, D.R. Experimental investigation of nanomechanics of the mineral-protein interface in nacre. Mech. Res. Commun. 2008, 35, 17–23. [Google Scholar] [CrossRef]
- Chen, K.; Tang, X.; Jia, B.; Chao, C.; Wei, Y.; Hou, J.; Dong, L.; Deng, X.; Xiao, T.H.; Goda, K.; et al. Graphene oxide bulk material reinforced by heterophase platelets with multiscale interface crosslinking. Nat. Mater. 2022, 21, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yue, Y.; Guo, L.; Wu, J.; Zhang, Y.; Li, X.; Mao, S.; Han, X. Cloning nacre’s 3D interlocking skeleton in engineering composites to achieve exceptional mechanical properties. Adv. Mater. 2016, 28, 5099–5105. [Google Scholar] [CrossRef]
- Zhao, H.; Yue, Y.; Zhang, Y.; Li, L.; Guo, L. Ternary artificial nacre reinforced by ultrathin amorphous alumina with exceptional mechanical properties. Adv. Mater. 2016, 28, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, B.; Katti, K.S.; Katti, D.R.; Verma, D. Dynamic nanomechanical response of nacre. J. Mater. Res. 2006, 21, 2045–2051. [Google Scholar] [CrossRef]
- Sun, J.; Ling, M.; Wang, Y.; Chen, D.; Zhang, S.; Tong, J.; Wang, S. Quasi-static and dynamic nanoindentation of some selected biomaterials. J. Bionic Eng. 2014, 11, 144–150. [Google Scholar] [CrossRef]
- McNally, T.; Pötschke, P.; Halley, P.; Murphy, M.; Martin, D.; Bell, S.E.; Brennan, G.P.; Bein, D.; Lemoine, P.; Quinn, J.P. Polyethylene multiwalled carbon nanotube composites. Polymer 2005, 46, 8222–8232. [Google Scholar] [CrossRef]
- Zhang, Q.; Archer, L.A. Poly (ethylene oxide)/silica nanocomposites: Structure and rheology. Langmuir 2002, 18, 10435–10442. [Google Scholar] [CrossRef]
- Ren, J.; Silva, A.S.; Krishnamoorti, R. Linear viscoelasticity of disordered polystyrene- polyisoprene block copolymer based layered-silicate nanocomposites. Macromolecules 2000, 33, 3739–3746. [Google Scholar] [CrossRef]
- Ranganathan, R.; Ozisik, R.; Keblinski, P. Viscoelastic damping in crystalline composites: A molecular dynamics study. Compos. Part B Eng. 2016, 93, 273–279. [Google Scholar] [CrossRef]
- Hattemer, G.D.; Arya, G. Viscoelastic properties of polymer-grafted nanoparticle composites from molecular dynamics simulations. Macromolecules 2015, 48, 1240–1255. [Google Scholar] [CrossRef]
- Peng, W.; Ranganathan, R.; Keblinski, P.; Akcora, P.; Ozisik, R. Viscoelastic and dynamic properties of polymer grafted nanocomposites with high glass transition temperature graft chains. J. Appl. Phys. 2019, 126, 195102. [Google Scholar] [CrossRef]
- Ranganathan, R.; Shi, Y.; Keblinski, P. Commonalities in frequency-dependent viscoelastic damping in glasses in the MHz to THz regime. J. Appl. Phys. 2017, 122, 145103. [Google Scholar] [CrossRef]
- Moghimikheirabadi, A.; Karatrantos, A.V.; Kröger, M. Ionic Polymer Nanocomposites Subjected to Uniaxial Extension: A Nonequilibrium Molecular Dynamics Study. Polymers 2021, 13, 4001. [Google Scholar] [CrossRef] [PubMed]
- Hagita, K.; Murashima, T.; Ohkuma, T.; Jinnai, H. Ring-Filling Effect on Stress–Strain Curves of Randomly End-Linked Tetra-Arm Prepolymers. Macromolecules 2022, 55, 6547–6561. [Google Scholar] [CrossRef]
- Borodin, O.; Bedrov, D.; Smith, G.D.; Nairn, J.; Bardenhagen, S. Multiscale modeling of viscoelastic properties of polymer nanocomposites. J. Polym. Sci. Part B Polym. Phys. 2005, 43, 1005–1013. [Google Scholar] [CrossRef]
- Riggleman, R.A.; Toepperwein, G.N.; Papakonstantopoulos, G.J.; de Pablo, J.J. Dynamics of a glassy polymer nanocomposite during active deformation. Macromolecules 2009, 42, 3632–3640. [Google Scholar] [CrossRef]
- Shen, J.; Liu, J.; Li, H.; Gao, Y.; Li, X.; Wu, Y.; Zhang, L. Molecular dynamics simulations of the structural, mechanical and visco-elastic properties of polymer nanocomposites filled with grafted nanoparticles. Phys. Chem. Chem. Phys. 2015, 17, 7196–7207. [Google Scholar] [CrossRef]
- Mathiazhagan, S.; Anup, S. Atomistic simulations of length-scale effect of bioinspired brittle-matrix nanocomposite models. J. Eng. Mech. 2018, 144, 04018104. [Google Scholar] [CrossRef]
- Weng, S.; Ning, H.; Fu, T.; Hu, N.; Zhao, Y.; Huang, C.; Peng, X. Molecular dynamics study of strengthening mechanism of nanolaminated graphene/Cu composites under compression. Sci. Rep. 2018, 8, 3089. [Google Scholar] [CrossRef]
- Alian, A.; Dewapriya, M.; Meguid, S. Molecular dynamics study of the reinforcement effect of graphene in multilayered polymer nanocomposites. Mater. Des. 2017, 124, 47–57. [Google Scholar] [CrossRef]
- Naveen, J.; Jawaid, M.; Goh, K.L.; Reddy, D.M.; Muthukumar, C.; Loganathan, T.M.; Reshwanth, K.N.G.L. Advancement in Graphene-Based Materials and Their Nacre Inspired Composites for Armour Applications—A Review. Nanomaterials 2021, 11, 1239. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; Yao, Y.; Zeng, X.; Sun, J.; Hu, J.; Sun, R.; Xu, J.B.; Wong, C.P. Learning from natural nacre: Constructing layered polymer composites with high thermal conductivity. ACS Appl. Mater. Interfaces 2017, 9, 33001–33010. [Google Scholar] [CrossRef] [PubMed]
- Gu, G.X.; Takaffoli, M.; Hsieh, A.J.; Buehler, M.J. Biomimetic additive manufactured polymer composites for improved impact resistance. Extrem. Mech. Lett. 2016, 9, 317–323. [Google Scholar] [CrossRef]
- Wang, J.; Cheng, Q.; Tang, Z. Layered nanocomposites inspired by the structure and mechanical properties of nacre. Chem. Soc. Rev. 2012, 41, 1111–1129. [Google Scholar] [CrossRef]
- Kilikevičius, S.; Kvietkaitė, S.; Mishnaevsky, L., Jr.; Omastová, M.; Aniskevich, A.; Zeleniakienė, D. Novel hybrid polymer composites with graphene and Mxene nano-reinforcements: Computational analysis. Polymers 2021, 13, 1013. [Google Scholar] [CrossRef]
- Peng, W.; Ranganathan, R.; Keblinski, P.; Ozisik, R. Viscoelastic and dynamic properties of well-mixed and phase-separated binary polymer blends: A molecular dynamics simulation study. Macromolecules 2017, 50, 6293–6302. [Google Scholar] [CrossRef]
- Shao, C.; Keten, S. Stiffness enhancement in nacre-inspired nanocomposites due to nanoconfinement. Sci. Rep. 2015, 5, 16452. [Google Scholar] [CrossRef]
- Xia, W.; Song, J.; Hsu, D.D.; Keten, S. Understanding the interfacial mechanical response of nanoscale polymer thin films via nanoindentation. Macromolecules 2016, 49, 3810–3817. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2009, 18, 15012. [Google Scholar] [CrossRef]
- Kremer, K.; Grest, G.S. Dynamics of entangled linear polymer melts: A molecular-dynamics simulation. J. Chem. Phys. 1990, 92, 5057–5086. [Google Scholar] [CrossRef]
- Hirel, P. Atomsk: A tool for manipulating and converting atomic data files. Comput. Phys. Commun. 2015, 197, 212–219. [Google Scholar] [CrossRef]
- Evans, D.J.; Morriss, G. Nonlinear-response theory for steady planar Couette flow. Phys. Rev. A 1984, 30, 1528. [Google Scholar] [CrossRef]
- Daivis, P.J.; Todd, B. A simple, direct derivation and proof of the validity of the SLLOD equations of motion for generalized homogeneous flows. J. Chem. Phys. 2006, 124, 194103. [Google Scholar] [CrossRef]
- Thompson, A.P.; Plimpton, S.J.; Mattson, W. General formulation of pressure and stress tensor for arbitrary many-body interaction potentials under periodic boundary conditions. J. Chem. Phys. 2009, 131, 154107. [Google Scholar] [CrossRef] [PubMed]
- Kee, R.; Rupley, F.; Miller, J.A. Chemkin-II: A Fortran Chemical Kinetics Package for the Analysis of Gas-Phase Chemical Kinetics; Sandia nAtional Laboratories Report, SAND 89-8009; Sandia National Lab: Livermore, CA, USA, 1990; p. 158. [Google Scholar]
- Zhang, L.; Lin, J.; Lin, S. Elastic properties of graft copolymers in the lamellar phase studied by self-consistent field theory. Soft Matter 2009, 5, 173–181. [Google Scholar] [CrossRef]
- Zhu, X.; Wang, L.; Lin, J. Distinct Elastic Response to Hierarchical Nanostructures. Macromolecules 2011, 44, 8314–8323. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, J.; Shen, J.; Zhang, L.; Guo, Z.; Cao, D. Uniaxial deformation of nanorod filled polymer nanocomposites: A coarse-grained molecular dynamics simulation. Phys. Chem. Chem. Phys. 2014, 16, 16039–16048. [Google Scholar] [CrossRef]
- Michalet, X. Mean square displacement analysis of single-particle trajectories with localization error: Brownian motion in an isotropic medium. Phys. Rev. E 2010, 82, 041914. [Google Scholar] [CrossRef]
- Kalathi, J.T.; Kumar, S.K.; Rubinstein, M.; Grest, G.S. Rouse mode analysis of chain relaxation in homopolymer melts. Macromolecules 2014, 47, 6925–6931. [Google Scholar] [CrossRef]
- Frigo, M.; Johnson, S.G. FFTW: An adaptive software architecture for the FFT. In Proceedings of the 1998 IEEE International Conference on Acoustics, Speech and Signal Processing, ICASSP’98 (Cat. No. 98CH36181), Seattle, WA, USA, 12–15 May 1998; Volume 3, pp. 1381–1384. [Google Scholar]
- Gonçalves, S.; Bonadeo, H. Vibrational densities of states from molecular-dynamics calculations. Phys. Rev. B 1992, 46, 12019. [Google Scholar] [CrossRef]
- Schnell, B.; Meyer, H.; Fond, C.; Wittmer, J.; Baschnagel, J. Simulated glass-forming polymer melts: Glass transition temperature and elastic constants of the glassy state. Eur. Phys. J. E 2011, 34, 97. [Google Scholar] [CrossRef]
- Tjong, S.C. Structural and mechanical properties of polymer nanocomposites. Mater. Sci. Eng. R Rep. 2006, 53, 73–197. [Google Scholar] [CrossRef]
- Hall, L.M.; Jayaraman, A.; Schweizer, K.S. Molecular theories of polymer nanocomposites. Curr. Opin. Solid State Mater. Sci. 2010, 14, 38–48. [Google Scholar] [CrossRef]
- Milkus, R.; Ness, C.; Palyulin, V.V.; Weber, J.; Lapkin, A.; Zaccone, A. Interpretation of the vibrational spectra of glassy polymers using coarse-grained simulations. Macromolecules 2018, 51, 1559–1572. [Google Scholar] [CrossRef]
- Mausbach, P.; Fingerhut, R.; Vrabec, J. Structure and dynamics of the Lennard-Jones fcc-solid focusing on melting precursors. J. Chem. Phys. 2020, 153, 104506. [Google Scholar] [CrossRef]
- Liu, N.; Pidaparti, R.; Wang, X. Mechanical performance of graphene-based artificial nacres under impact loads: A coarse-grained molecular dynamic study. Polymers 2017, 9, 134. [Google Scholar] [CrossRef]
- Peng, C.; Tran, P. Bioinspired functionally graded gyroid sandwich panel subjected to impulsive loadings. Compos. Part B Eng. 2020, 188, 107773. [Google Scholar] [CrossRef]
- San Ha, N.; Lu, G. A review of recent research on bio-inspired structures and materials for energy absorption applications. Compos. Part B Eng. 2020, 181, 107496. [Google Scholar]
- Sen, D.; Buehler, M.J. Shock loading of bone-inspired metallic nanocomposites. In Solid State Phenomena; Trans Tech Publ.: Bäch, Switzerland, 2008; Volume 139, pp. 11–22. [Google Scholar]
- Lazarus, B.S.; Velasco-Hogan, A.; Gómez-del Río, T.; Meyers, M.A.; Jasiuk, I. A review of impact resistant biological and bioinspired materials and structures. J. Mater. Res. Technol. 2020, 9, 5705–15738. [Google Scholar] [CrossRef]
- Nova, A.; Keten, S.; Pugno, N.M.; Redaelli, A.; Buehler, M.J. Molecular and nanostructural mechanisms of deformation, strength and toughness of spider silk fibrils. Nano Lett. 2010, 10, 2626–2634. [Google Scholar] [CrossRef]
- Nair, A.K.; Libonati, F.; Qin, Z.; Dimas, L.S.; Buehler, M.J. Mechanical and interface properties of biominerals: Atomistic to coarse grained modeling. In Biomineralization Sourcebook, 1st ed.; CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar]
- Egan, P.; Sinko, R.; LeDuc, P.R.; Keten, S. The role of mechanics in biological and bio-inspired systems. Nat. Commun. 2015, 6, 7418. [Google Scholar]
- Ruiz, L.; Xia, W.; Meng, Z.; Keten, S. A coarse-grained model for the mechanical behavior of multi-layer graphene. Carbon 2015, 82, 103–115. [Google Scholar] [CrossRef]
- Xia, W.; Song, J.; Meng, Z.; Shao, C.; Keten, S. Designing multi-layer graphene-based assemblies for enhanced toughness in nacre-inspired nanocomposites. Mol. Syst. Des. Eng. 2016, 1, 40–47. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Mean () | Std. () | |
|---|---|---|---|
| Base polymer | 0.42 | 25.26 | 8.94 |
| Columnar composite | 0.55 | 48.01 | 15.37 |
| Staggered composite | 0.72 | 54.62 | 20.14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, P.P.; Ranganathan, R. Tensile and Viscoelastic Behavior in Nacre-Inspired Nanocomposites: A Coarse-Grained Molecular Dynamics Study. Nanomaterials 2022, 12, 3333. https://doi.org/10.3390/nano12193333
Singh PP, Ranganathan R. Tensile and Viscoelastic Behavior in Nacre-Inspired Nanocomposites: A Coarse-Grained Molecular Dynamics Study. Nanomaterials. 2022; 12(19):3333. https://doi.org/10.3390/nano12193333
Chicago/Turabian StyleSingh, Param Punj, and Raghavan Ranganathan. 2022. "Tensile and Viscoelastic Behavior in Nacre-Inspired Nanocomposites: A Coarse-Grained Molecular Dynamics Study" Nanomaterials 12, no. 19: 3333. https://doi.org/10.3390/nano12193333
APA StyleSingh, P. P., & Ranganathan, R. (2022). Tensile and Viscoelastic Behavior in Nacre-Inspired Nanocomposites: A Coarse-Grained Molecular Dynamics Study. Nanomaterials, 12(19), 3333. https://doi.org/10.3390/nano12193333