
Hybrid MXene-Graphene/Hexagonal Boron Nitride Structures: Electronic and Molecular Adsorption Properties


Abstract
:1. Introduction
2. Computational Method
3. Results and Discussions
Gas Adsorption on MXenes
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Novoselov, K.; Geim, A.; Morozov, S.; Jiang, D.; Zhang, Y.; Dubonos, S.; Grigorieva, I.; Firsov, A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liu, F.; Fu, W.; Fang, Z.; Zhou, W.; Liu, Z. Two-Dimensional Heterostructures: Fabrication, Characterization, and Application. Nanoscale 2014, 6, 12250–12272. [Google Scholar] [CrossRef]
- Chen, C.; Ji, X.; Xu, K.; Zhang, B.; Miao, L.; Jiang, J. Prediction of T-and H-Phase Two-Dimensional Transition-Metal Carbides/Nitrides and Their Semiconducting-Metallic Phase Transition. ChemPhysChem 2017, 18, 1897–1902. [Google Scholar] [CrossRef]
- Winter, A.; George, A.; Neumann, C.; Tang, Z.; Mohn, M.J.; Biskupek, J.; Masurkar, N.; Reddy, A.L.M.; Weimann, T.; Hubner, U.; et al. Lateral Heterostructures of Two-Dimensional Materials by Electron-Beam Induced Stitching. Carbon 2018, 128, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Oughaddou, H.; Enriquez, H.; Tchalala, M.R.; Yildirim, H.; Mayne, A.J.; Bendounan, A.; Dujardin, G.; Ait Ali, M.; Kara, A. Silicene, a Promising New 2D Material. Prog. Surf. Sci. 2015, 90, 46–83. [Google Scholar] [CrossRef] [Green Version]
- Vogt, P. Silicene, Germanene and Other Group IV 2D Materials. Beilstein J. Nanotechnol. 2018, 9, 2665–2667. [Google Scholar] [CrossRef]
- Onodera, M.; Taniguchi, T.; Watanabe, K.; Isayama, M.; Masubuchi, S.; Moriya, R.; Machida, T. Hexagonal Boron Nitride Synthesized at Atmospheric Pressure Using Metal Alloy Solvents: Evaluation as a Substrate for 2D Materials. Nano Lett. 2020, 20, 735–740. [Google Scholar] [CrossRef]
- Zhao, G.; Cheng, Y.; Wu, Y.; Xu, X.; Hao, X. New 2D Carbon Nitride Organic Materials Synthesis with Huge-Application Prospects in CN Photocatalyst. Small 2018, 14, 1704138. [Google Scholar] [CrossRef]
- Bafekry, A.; Stampfl, C.; Faraji, M.; Yagmurcukardes, M.; Fadlallah, M.M.; Jappor, H.R.; Ghergherehchi, M.; Feghhi, S.A.H. A Dirac-semimetal two-dimensional BeN4: Thickness-dependent electronic and optical properties. Appl. Phys. Lett. 2021, 118, 203103. [Google Scholar] [CrossRef]
- Bafekry, A.; Faraji, M.; Fadlallah, M.M.; Hoat, D.M.; Khatibani, A.B.; Sarsari, I.A.; Ghergherehchi, M. Effect of adsorption and substitutional B doping at different concentrations on the electronic and magnetic properties of a BeO monolayer: A first-principles study. Phys. Chem. Chem. Phys. 2021, 23, 24922–24931. [Google Scholar] [CrossRef]
- Heine, T. Transition Metal Chalcogenides: Ultrathin Inorganic Materials with Tunable Electronic Properties. Acc. Chem. Res. 2015, 48, 65–72. [Google Scholar] [CrossRef]
- Helal, M.A.; El-Sayed, H.M.; Maarouf, A.A.; Fadlallah, M.M. Metal dichalcogenide nanomeshes: Structural, electronic and magnetic properties. Phys. Chem. Chem. Phys. 2021, 23, 21183–21195. [Google Scholar] [CrossRef]
- Naseri, M.; Bafekry, A.; Faraji, M.; Hoat, D.M.; Fadlallah, M.M.; Ghergherehchi, M.; Sabbaghi, N.; Gogova, D. Two-dimensional buckled tetragonal cadmium chalcogenides including CdS, CdSe, and CdTe monolayers as photo-catalysts for water splitting. Phys. Chem. Chem. Phys. 2021, 23, 12226–12232. [Google Scholar] [CrossRef]
- Liu, L.; Kou, L.; Wang, Y.; Lu, C.; Hu, X. Electronic and Effective Mass Modulation in Two-Dimensional BCN by Strain Engineering. Nanotechnology 2020, 31, 455702. [Google Scholar] [CrossRef]
- Bafekry, A.; Naseri, M.; Fadlallah, M.M.; Abdolhosseini Sarsari, I.; Faraji, M.; Bagheri Khatibani, A.; Ghergherehchi, M.; Gogova, D. A novel two-dimensional boron–carbon–nitride (BCN) monolayer: A first-principles insight. J. Appl. Phys. 2021, 130, 114301. [Google Scholar] [CrossRef]
- Bafekry, A.; Faraji, M.; Fadlallah, M.M.; Mortazavi, B.; Ziabari, A.A.; Khatibani, A.B.; Nguyen, C.V.; Ghergherehchi, M.; Gogova, D. Point Defects in a Two-Dimensional ZnSnN2 Nanosheet: A First-Principles Study on the Electronic and Magnetic Properties. J. Phys. Chem. C 2021, 125, 13067–13075. [Google Scholar] [CrossRef]
- Hong, Y.L.; Liu, Z.; Wang, L.; Zhou, T.; Ma, W.; Xu, C.; Feng, S.; Chen, L.; Chen, M.L.; Sun, D.M.; et al. Chemical Vapor Deposition of Layered Two-Dimensional MoSi2N4 Materials. Science 2020, 369, 670–674. [Google Scholar] [CrossRef]
- Bafekry, A.; Faraji, M.; Hoat, D.M.; Shahrokhi, M.; Fadlallah, M.M.; Shojaei, F.; Feghhi, S.A.H.; Ghergherehchi, M.; Gogova, D. MoSi2N4 single-layer: A novel two-dimensional material with outstanding mechanical, thermal, electronic and optical properties. J. Phys. D Appl. Phys. 2021, 54, 155303. [Google Scholar] [CrossRef]
- Abdelati, M.A.; Maarouf, A.A.; Fadlallah, M.M. Substitutional transition metal doping in MoSi2N4 monolayer: Structural, electronic and magnetic properties. Phys. Chem. Chem. Phys. 2022, 24, 3035–3042. [Google Scholar] [CrossRef]
- Zhang, C.J.; Ma, Y.; Zhang, X.; Abdolhosseinzadeh, S.; Sheng, H.; Lan, W.; Pakdel, A.; Heier, J.; Nüesch, F. Two-Dimensional Transition Metal Carbides and Nitrides (MXenes): Synthesis, Properties, and Electrochemical Energy Storage Applications. Energy Environ. Mater. 2020, 3, 29–55. [Google Scholar] [CrossRef]
- Hantanasirisakul, K.; Gogotsi, Y. Electronic and Optical Properties of 2D Transition Metal Carbides and Nitrides (MXenes). Adv. Mater. 2018, 30, 1804779. [Google Scholar] [CrossRef]
- Faraji, M.; Bafekry, A.; Fadlallah, M.M.; Molaei, F.; Hieu, N.N.; Qian, P.; Ghergherehchi, M.; Gogova, D. Surface modification of titanium carbide MXene monolayers (Ti2C and Ti3C2) via chalcogenide and halogenide atoms. Phys. Chem. Chem. Phys. 2021, 23, 15319–15328. [Google Scholar] [CrossRef]
- Akgenc, B. New Predicted Two-Dimensional MXenes and Their Structural, Electronic and Lattice Dynamical Properties. Solid State Commun. 2019, 303-304, 113739. [Google Scholar] [CrossRef]
- Anasori, B.; Lukatskaya, M.R.; Gogotsi, Y. 2D metal carbides and nitrides (MXenes) for energy storage. Nat. Rev. Mater. 2017, 2, 1–17. [Google Scholar] [CrossRef]
- Jeon, J.; Yang, Y.; Choi, H.; Park, J.H.; Lee, B.H.; Lee, S. MXenes for Future Nanophotonic Device Applications. Nanophotonics 2020, 9, 1831–1853. [Google Scholar] [CrossRef]
- Khazaei, M.; Ranjbar, A.; Arai, M.; Sasaki, T.; Yunoki, S. Electronic Properties and Applications of MXenes: A Theoretical Review. J. Mater. Chem. C 2017, 5, 2488–2503. [Google Scholar] [CrossRef] [Green Version]
- Lipatov, A.; Sinitskii, A. Electronic and Mechanical Properties of MXenes Derived from Single-Flake Measurements. In 2D Metal Carbides and Nitrides (MXenes): Structure, Properties and Applications; Springer: Berlin, Germany, 2019; pp. 301–325. [Google Scholar]
- Li, X.; Bai, Y.; Shi, X.; Su, N.; Nie, G.; Zhang, R.; Nie, H.; Ye, L. Applications of MXene (Ti3C2Tx) in photocatalysis: A review. Mater. Adv. 2021, 2, 1570–1594. [Google Scholar] [CrossRef]
- Peng, C.; Zhou, T.; Wei, P.; Xu, W.; Pan, H.; Peng, F.; Jia, J.; Zhang, K.; Yu, H. Photocatalysis over MXene-based hybrids: Synthesis, surface chemistry, and interfacial charge kinetics. APL Mater. 2021, 9, 070703. [Google Scholar] [CrossRef]
- Levendorf, M.; Kim, C.J.; Brown, L.; Huang, P.; Havener, R.; Muller, D.; Park, J. Graphene and Boron Nitride Lateral Heterostructures for Atomically Thin Circuitry. Nature 2012, 488, 627–632. [Google Scholar] [CrossRef]
- Saber, M.R.; Khabiri, G.; Maarouf, A.A.; Ulbricht, M.; Khalil, A.S.G. A Comparative Study on the Photocatalytic Degradation of Organic Dyes Using Hybridized 1T/2H, 1T/3R and 2H MoS2 Nano-Sheets. RSC Adv. 2018, 8, 26364–26370. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Tian, H.; Ren, C.; Yu, J.; Sun, M. Electronic and Optical Properties of Heterostructures Based on Transition Metal Dichalcogenides and Graphene-Like Zinc Oxide. Sci. Rep. 2018, 8, 12009. [Google Scholar] [CrossRef] [PubMed]
- Winkler, C.; Harivyasi, S.S.; Zojer, E. Controlling the Electronic Properties of van der Waals Heterostructures by Applying Electrostatic Design. 2D Mater. 2018, 5, 035019. [Google Scholar] [CrossRef]
- Bafekry, A.; Faraji, M.; Abdollahzadeh Ziabari, A.; Fadlallah, M.M.; Nguyen, C.V.; Ghergherehchi, M.; Feghhi, S.A.H. A van der Waals heterostructure of MoS2/MoSi2N4: A first-principles study. New J. Chem. 2021, 45, 8291–8296. [Google Scholar] [CrossRef]
- Du, Y.T.; Kan, X.; Yang, F.; Gan, L.Y.; Schwingenschlogl, U. MXene/Graphene Heterostructures as High-Performance Electrodes for Li-Ion Batteries. ACS Appl. Mater. Interfaces 2018, 10, 32867–32873. [Google Scholar] [CrossRef] [PubMed]
- Demiroglu, I.; Peeters, F.M.; Gülseren, O.; Cakır, D.; Sevik, C. Alkali Metal Intercalation in MXene/Graphene Heterostructures: A New Platform for Ion Battery Applications. J. Phys. Chem. Lett. 2019, 10, 727–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, D.X.; Du, Y.Z.; Ma, W.G.; Zhang, X. High Rate and Capacity Performances of Functionalized MXene/Graphene Heterostructure Anodes for Magnesium-Ion Batteries. Int. J. Energy Res. 2021, 45, 3421–3429. [Google Scholar] [CrossRef]
- Yang, W.; Byun, J.J.; Yang, J.; Moissinac, F.P.; Ma, Y.; Ding, H.; Sun, W.; Dryfe, R.A.W.; Barg, S. All-In-One MXene–Boron Nitride–MXene “OREO” with Vertically Aligned Channels for Flexible Structural Supercapacitor Design. ACS Appl. Energy Mater. 2021, 4, 7959–7972. [Google Scholar] [CrossRef]
- Zeng, Z.H.; Wu, N.; Wei, J.J.; Yang, Y.F.; Wu, T.T.; Li, B.; Hauser, S.B.; Yang, W.D.; Liu, J.R.; Zhao, S.Y. Porous and Ultra-Flexible Crosslinked MXene/Polyimide Composites for Multifunctional Electromagnetic Interference Shielding. Nano-Micro Lett. 2022, 14, 1–16. [Google Scholar] [CrossRef]
- Zeng, Z.; Wang, C.; Siqueira, G.; Han, D.; Huch, A.; Abdolhosseinzadeh, S.; Heier, J.; Nüesch, F.; Zhang, C.; Nyström, G. Nanocellulose-MXene Biomimetic Aerogels with Orientation-Tunable Electromagnetic Interference Shielding Performance. Adv. Sci. 2020, 7, 2000979. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Sun, W.; Zhan, C.; Kent, P.R.C.; Jiang, D.E. Interfacial and Electronic Properties of Heterostructures of MXene and Graphene. Phys. Rev. B 2019, 99, 085429. [Google Scholar] [CrossRef] [Green Version]
- Aierken, Y.; Sevik, C.; Gülseren, O.; Peeters, F.; Cakır, D. MXenes/Graphene Heterostructures for Li Battery Applications: A First Principles Study. J. Mater. Chem. 2018, 6, 2337–2345. [Google Scholar] [CrossRef]
- Zhou, J.; Li, D.; Zhao, W.; Jing, B.; Ao, Z.; An, T. First-Principles Evaluation of Volatile Organic Compounds Degradation in Z-Scheme Photocatalytic Systems: MXene and Graphitic-CN Heterostructures. ACS Appl. Mater. Interfaces 2021, 13, 23843–23852. [Google Scholar] [CrossRef]
- Lee, S.H.; Eom, W.; Shin, H.; Ambade, R.B.; Bang, J.H.; Kim, H.W.; Han, T.H. Room-Temperature, Highly Durable Ti3C2Tx MXene/Graphene Hybrid Fibers for NH3 Gas Sensing. ACS Appl. Mater. Interfaces 2020, 12, 10434–10442. [Google Scholar] [CrossRef]
- Wang, B.; Wu, X.; Zhang, X.; Pang, G.; Li, S. Mo2C-Embedded Biomass-Derived Honeycomb-Like Nitrogen-Doped Carbon Nanosheet/Graphene Aerogel Films for Highly Efficient Electrocatalytic Hydrogen Evolution. New J. Chem. 2020, 44, 1147–1156. [Google Scholar] [CrossRef]
- Wang, B.; Wang, G.; Wang, H. Hybrids of Mo2C Nanoparticles Anchored on Graphene Sheets as Anode Materials for High Performance Lithium-Ion Batteries. J. Mater. Chem. A 2015, 3, 17403–17411. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A Modular and Open-Source Software Project for Quantum Simulations of Materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thonhauser, T.; Cooper, V.R.; Li, S.; Puzder, A.; Hyldgaard, P.; Langreth, D.C. van der Waals Density Functional: Self-Consistent Potential and the Nature of the van der Waals Bond. Phys. Rev. B 2007, 76, 125112. [Google Scholar] [CrossRef] [Green Version]
- Thonhauser, T.; Zuluaga, S.; Arter, C.; Berland, K.; Schröder, E.; Hyldgaard, P. Spin Signature of Nonlocal Correlation Binding in Metal-Organic Frameworks. Phys. Rev. Lett. 2015, 115, 136402. [Google Scholar] [CrossRef] [Green Version]
- Khazaei, M.; Arai, M.; Sasaki, T.; Estili, M.; Sakka, Y. Trends in Electronic Structures and Structural Properties of MAX Phases: A First-Principles Study on M2AlC (M = Sc, Ti, Cr, Zr, Nb, Mo, Hf, or Ta), M2AlN, and Hypothetical M2AlB Phases. J. Phys. Condens. Matter 2014, 26, 505503. [Google Scholar] [CrossRef]
- Yorulmaz, U.; Özden, A.; Perkgoz, N.K.; Ay, F.; Sevik, C. Vibrational and Mechanical Properties of Single Layer MXene Structures: A First-Principles Investigation. Nanotechnology 2016, 27, 335702. [Google Scholar] [CrossRef]
- Junkaew, A.; Arróyave, R. Enhancement of Selectivity of MXenes (M2C, M = Ti, V, Nb, Mo) Via Oxygen-Functionalization: Promising Materials for Gas-Sensing and -Separation. Phys. Chem. Chem. Phys. 2018, 20, 6073–6082. [Google Scholar] [CrossRef]
- Lei, J.; Kutana, A.; Yakobson, B.I. Predicting Stable Phase Monolayer Mo2C (MXene), a Superconductor with Chemically-Tunable Critical Temperature. J. Mater. Chem. C 2017, 5, 3438–3444. [Google Scholar] [CrossRef] [Green Version]
- Pang, D.; Alhabeb, M.; Mu, X.; Dall’Agnese, Y.; Gogotsi, Y.; Gao, Y. Electrochemical actuators based on two-dimensional Ti3C2Tx (MXene). Nano Lett. 2019, 19, 7443–7448. [Google Scholar] [CrossRef]
- Demaison, J.; Herman, M.; Liévin, J. The Equilibrium OH Bond Length. Int. Rev. Phys. Chem. 2007, 26, 391–420. [Google Scholar] [CrossRef]
- Carey, F.A.; Sundberg, R.J. Chemical bonding and molecular structure. In Advanced Organic Chemistry; Springer: Berlin, Germany, 2007; pp. 1–117. [Google Scholar]
- Demaison, J.; Császár, A.G. Equilibrium CO Bond Lengths. J. Mol. Struct. 2012, 1023, 7–14. [Google Scholar] [CrossRef]
- Wurth, W.; Stöhr, J.; Feulner, P.; Pan, X.; Bauchspiess, K.R.; Baba, Y.; Hudel, E.; Rocker, G.; Menzel, D. Bonding, Structure, and Magnetism of Physisorbed and Chemisorbed O2 on Pt(111). Phys. Rev. Lett. 1990, 65, 2426–2429. [Google Scholar] [CrossRef]
- Grosjean, B.; Pean, C.; Siria, A.; Bocquet, L.; Vuilleumier, R.; Bocquet, M.L. Chemisorption of Hydroxide on 2D Materials from DFT Calculations: Graphene Versus Hexagonal Boron Nitride. J. Phys. Chem. Lett. 2016, 7, 4695–4700. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.; Jung, S.C.; Han, Y.K. Selectively Strong Molecular Adsorption on Boron Nitride Monolayer Induced by Transition Metal Substrate. Curr. Appl. Phys. 2013, 13, 2059–2063. [Google Scholar] [CrossRef]
- Yang, S.; Lei, G.; Xu, H.; Xu, B.; Li, H.; Lan, Z.; Wang, Z.; Gu, H. A DFT Study of CO Adsorption on the Pristine, Defective, In-Doped and Sb-Doped Graphene and the Effect of Applied Electric Field. Appl. Surf. Sci. 2019, 480, 205–211. [Google Scholar] [CrossRef]
- Enriquez, J.I.G.; Al Rey, C.V. Hydrogen Adsorption on Pristine, Defected, and 3d-Block Transition Metal-Doped Penta-Graphene. Int. J. Hydrogen Energy 2016, 41, 12157–12166. [Google Scholar] [CrossRef]
- Chettri, B.; Patra, P.; Hieu, N.N.; Rai, D. Hexagonal Boron Nitride (h-BN) Nanosheet as a Potential Hydrogen Adsorption Material: A Density Functional Theory (DFT) Study. Surf. Interfaces 2021, 24, 101043. [Google Scholar] [CrossRef]
- El-Barbary, A.; Eid, K.M.; Kamel, M.; Taha, H.; Ismail, G. Adsorption of CO, CO2, NO and NO2 on Boron Nitride Nanotubes: DFT Study. J. Surf. Eng. Mater. Adv. Technol. 2015, 5, 154. [Google Scholar]
- Behmagham, F.; Vessally, E.; Massoumi, B.; Hosseinian, A.; Edjlali, L. A Computational Study on the SO2 Adsorption by the Pristine, Al, and Si Doped BN Nanosheets. Superlattices Microstruct. 2016, 100, 350–357. [Google Scholar] [CrossRef]
- Carmichael, I.; Bentley, J. Comparison of the Magnetic Properties and Harmonic Force Fields of Nitrogen Dioxide and Carbon Dioxide (1-)(CO2-) by ab initio calculation. J. Phys. Chem. 1985, 89, 2951–2954. [Google Scholar] [CrossRef]
- Grabowsky, S.; Luger, P.; Buschmann, J.; Schneider, T.; Schirmeister, T.; Sobolev, A.N.; Jayatilaka, D. The Significance of Ionic Bonding in Sulfur Dioxide: Bond Orders from X-ray Diffraction Data. Angew. Chem. Int. Ed. 2012, 51, 6776–6779. [Google Scholar] [CrossRef]
- Hellmann, R.; Bich, E.; Vogel, E.; Vesovic, V. Ab Initio Intermolecular Potential Energy Surface and Thermophysical Properties of Hydrogen Sulfide. Phys. Chem. Chem. Phys. 2011, 13, 13749–13758. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, J.M.; Xu, K.W.; Ji, V. A First-Principles Study on Gas Sensing Properties of Graphene and Pd-Doped Graphene. Appl. Surf. Sci. 2015, 343, 121–127. [Google Scholar] [CrossRef]
- Shokuhi Rad, A.; Esfahanian, M.; Maleki, S.; Gharati, G. Application of Carbon Nanostructures toward SO2 and SO3 Adsorption: A Comparison Between Pristine Graphene and N-Doped Graphene by DFT Calculations. J. Sulfur Chem. 2016, 37, 176–188. [Google Scholar] [CrossRef]
- Faye, O.; Raj, A.; Mittal, V.; Beye, A.C. H2S Adsorption on Graphene in the Presence of Sulfur: A Density Functional Theory Study. Comput. Mater. Sci. 2016, 117, 110–119. [Google Scholar] [CrossRef]
- Sagynbaeva, M.; Hussain, T.; Panigrahi, P.; Johansson, B.; Ahuja, R. Complementing the Adsorption Energies of CO2, H2S and NO2 to h-BN Sheets by Doping with Carbon. EPL Europhys. Lett. 2015, 109, 57008. [Google Scholar] [CrossRef]
- Osouleddini, N.; Rastegar, S.F. DFT Study of the CO2 and CH4 Assisted Adsorption on the Surface of Graphene. J. Electron Spectrosc. Relat. Phenom. 2019, 232, 105–110. [Google Scholar] [CrossRef]
- Ullah, H.; Ayub, K.; Ullah, Z.; Hanif, M.; Nawaz, R.; Bilal, S.; Ali Shah, A.U.H. Theoretical Insight of Polypyrrole Ammonia Gas Sensor. Synth. Met. 2013, 172, 14–20. [Google Scholar] [CrossRef]
- Au, C.T.; Zhou, T.J.; Lai, W.J.; Ng, C.F. An ab Initio Study of Methane Activation on Lanthanide Oxide. Catal. Lett. 1997, 49, 53–58. [Google Scholar] [CrossRef]
- Li, H.; Chen, Z.; Fang, X.; Tie, D. Absorption of NH3 on Pristine and Defected Boron Nitride Nanosheets: A First Principle Study. Superlattices Microstruct. 2015, 88, 371–376. [Google Scholar] [CrossRef]
- Seyed-Talebi, S.M.; Neek-Amal, M. The Different Adsorption Mechanism of Methane Molecule onto a Boron Nitride and a Graphene Flakes. J. Appl. Phys. 2014, 116, 153507. [Google Scholar] [CrossRef]
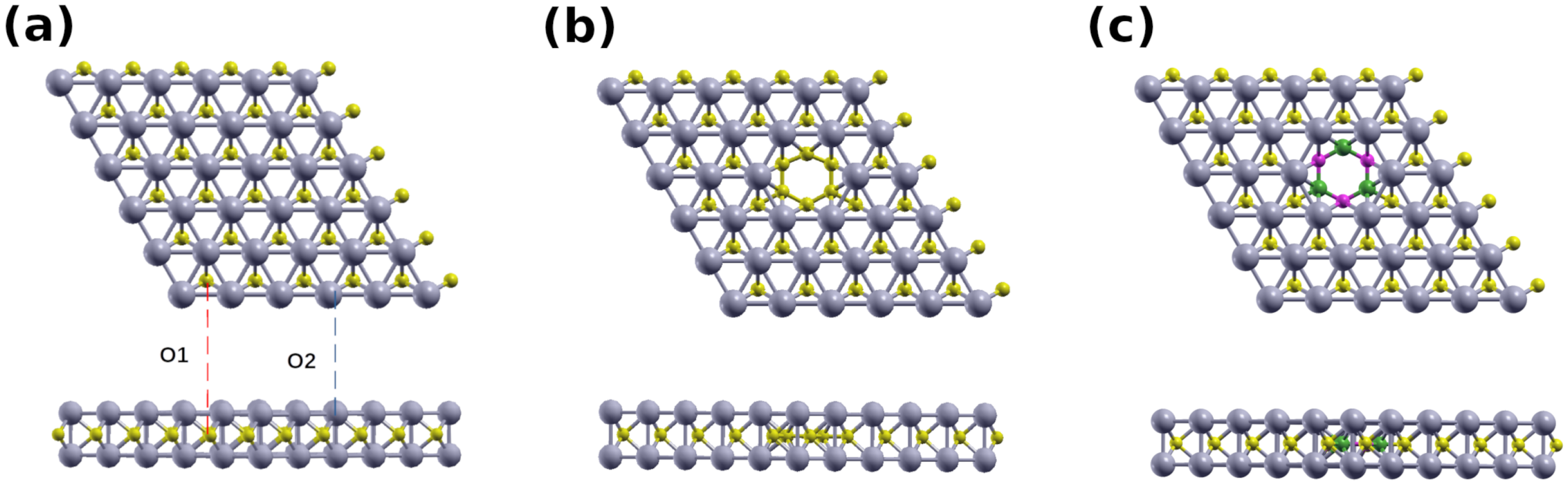
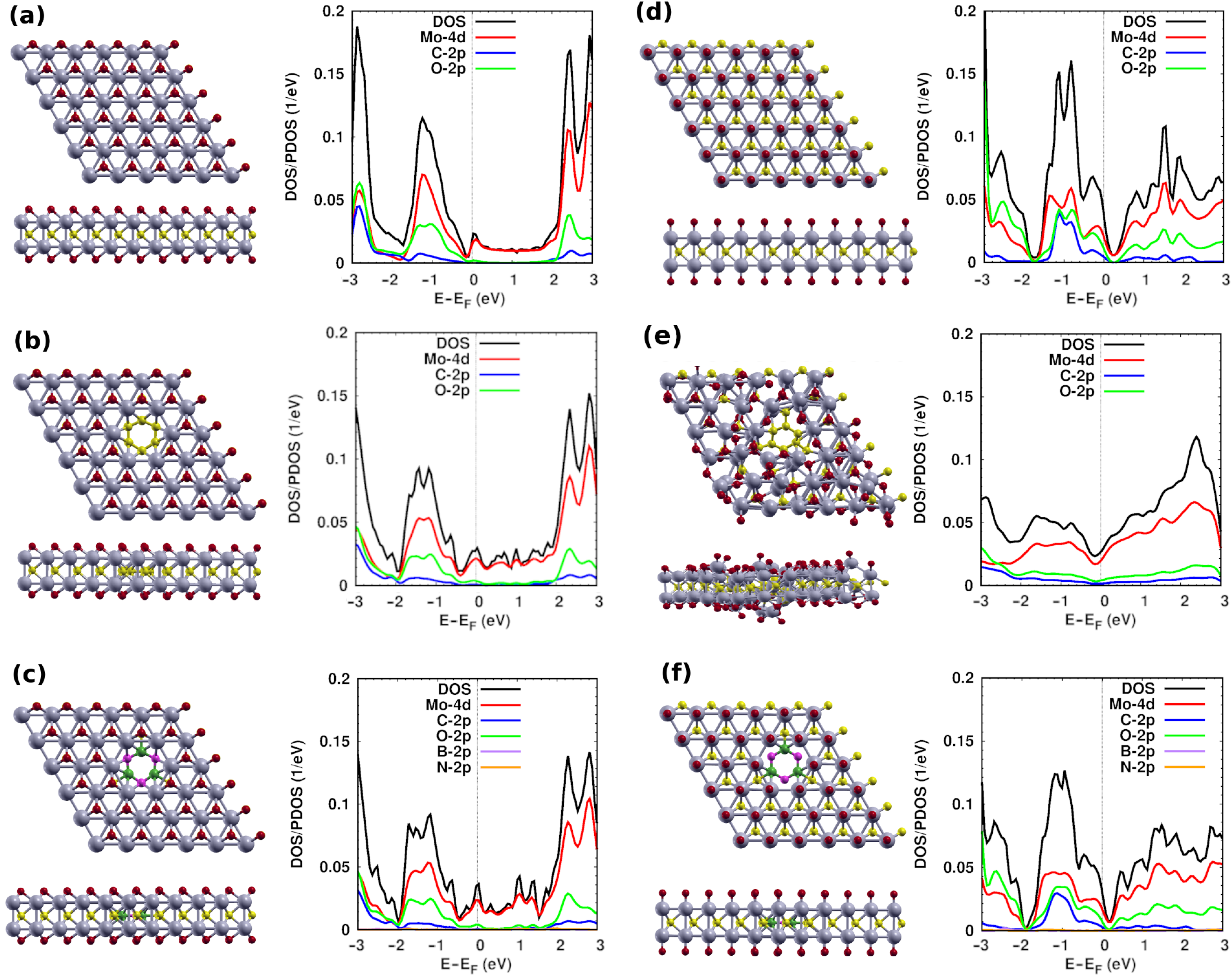

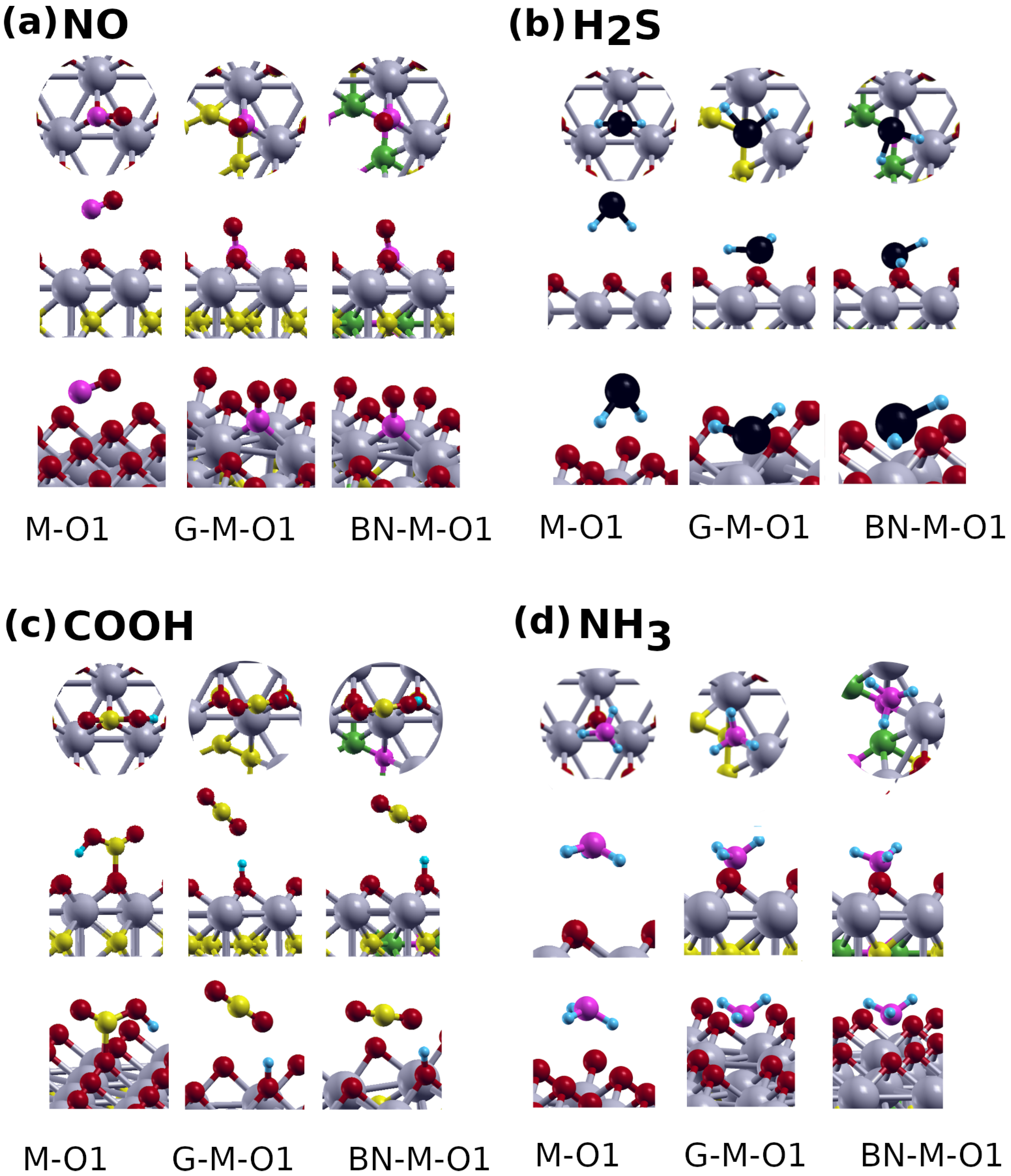

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sys. | Lattice Constant | |||||||
|---|---|---|---|---|---|---|---|---|
| M-O1 | −9.16 | 6.07 | 2.84 | 2.17 | 2.05 | - | - | 72 |
| G-M-O1 | −9.11 | 6.07 | 2.83 | 2.17 | 2.08 | 2.26 | 2.35 | 66 |
| BN-M-O1 | −9.09 | 6.07 | 2.85 | 2.20 | 2.04 | 2.26 | 1.73 | 70 |
| M-O2 | −8.30 | 6.07 | 2.93 | 2.23 | 1.72 | - | - | 72 |
| G-M-O2 | −8.82 | 6.07 | 2.86 | 2.18 | 1.95 | 2.20 | 1.44 | 66 |
| BN-M-O2 | −8.30 | 6.07 | 2.86 | 2.24 | 1.72 | 2.40 | 1.65 | 70 |
| Gas | M-O1 | G-M-O1 | BN-M-O1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| X-X | E | Q | d(Y-Z) | X-X | E | Q | d(Y-Z) | X-X | E | Q | d(Y-Z) | |
| OH | O1-O1 | −1.15 | −0.21 | 1.45 (O-O) | C1-C1 | −5.48/−0.5 [61] | 0.07 | 2.11 (Mo-O) | N1-N1 | −5.45 | 0.07/−2.3 [61] | 2.11 (Mo-O) |
| – | – | – | – | Mo1-C1 | −5.48 | 0.06 | 2.11 (Mo-O) | Mo1-N1 | −5.45 | 0.07 | 2.11 (Mo-O) | |
| NO | O1-O1 | −1.10 | −0.56 | 2.05 (O-N) | C1-C1 | −2.58 | −0.06 | 2.07 (Mo-N) | N1-N1 | −3.15/−0.026 [62] | −0.06 | 2.09 (Mo-N) |
| – | – | – | – | Mo1-C1 | −2.58 | −0.07 | 2.07 (Mo-N) | Mo1-N1 | −3.15 | −0.07 | 2.09 (Mo-N) | |
| CO | O1-O1 | −0.10 | −0.07 | 4.17 (O-C) | C1-C1 | −1.26/−1.13 [63] | −0.21 | 2.21 (Mo-C) | N1-N1 | −1.81/−0.02 [62] | −0.21 | 2.21 (Mo-C) |
| – | – | – | – | Mo1-C1 | −1.27 | −0.19 | 2.21 (Mo-C) | Mo1-N1 | −1.81 | −0.19 | 2.22 (Mo-C) | |
| N | O1-O1 | −0.09 | −0.07 | 3.08 (O-N) | C1-C1 | −0.23 | −0.20 | 3.12 (Mo-N) | N1-N1 | −0.64 | −0.21 | 3.28 (O-N) |
| – | – | – | – | Mo1-C1 | −0.24 | −0.17 | 3.03 (Mo-N) | Mo1-N1 | −0.64 | −0.18 | 3.26 (Mo-N) | |
| H | O1-O1 | −0.05 | −0.014 | 2.46 (O-H) | C1-C1 | −0.07/−0.08 [64] | −0.02 | 3.58 (O-H) | N1-N1 | −0.09/−0.21 [65] | −0.02 | 2.90 (Mo-H) |
| – | – | – | – | Mo1-Mo1 | −0.08 | −0.06 | 2.69 (O-H) | Mo1-Mo1 | −0.06 | −0.02 | 2.74 (O-H) | |
| Gas | M-O1 | G-M-O1 | BN-M-O1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| X-X | E | Q | d (Y-Z) | X-X | E | Q | d (Y-Z) | X-X | E | Q | d (Y-Z) | |
| NO | O1-O1 | −0.58 | −0.59 | 1.97 (O-N) | C1-(C1,C6) | −2.46/−2.17 [71] | −0.03 | 2.21 (Mo-N) | N1-(B1,N1) | −2.91/−0.03 [62] | −0.03 | 2.21 (Mo-N) |
| – | – | – | – | Mo1-(C1,C6) | −2.46 | −0.04 | 2.21 (Mo-N) | Mo1-(B1,N1) | −2.91 | −0.04 | 2.20 (Mo-N) | |
| SO | O1-O1 | −0.49 | −0.06 | 3.66 (O-O) | C1-C | −2.31/−1.97 [72] | −0.12 | 2.13 (Mo-O) | N1-(B,N) | −2.78/−0.08 [67] | −0.13 | 2.14 (Mo-O) |
| – | – | – | – | Mo1-C | −2.28 | −0.13 | 2.15 (Mo-O) | Mo1-N1 | −1.93 | −0.24 | 2.15 (Mo-O) | |
| HS | O1-O1 | −0.26 | −0.27 | 3.46 (O-H) | C1-C1 | −1.19/−0.45 [73] | −0.61 | 2.78 (Mo-S) | N1-N1 | −1.65/−0.11 [74] | −0.59 | 2.69 (Mo-S) |
| – | – | – | – | Mo1-C1 | −1.22 | −0.62 | 2.72 (Mo-S) | Mo1-N1 | −1.65 | −0.61 | 2.71 (Mo-S) | |
| CO | O1-O1 | −0.19 | −0.06 | 2.80 (O-C) | C1-C3 | −0.56/-0.32 [75] | −0.32 | 2.54 (Mo-O) | N1-B2 | −0.78/−0.02 [62] | −0.05 | 2.95 (Mo-O) |
| – | – | – | – | Mo1-Mo1 | −0.12 | −0.22 | 3.07 (O-O) | Mo1-Mo1 | −0.55 | −0.05 | 3.05 (O-O) | |
| Gas | M-O1 | G-M-O1 | BN-M-O1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| X-X | E | Q | d (Y-Z) | X-X | E | Q | d (Y-Z) | X-X | E | Q | d (Y-Z) | |
| COOH | O1-O1 | −2.95 | −0.45 | 1.54 (O-C) | C1-C1 | −3.32 | −0.24 | 2.26 (Mo-O) | N1-N1 | −3.11 | −0.53 | 2.41 (Mo-C) |
| – | – | – | – | Mo1-d | D | D | D | Mo1-d | D | D | D | |
| NH | O1-O1 | −0.40 | −0.30 | 2.66 (O-N) | C1-C1 | −1.78/−1.09 [71] | −0.45 | 2.95 (O-N) | N1-N1 | −2.18/−0.17 [78] | −0.45 | 2.80 (O-N) |
| – | – | – | – | Mo1-C1 | −1.78 | −0.46 | 2.76 (Mo-N) | Mo1-N1 | −2.66 | −0.46 | 2.75 (Mo-N) | |
| CH | O1-O1 | −0.21 | −0.03 | 2.81 (O-H) | C1-C1 | −0.41/−0.33 [75] | −0.13 | 2.36 (O-H) | N1-N1 | −0.80/−0.12 [79] | −0.03 | 2.40 (O-H) |
| – | – | – | – | Mo1-Mo1 | −0.24 | −0.13 | 2.60 (O-H) | Mo1-Mo1 | −0.66 | −0.04 | 2.60 (O-H) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhajri, F.; Fadlallah, M.M.; Alkhaldi, A.; Maarouf, A.A. Hybrid MXene-Graphene/Hexagonal Boron Nitride Structures: Electronic and Molecular Adsorption Properties. Nanomaterials 2022, 12, 2739. https://doi.org/10.3390/nano12162739
Alhajri F, Fadlallah MM, Alkhaldi A, Maarouf AA. Hybrid MXene-Graphene/Hexagonal Boron Nitride Structures: Electronic and Molecular Adsorption Properties. Nanomaterials. 2022; 12(16):2739. https://doi.org/10.3390/nano12162739
Chicago/Turabian StyleAlhajri, Fawziah, Mohamed M. Fadlallah, Amal Alkhaldi, and Ahmed A. Maarouf. 2022. "Hybrid MXene-Graphene/Hexagonal Boron Nitride Structures: Electronic and Molecular Adsorption Properties" Nanomaterials 12, no. 16: 2739. https://doi.org/10.3390/nano12162739
APA StyleAlhajri, F., Fadlallah, M. M., Alkhaldi, A., & Maarouf, A. A. (2022). Hybrid MXene-Graphene/Hexagonal Boron Nitride Structures: Electronic and Molecular Adsorption Properties. Nanomaterials, 12(16), 2739. https://doi.org/10.3390/nano12162739